Tropospheric Ozone at Northern Mid-Latitudes: Modeled and Measured Long-Term Changes


Abstract
:1. Introduction
2. Ozone Measurements
3. Description of the SOCOL Chemistry-Climate Model and Simulations
3.1. The SOCOLv3 Chemistry-Climate Model
3.2. Simulations
3.3. Tropospheric Ozone Chemistry
H + O2 + M → HO2 + M
HO2 + NO → NO2 + OH
NO2 + hν → NO + O
O + O2 + M → O3 + M
∑ CO + 2O2 → CO2 + O3
O(1D) + H2O → 2OH
∑ O3 + H2O + hν → O2 + 2OH
H + O2 + M → HO2 + M
HO2 + O3 → OH + 2O2
∑ CO + O3 → CO2 + O2
HO2 + O3 → OH + 2O2
∑ 2O3 → 3O2
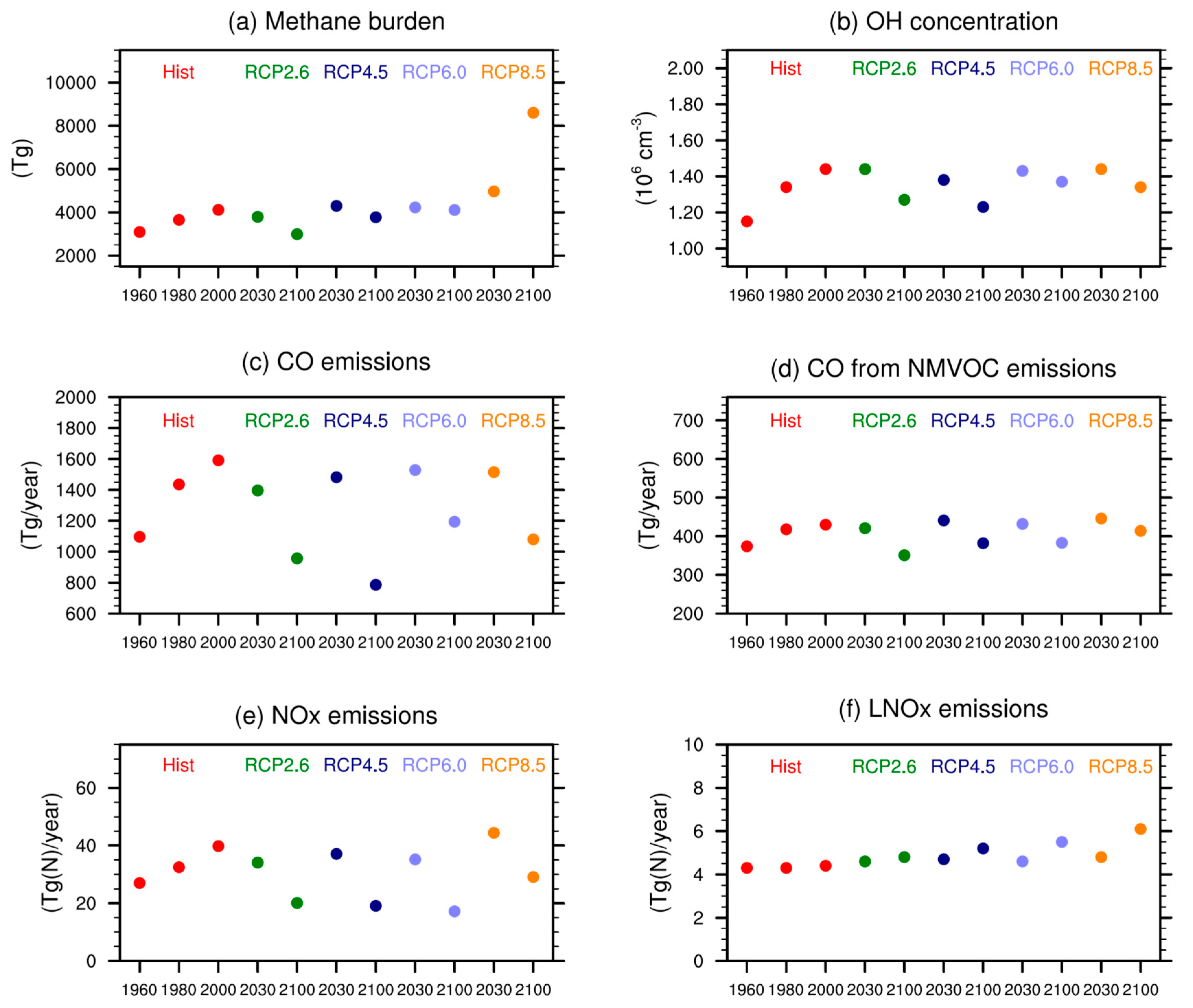
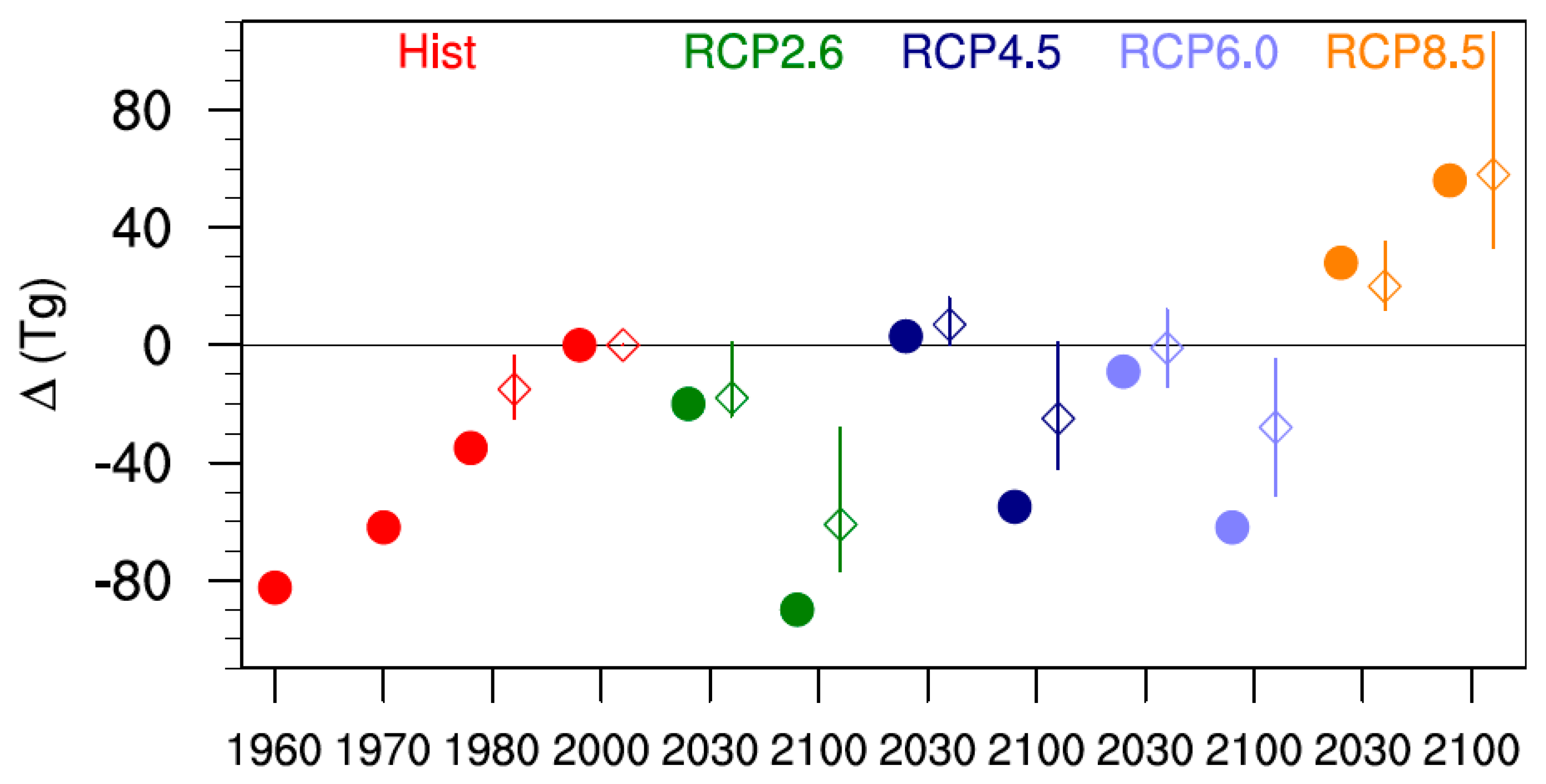
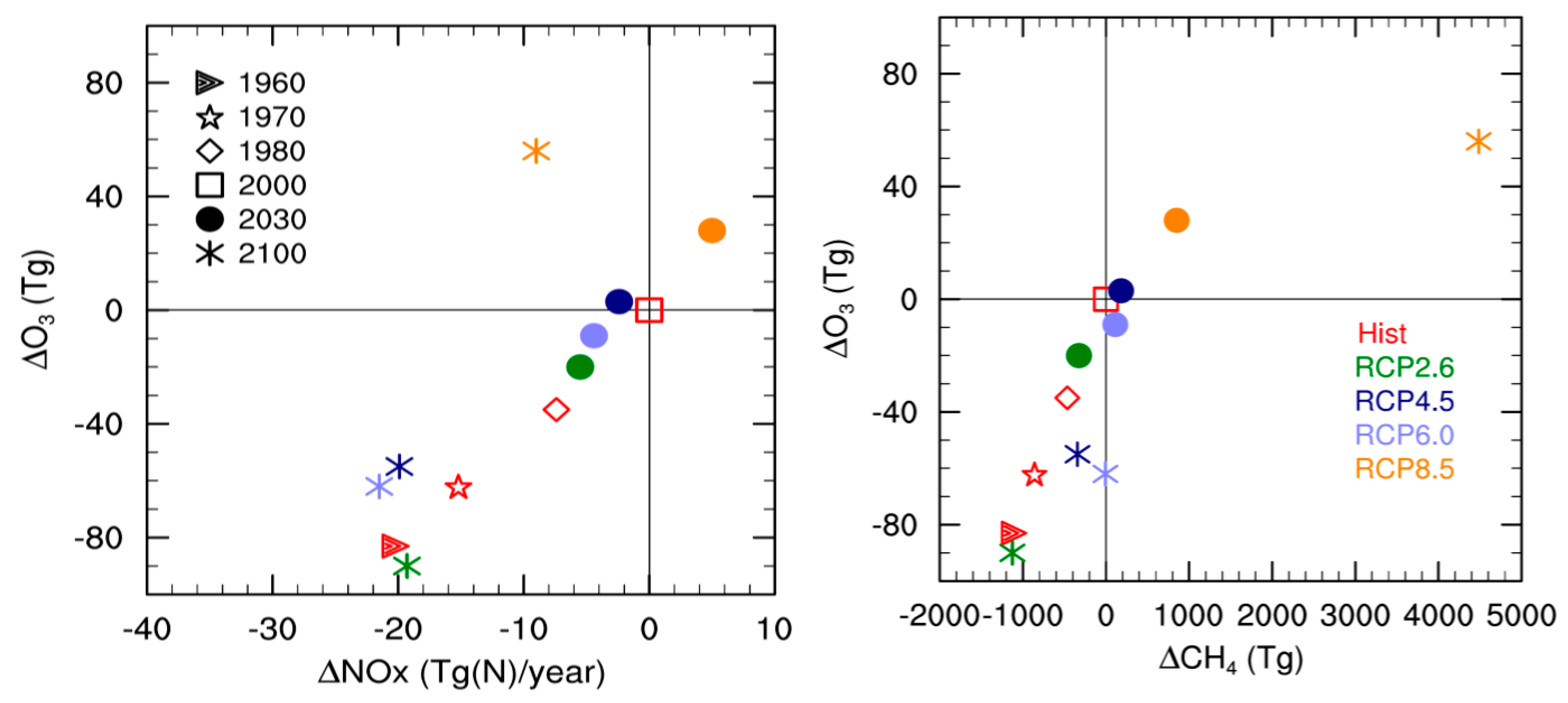
4. Tropospheric Ozone Budget: Comparison of SOCOLv3 with Other Models
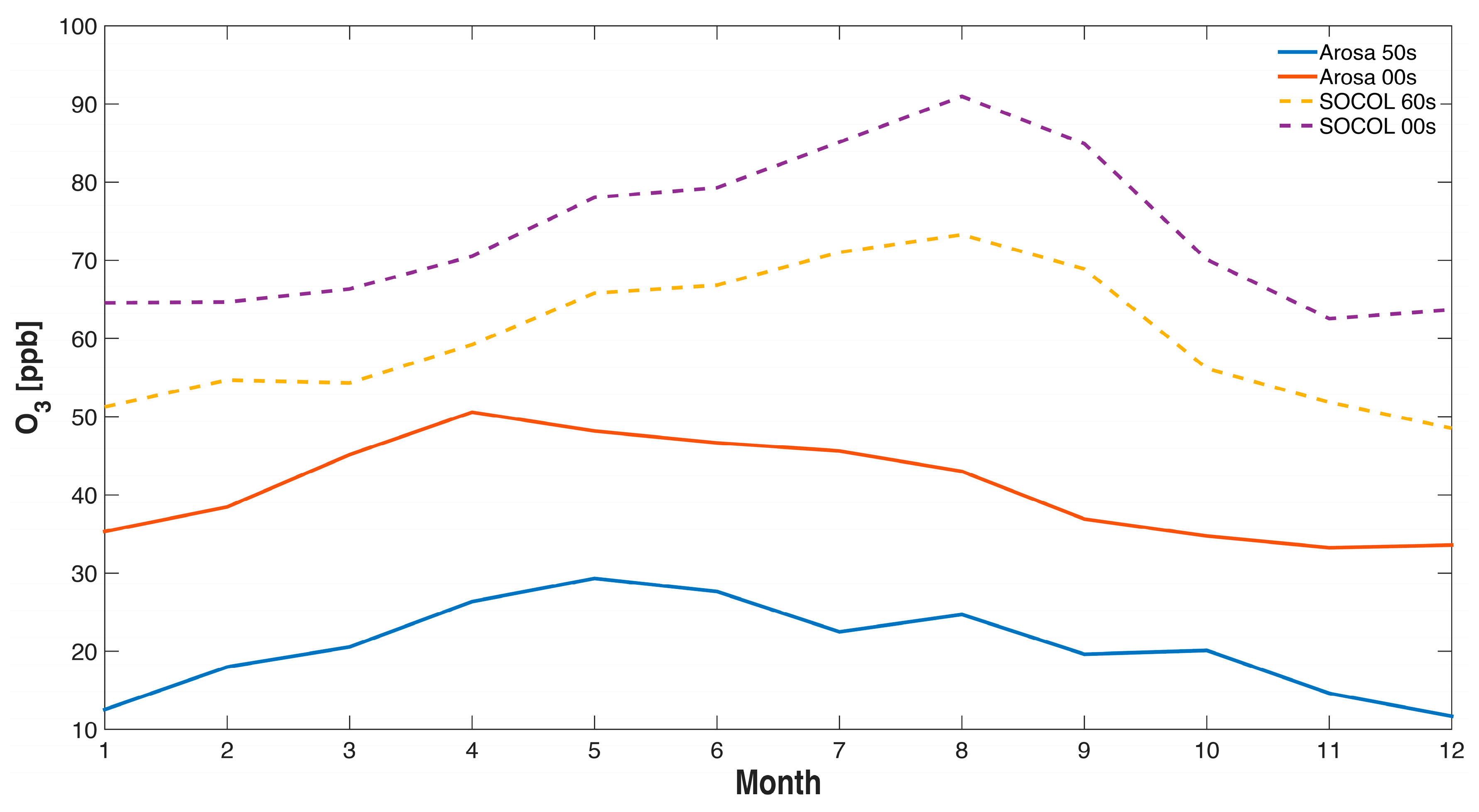
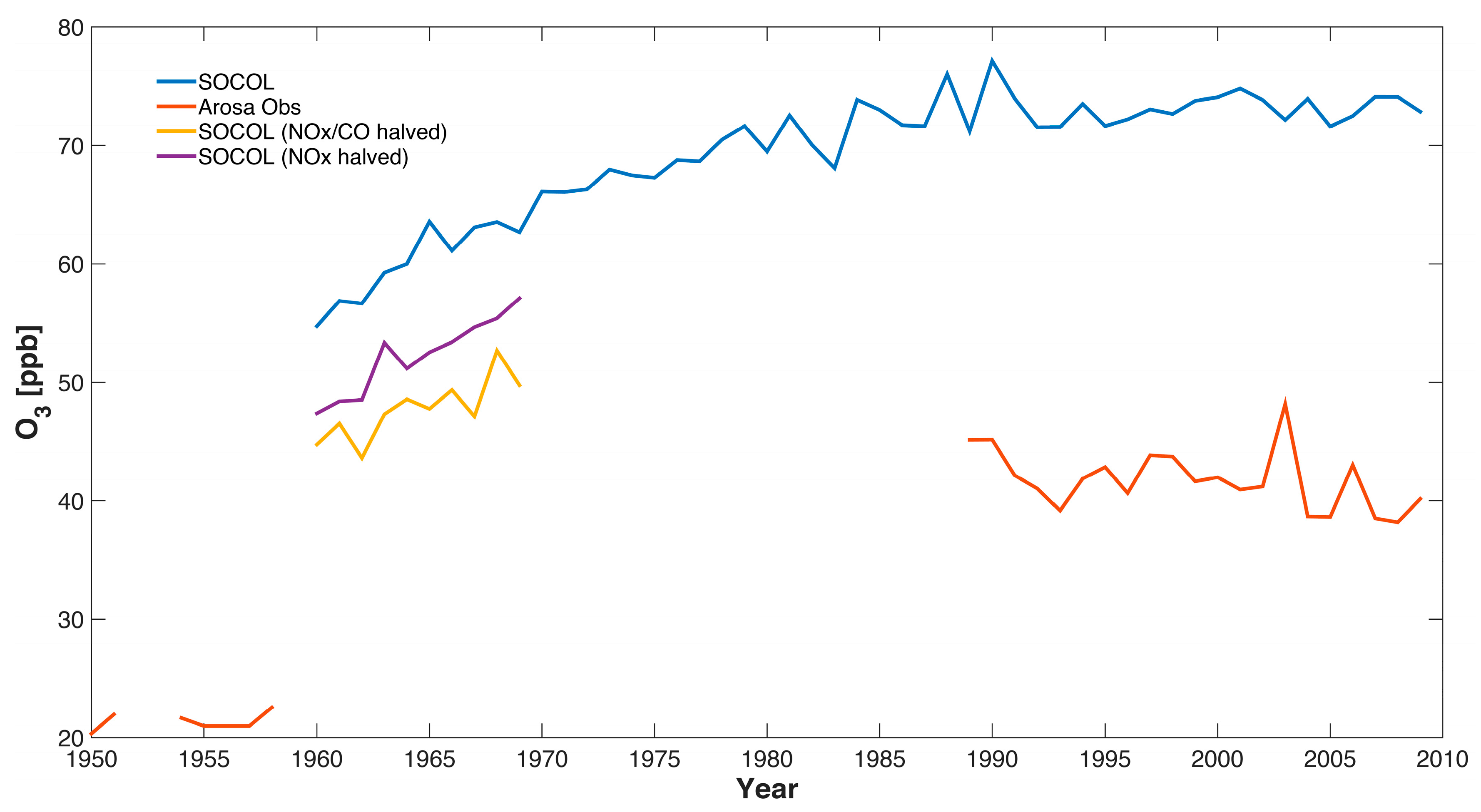
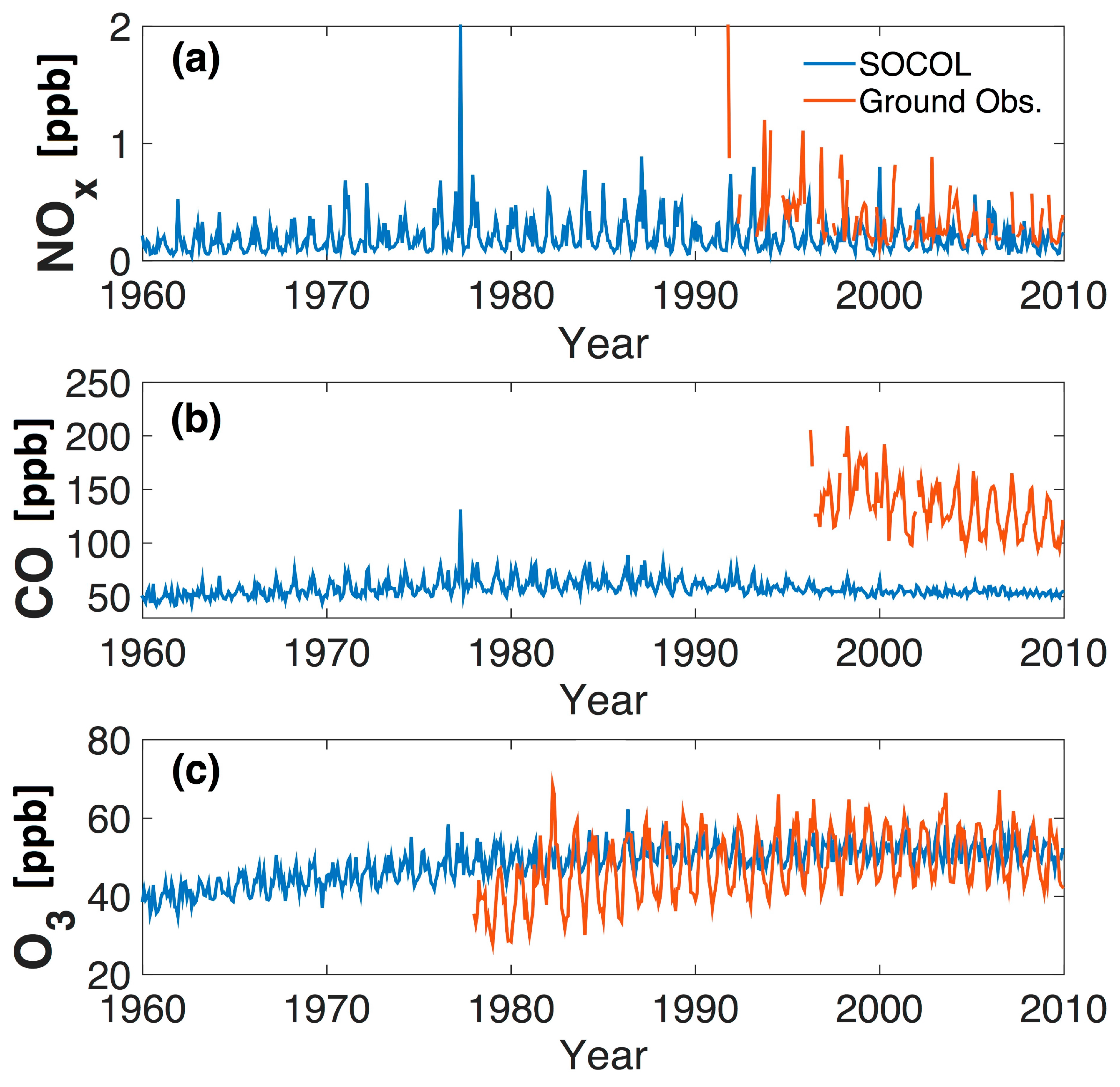
