1. Introduction
Atmospheric aerosols are solid or liquid particles suspended in air and are comprised of mixtures of organic and/or inorganic species: organic molecules, salts, soot, minerals, and metals [
1]. Aerosols have highly uncertain effects on radiative forcing [
2]. Aerosol forcing occurs via two mechanisms: light can be scattered or absorbed directly by the aerosol particles (the “direct effect”, also aerosol-radiation interactions) or indirectly through aerosol effects on cloud properties (the “indirect effect”, also aerosol-cloud interactions) [
3]. The latest Intergovernmental Panel on Climate Change (IPCC) report, released in 2013