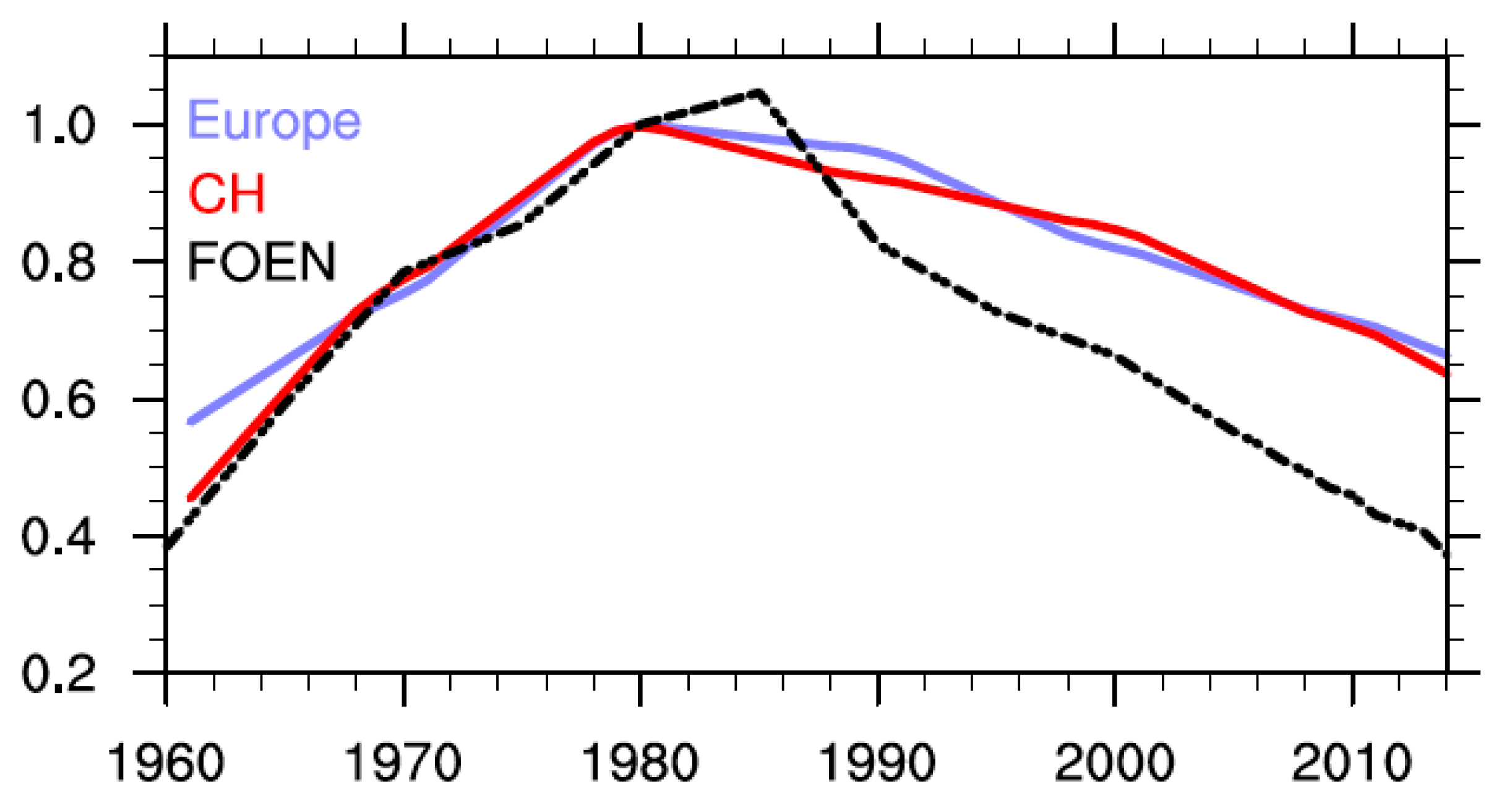
5. Comparison of SOCOL Simulations with Measurements
6. Discussion and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Grosjean, D. Ambient PAN and PPN in southern California from 1960 to the SCOS97-NASTRO. Atmos. Environ. 2003, 37 (Suppl. 2), S221–S238. [Google Scholar] [CrossRef]
- Haagen-Smit, A. Chemistry and Physiology of Los Angeles Angeles Smog. Ind. Eng. Chem. Res. 1952, 44, 1342–1346. [Google Scholar] [CrossRef]
- Martien, P.T.; Harley, R.A. Adjoint sensitivity analysis for a three dimensional photochemical model: Application to Southern California. Environ. Sci. Technol. 2006, 40, 4200–4210. [Google Scholar] [CrossRef] [PubMed]
- South Coast Air Quality Management District. Available online: http://www.aqmd.gov/home/library/air-quality-data-studies/historic-ozone-air-quality-trends (accessed on 21 August 2017).
- World Meteorological Organization: Scientific Assessment of Ozone Depletion: 2014; Report No. 55; WMO Global Ozone Research and Monitoring Project: Geneva, Switzerland, 2014.
- Stocker, T.F.; Qin, D. Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2013. [Google Scholar]
- Task Force on Hemispheric Transport of air pollution (HTAP). Available online: www.htap.org (accessed on 21 August 2017).
- Cooper, O.R.; Parrish, D.D.; Stohl, A.; Trainer, M.; Nédélec, P.; Thouret, V.; Cammas, J.P.; Oltmans, S.J.; Johnson, B.J.; Tarasick, D.; et al. Increasing springtime ozone mixing ratio in the free troposphere over western North America. Nature 2010, 463, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Fiore, A.M.; Dentener, F.J.; Wild, O.; Cuvelier, C.; Schultz, M.G.; Hess, P.; Textor, C.; Schulz, M.; Doherty, R.M.; Horowitz, W.; et al. Multimodel estimates of intercontinental source-receptor relationships for ozone pollution. J. Geophys. Res. 2009, 114, D04301. [Google Scholar] [CrossRef]
- Ordonez, C.; Brunner, D.; Staehelin, J.; Hadjinicolaou, P.; Pyle, J.A.; Jonas, M.; Wernli, H.; Prevot, A.S.H. Strong influence of lowermost stratospheric ozone on lower tropospheric background ozone changes over Europe. Geophys. Res. Lett. 2007, 34. [Google Scholar] [CrossRef]
- Young, P.J.; Archibald, A.T.; Bowman, K.W.; Lamarque, J.-F.; Naik, V.; Stevenson, D.S.; Tilmes, S.; Voulgarakis, A.; Wild, O.; Bergmann, D.; et al. Pre-industrial to end 21st century projections of tropospheric ozone from the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP). Atmos. Chem. Phys. 2013, 13, 2063–2090. [Google Scholar] [CrossRef] [Green Version]
- Lamarque, J.F.; Bond, T.C.; Eyring, V.; Granier, C.; Heil, A.; Klimont, Z.; Lee, D.; Liousse, C.; Mieville, A.; Owen, B.; et al. Historical (1850–2000) gridded anthropogenic and biomass burning emissions of reactive gases and aerosols: Methodology and application. Atmos. Chem. Phys. 2010, 10, 7017–7039. [Google Scholar] [CrossRef] [Green Version]
- Lamarque, J.-F.; Shindell, D.T.; Josse, B.; Young, P.J.; Cionni, I.; Eyring, V.; Bergmann, D.; Cameron-Smith, P.; Collins, W.J.; Doherty, R.; et al. The Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP): Overview and description of models, simulations and climate diagnostics. Geosci. Model Dev. 2013, 6, 179–206. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, D.S.; Young, P.J.; Naik, V.; Lamarque, J.-F.; Shindell, D.T.; Voulgarakis, A.; Skeie, R.B.; Dalsoren, S.B.; Myhre, G.; Berntsen, T.K.; et al. Tropospheric ozone changes, radiative forcing and attribution to emissions in the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP). Atmos. Chem. Phys. 2013, 13, 3063–3085. [Google Scholar] [CrossRef] [Green Version]
- Parrish, D.D.; Lamarque, J.-F.; Naik, V.; Horowitz, L.; Shindell, D.T.; Staehelin, J.; Derwent, R.; Cooper, O.R.; Tanimoto, H.; Volz-Thomas, A.; et al. Long-term changes in lower tropospheric baseline ozone concentrations: Comparing chemistry-climate models and observations at northern mid-latitudes. J. Geophys. Res. 2014, 119, 5719–5736. [Google Scholar] [CrossRef]
- Parrish, D.D.; Law, K.S.; Staehelin, J.; Derwent, R.; Cooper, O.R.; Tanimoto, H.; Volz Thomas, A.; Gilge, S.; Scheel, H.-E.; Steinbacher, M.; et al. Long-term changes in lower tropospheric baseline ozone concentrations at northern mid-latitudes. Atmos. Chem. Phys. 2012, 12, 11485–11504. [Google Scholar] [CrossRef] [Green Version]
- Parrish, D.D.; Law, K.S.; Staehelin, J.; Derwent, R.; Cooper, O.R.; Tanimoto, H.; Volz Thomas, A.; Gilge, S.; Scheel, H.-E.; Steinbacher, M.; et al. Lower tropospheric ozone at northern mid-latitudes: Changing seasonal cycle. Geophys. Res. Lett. 2013, 40, 1631–1636. [Google Scholar] [CrossRef]
- Revell, L.E.; Tummon, F.; Stenke, A.; Sukhodolov, T.; Coulon, A.; Rozanov, E.; Garny, H.; Grewe, V.; Peter, T. Drivers of the tropospheric ozone budget throughout the 21st century under the medium-high climate scenario RCP 6.0. Atmos. Chem. Phys. 2015, 15, 5887–5902. [Google Scholar] [CrossRef] [Green Version]
- Stenke, A.; Schraner, M.; Rozanov, E.; Egorova, T.; Luo, B.; Peter, T. The SOCOL version 3.0 chemistry-climate model: Description, evaluation, and implications from an advanced transport algorithm. Geosci. Model Dev. 2013, 6, 1407–1427. [Google Scholar] [CrossRef]
- Tummon, F.; Revell, L.; Stenke, A.; Staehelin, J.; Peter, T. Diagnosing Changes in European Free Tropospheric Ozone Over the Past 35 Years. 2017; in preparation. [Google Scholar]
- TOAR. Available online: http://www.igacproject.org/activities/TOAR (accessed on 21 August 2017).
- Rubin, M.B. The history of ozone. The Schönbein period, 1839–1868. Bull. Hist. Chem. 2001, 26, 40–56. [Google Scholar]
- Wolf, R. Ozongehalt der Luft und seinen Zusammenhang mit der Mortalität. In Vorträge in der Bernischen Naturforschenden Gesellschaft, Bern; Verlag Huber & Comp.: Bern, Switzerland, 1855. [Google Scholar]
- Fox, C.B. Ozone and Antozone; Churchill Publisher: London, UK, 1873. [Google Scholar]
- Kley, D.; Volz, A.; Mülheim, F. Ozone measurements in historic perspective. In Tropospheric Ozone; Isaksen, I.S.A., Ed.; D. Reidel: Kufstein, Österreich, 1988; pp. 63–73. [Google Scholar]
- Pavelin, E.G.; Johnson, C.E.; Rughooputh, S.; Toumi, R. Evaluation of pre-industrial surface ozone measurements made using Schönbein’s method. Atmos. Environ. 1999, 33, 919–929. [Google Scholar] [CrossRef]
- Volz, A.; Kley, D. Evaluation of the Montsouris series of ozone measurements made in the nineteenth century. Nature 1988, 332, 240–242. [Google Scholar] [CrossRef]
- Staehelin, J.; Brönnimann, S.; Peter, T.; Stübi, R.; Viatte, P.; Tummon, F. The value of Swiss long-term ozone observations for international atmospheric research. In From Weather Observations to Atmospheric and Climate Sciences in Switzerland—Celebrating 100 Years of the Swiss Society for Meteorology; Willemse, S., Furger, M., Eds.; vdf Hochschulverlag AG: Zürich, Switzerland, 2016; pp. 325–349. [Google Scholar]
- Staehelin, J.; Thudium, J.; Bühler, R.; Volz-Thomas, A.; Graber, W. Surface ozone trends at Arosa (Switzerland). Atmos. Environ. 1994, 28, 75–87. [Google Scholar] [CrossRef]
- Derwent, R.G.; Simmonds, P.G.; Manning, A.J.; Spain, T.G. Trends over a 20-year period from 1987 to 2007 in surface ozone at the atmospheric research station, Mace Head, Ireland. Atmos. Environ. 2007, 41, 9091–9098. [Google Scholar] [CrossRef]
- Cui, J.; Pandey Deolal, S.; Sprenger, M.; Henne, S.; Staehelin, J.; Steinbacher, M.; Nedelec, P. Free tropospheric ozone changes over Europe as observed at Jungfraujoch (1990–2008): An analysis based on backward trajectories. J. Geophys. Res. 2011, 116, D10304. [Google Scholar] [CrossRef]
- Atmospheric Ozone 1985, Assessment of Our Understanding of the Processes Controlling Its Present Distribution and Change; Global Ozone Research and Monitoring Project 16; World Meteorological Organization (WMO): Geneva, Switzerland, 1985; Volume II, pp. 410–411.
- Logan, J.A.; Staehelin, J.; Megretskaia, I.A.; Cammas, J.-P.; Thouret, V.; Claude, H.; De Backer, H.; Steinbacher, M.; Scheel, H.-E.; Stübi, R.; et al. Changes in ozone over Europe: Analysis of ozone measurements from sondes, regular aircraft (MOZAIC) and alpine surface sites. J. Geophys. Res. 2012, 117, D09301. [Google Scholar] [CrossRef]
- Smit, H. The Panel for Assessment of Standard Operating Procedures for Ozonsondes (ASOPOS), 2011: Quality Assurance and Quality Control for Ozonesonde Measurements in GAW; GAW Report 201; World Meteorological Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Wild, O. Modelling the global tropospheric ozone budget: Exploring the variability in current models. Atmos. Chem. Phys. 2007, 7, 2643–2660. [Google Scholar] [CrossRef]
- Schnadt Poberaj, C.; Staehelin, J.; Brunner, D.; Thouret, V.; Mohnen, V. A UT/LS ozone climatology of the nineteen seventies deduced from the GASP aircraft measurement program. Atmos. Chem. Phys. 2007, 7, 5917–5936. [Google Scholar] [CrossRef]
- Dias-Lalcaca, P.; Brunner, D.; Imfeld, W.; Moser, W.; Staehelin, J. An automated system for the measurement of nitrogen oxides and ozone concentrations from a passenger aircraft: Instrumentation and first results of the project NOXAR. Environ. Sci. Technol. 1998, 32, 3228–3236. [Google Scholar] [CrossRef]
- Brunner, D.; Staehelin, J.; Jeker, D.; Wernli, H.; Schumannm, U. Nitrogen oxides and ozone in the tropopause region of the Northern Hemisphere: Measurements from commercial aircraft in 1995/96 and 1997. J. Geophys. Res. 2001, 106, 27673–27699. [Google Scholar] [CrossRef]
- Thouret, V.; Cammas, J.-P.; Sauvage, B.; Athier, B.; Zbinden, R.M.; Nédélec, P.; Simmon, P.; Karcher, F. Tropopause referenced ozone climatology and inter-annual variability (1994–2003) from MOZAIC programme. Atmos. Chem. Phys. 2006, 6, 1033–1051. [Google Scholar] [CrossRef]
- Brenninkmeijer, C.A.M.; Crutzen, P.; Boumard, F.; Dauer, T.; Dix, B.; Ebinghaus, R.; Filippi, D.; Fischer, H.; Franke, H.; Frie, B.; et al. Civil Aircraft for the regular investigation of the atmosphere based on an instrumented container: The new CARIBIC system. Atmos. Chem. Phys. 2007, 7, 4953–4976. [Google Scholar] [CrossRef] [Green Version]
- IAGOS Data Portal. Available online: http://iagos.sedoo.fr/ (accessed on 21 August 2017).
- Schnadt Poberaj, C.; Staehelin, J.; Brunner, D.; Thouret, V.; De Backer, H.; Stübi, R. Long-term changes in UT/LS ozone between the late 1970s and the 1990s deduced from the GASP and MOZAIC aircraft programs and from ozonesonde. Atmos. Chem. Phys. 2009, 9, 5343–5369. [Google Scholar] [CrossRef]
- Staufer, J.; Staehelin, J.; Stübi, R.; Peter, T.; Tummon, F.; Thouret, V. Trajectory matching of ozonesondes and MOZAIC measurements in the UTLS, Part II: Application to the global ozonesonde network. Atmos. Meas. Technol. 2014, 7, 241–266. [Google Scholar] [CrossRef] [Green Version]
- Van der A, R.J.; Eskes, H.J.; Boersma, K.F.; van Noije, T.P.C.; Van Roozendael, M.; De Smedt, I.; Peters, D.H.M.U.; Meijer, E.W. Trends, seasonal variability and dominant NOx source derived for a ten year record of NO2 measured from space. J. Geophys. Res. 2008, 113, D04302. [Google Scholar] [CrossRef]
- Oltmans, S.J.; Lefohn, A.S.; Harris, J.M.; Galbally, J.; Scheel, H.S.; Bodecker, G.; Brunke, E.; Claude, H.; Tarasick, D.; Johnson, B.J.; et al. Long-term changes in tropospheric ozone. Atmos. Environ. 2006, 40, 3156–3173. [Google Scholar] [CrossRef]
- Cooper, O.R.; Parrish, D.D.; Ziemke, J.; Balashov, N.V.; Cupeiro, M.; Galbally, I.E.; Gilge, S.; Horowitz, L.; Jensen, N.R.; Lamarque, J.F.; et al. Global distribution and trends of tropospheric ozone: An observation based review. Elementa 2014, 2, 000029. [Google Scholar] [CrossRef]
- Roeckner, E.; Bäuml, G.; Bonaventura, L.; Brokopf, R.; Esch, M.; Giorgetta, M.; Hagemann, S.; Kirchner, I.; Kornblueh, L.; Manzini, E.; et al. The Atmospheric General Circulation Model ECHAM 5. Part I: Model Description; Report No. 349; Max-Planck-Institut für Meteorologie: Hamburg, Germany, 2003; Available online: http://pubman.mpdl.mpg.de/pubman/faces/viewItemFullPage.jsp?itemId=escidoc:995269:2 (accessed on 4 March 2017).
- Egorova, T.A.; Rozanov, E.V.; Zubov, V.A.; Karol, I.L. Model for investigating ozone trends (MEZON). Izv. Atmos. Ocean. Phys. 2003, 39, 277–292. [Google Scholar]
- Chang, J.S.; Brost, R.A.; Isaksen, I.S.A.; Madronich, S.; Middleton, P.; Stockwell, W.R.; Walcek, C.J. A three dimensional Eulerian acid deposition model: Physical concepts and formulation. J. Geophys. Res. 1987, 92, 14681–14700. [Google Scholar] [CrossRef]
- Poeschl, U.; von Kuhlmann, R.; Poisson, N.; Crutzen, P.J. Development and intercomparison of condensed isoprene oxidation mechanisms for global atmospheric modeling. J. Atmos. Chem. 2000, 37, 29–52. [Google Scholar] [CrossRef]
- Ehhalt, D.; Prather, M.; Dentener, F.; Derwent, R.; Dlugokencky, E.; Holland, E.; Isaksen, I.; Katima, J.; Kirchhoff, V.; Matson, P.; et al. Atmospheric chemistry and greenhouse gases. In Climate Change 2001: The Scientific Basis. Contribution of Working Group I to the Third Assessment Report of the Intergovernmental Panel on Climate Change; Houghton, J.T., Ding, Y., Griggs, D.J., Noguer, M., van der Linden, P.J., Dai, X., Maskell, K., Johnson, C.A., Eds.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2001. [Google Scholar]
- Price, C.; Rind, D. A simple lightning parameterization for calculating global lightning distributions. J. Geophys. Res. 1992, 97, 9919–9933. [Google Scholar] [CrossRef]
- Grewe, V. The origin of ozone. Atmos. Chem. Phys. 2006, 6, 1495–1511. [Google Scholar] [CrossRef]
- Garny, H.; Grewe, V.; Dameris, M.; Bodeker, G.E.; Stenke, A. Attribution of ozone changes to dynamical and chemical processes in CCMs and CTMs. Geosci. Model Dev. 2011, 4, 271–286. [Google Scholar] [CrossRef] [Green Version]
- Meinshausen, M.; Smith, S.J.; Calvin, K.; Daniel, J.S.; Kainuma, M.L.T.; Lamarque, J.-F.; Matsumoto, K.; Montzka, S.A.; Raper, S.C.B.; Riahi, K.; et al. The RCP greenhouse gas concentrations and their extensions from 1765 to 2300. Clim. Chang. 2011, 109, 213–241. [Google Scholar] [CrossRef]
- Riahi, K.; Rao, S.; Krey, V.; Cho, C.; Chirkov, V.; Fischer, G.; Kindermann, G.; Nakicenovic, N.; Rafaj, P. RCP 8.5—A scenario of comparatively high greenhouse gas emissions. Clim. Chang. 2011, 109, 33–57. [Google Scholar] [CrossRef]
- Rayner, N.A.; Parker, D.E.; Horton, E.B.; Folland, C.K.; Alexander, L.V.; Rowell, D.P.; Kent, E.C.; Kaplan, A. Global analyses of sea surface temperature, sea ice, and night marine air temperature since the late nineteenth century. J. Geophys. Res. 2003, 108, 4407. [Google Scholar] [CrossRef]
- Van Vuuren, D.P.; Stehfest, E.; den Elzen, M.G.J.; Kram, T.; van Vliet, J.; Deetman, S.; Isaac, M.; Klein Goldewijk, K.; Hof, A.; Mendoza Beltram, A.; et al. RCP2.6: Exploring the possibility to keep global mean temperature increase below 2 °C. Clim. Chang. 2011, 109, 95–116. [Google Scholar] [CrossRef]
- Meehl, G.A.; Washington, W.M.; Arblaster, J.M.; Hu, A.; Teng, H.; Kay, J.E.; Gettelman, A.; Lawrence, D.M.; Sanderson, B.M.; Strand, W.G. Climate change projections in CESM1(CAM5) compared to CCSM4. J. Clim. 2013, 26, 6287–6308. [Google Scholar] [CrossRef]
- Thomson, A.M.; Calvin, K.V.; Smith, S.J.; Page Kyle, G.; Volke, A.; Patel, P.; Delgado-Arias, S.; Bond-Lamberty, B.; Wise, M.A.; Clarke, L.E.; et al. RCP 4.5: A pathway for stabilization of radiative forcing by 2100. Clim. Chang. 2011, 109, 77–94. [Google Scholar] [CrossRef]
- Masui, T.; Matsumoto, K.; Hijioka, Y.; Kinoshita, T.; Nozawa, T.; Ishiwatari, S.; Kato, E.; Shukla, P.R.; Yamagata, Y.; Kainuma, M. An emission pathway for stabilization at 6 Wm−2 radiative forcing. Clim. Chang. 2011, 109, 59–76. [Google Scholar] [CrossRef]
- Morgenstern, O.; Hegglin, M.I.; Rozanov, E.; O’Connor, F.M.; Abraham, N.L.; Akiyoshi, H.; Archibald, A.T.; Bekki, S.; Butchart, N.; Chipperfield, M.P.; et al. Review of the global models used within phase 1 of the Chemistry–Climate Model Initiative (CCMI). Geosci. Model Dev. 2017, 10, 639–671. [Google Scholar] [CrossRef]
- Van Vuuren, D.P.; Edmonds, J.; Kainuma, M.; Riahi, K.; Thomson, A.; Hibbard, K.; Hurtt, C.G.; Kram, T.; Krey, V.; Lamarque, J.-F.; et al. The representative concentration pathways: An overview. Clim. Chang. 2011, 109, 5–31. [Google Scholar] [CrossRef]
- Scientific Assessment of Ozone Depletion: 2010; Global Ozone Research and Monitoring Project—Report No. 52; World Meteorological Organization (WMO): Geneva, Switzerland, 2011.
- Arfeuille, F.; Luo, B.P.; Heckendorn, P.; Weisenstein, D.; Sheng, J.X.; Rozanov, E.; Schraner, M.; Brönnimann, S.; Thomason, L.W.; Peter, T. Modeling the stratospheric warming following the Mt. Pinatubo eruption: Uncertainties in aerosol extinctions. Atmos. Chem. Phys. 2013, 13, 11221–11234. [Google Scholar]
- Luo, B.P. Stratospheric Aerosol Data for Use in CCMI Models. 2013. Available online: ftp://iacftp.ethz.ch/pub_read/luo/ccmi/ (accessed on 23 August 2017).
- Stevenson, D.S.; Dentener, F.J.; Schultz, M.G.; Ellingsen, K.; van Noije, T.P.C.; Wild, O.; Zeng, G.; Amann, M.; Atherton, M.; Bell, N.; et al. Multimodel ensemble simulations of present-day and near-future tropospheric ozone. J. Geophys. Res. 2006, 111, D08301. [Google Scholar] [CrossRef]
- Naik, V.; Voulgarakis, A.; Fiore, A.M.; Horowitz, L.W.; Lamarque, J.-F.; Lin, M.; Prather, M.J.; Young, P.J.; Bergmann, D.; Cameron-Smith, P.J.; et al. Preindustrial to present-day changes in tropospheric hydroxyl radical and methane lifetime from the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP). Atmos. Chem. Phys. 2013, 13, 5277–5298. [Google Scholar] [CrossRef] [Green Version]
- Wild, O.; Palmer, P.I. How sensitive is tropospheric oxidation to anthropogenic emissions? Geophys. Res. Lett. 2008, 35, L22802. [Google Scholar] [CrossRef]
- Henne, S.; Dommen, D.; Neininger, B.; Reimann, S.; Staehelin, J.; Prévôt, A.S.H. Influence of mountain venting in the Alps on the ozone chemistry of the lower free troposphere and the European pollution export. J. Geophys. Res. 2005, 110, D22307. [Google Scholar] [CrossRef]
- Voulgarakis, A.; Naik, V.; Lamarque, J.-F.; Shindell, D.T.; Young, P.J.; Prather, M.J.; Wild, O.; Field, R.D.; Bergmann, D.; Cameron-Smith, P.; et al. Analysis of present day and future OH and methane lifetime in the ACCMIP simulations. Atmos. Chem. Phys. 2013, 13, 2563–2587. [Google Scholar] [CrossRef] [Green Version]
- Vom Menschen Verursachte Luftschadstoff-Emissionen in der Schweiz von 1900 bis 2010; Schriftenreihe Umwelt No. 255; Bundesamt für Umwelt, Wald und Landschaft (BUWAL/FOEN): Bern, Switzerland, 1995.
- Hassler, B.; McDonald, B.C.; Frost, G.J.; Borbon, A.; Carslaw, D.C.; Civerolo, K.; Granier, C.; Monks, P.S.; Monks, S.; Parrish, D.D.; et al. Analysis of long-term observation of NOx and CO in megacities and application to constraining emission inventories. Geophys. Res. Lett. 2016, 43, 9920–9930. [Google Scholar] [CrossRef]
- Van Aardenne, J.A.; Dentener, F.J.; Olivier, J.G.J.; Klein Goldewijk, C.G.M.; Lelieveld, J. A 1ox1o resolution data set of historical anthropogenic trace gas emissions for the period 1890–1990. Glob. Beogeochem. Cycles 2001, 15, 909–928. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Period | Flight Routes | Species |
|---|---|---|---|
| GASP | 1975–1979 | Pacific, Europe (Far East) | O3, others questionable |
| MOZAIC/IAGOS | Since 1994 | Various, starting from Europe | O3, H2O, and starting in 2001 also NOy (NOx + HNO3) and others |
| NOXAR | 1995/96, 97 | Zürich, Switzerland, to USA and Far East | O3 and NOx (=NO + NO2) |
| CARIBIC | Since 1997 | Various, from Frankfurt, Germany, monthly frequency | O3 and a large number of other species |
| Simulation | Period | Greenhouse Gases (CO2, N2O, CH4, ODSs) | Ozone Precursor Emissions (NOx, CO, NMVOCs) | Sea Surface Temperatures |
|---|---|---|---|---|
| REF-C1 | 1960–2010 | Observations until 2005 a then RCP 8.5 b | Historical emissions until 2000 c, then RCP6.0 | HadISST1 observations d |
| RCP 2.6 | 2000–2100 | Observations until 2005 then RCP 2.6 e | RCP 2.6 | CESM1(CAM5) f RCP 2.6 |
| RCP 4.5 | 2000–2100 | Observations until 2005 then RCP 4.5 g | RCP 4.5 | CESM1(CAM5) RCP 4.5 |
| RCP 6.0 | 1960–2100 | Observations until 2005 then RCP 6.0 h | Historical emissions until 2000 c, then RCP 6.0 | CESM1(CAM5) RCP 6.0 |
| RCP 8.5 | 2000–2100 | Observations until 2005 then RCP 8.5 b | RCP 8.5 | CESM1(CAM5) RCP 8.5 |
| NOx halved | 1960–1970 | As REF-C1 | As REF-C1, but NOx emissions halved | As REF-C1 |
| NOx & CO halved | 1960–1970 | As REF-C1 | As REF-C1, but NOx and CO emissions halved | As REF-C1 |
| Model | Burden (Tg(O3)) | Flux Terms (Tg(O3) year−1) | τ (Days) | |||
|---|---|---|---|---|---|---|
| P | L | D | S | |||
| SOCOL | 413 | 6857 a | 5597 b | 1500 | 240 (491 c) | 22.1 |
| ACCMIP | 337 ± 23 | 4877 ± 853 | 4260 ± 645 | 1094 ± 264 | 477 ± 96 | 23.4 ± 2.2 |
| ACCENT | 336 ± 27 | 5110 ± 606 | 4668 ± 27 | 1003 ± 200 | 552 ± 168 | 22.3 ± 2.0 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Staehelin, J.; Tummon, F.; Revell, L.; Stenke, A.; Peter, T. Tropospheric Ozone at Northern Mid-Latitudes: Modeled and Measured Long-Term Changes. Atmosphere 2017, 8, 163. https://doi.org/10.3390/atmos8090163
Staehelin J, Tummon F, Revell L, Stenke A, Peter T. Tropospheric Ozone at Northern Mid-Latitudes: Modeled and Measured Long-Term Changes. Atmosphere. 2017; 8(9):163. https://doi.org/10.3390/atmos8090163
Chicago/Turabian StyleStaehelin, Johannes, Fiona Tummon, Laura Revell, Andrea Stenke, and Thomas Peter. 2017. "Tropospheric Ozone at Northern Mid-Latitudes: Modeled and Measured Long-Term Changes" Atmosphere 8, no. 9: 163. https://doi.org/10.3390/atmos8090163
APA StyleStaehelin, J., Tummon, F., Revell, L., Stenke, A., & Peter, T. (2017). Tropospheric Ozone at Northern Mid-Latitudes: Modeled and Measured Long-Term Changes. Atmosphere, 8(9), 163. https://doi.org/10.3390/atmos8090163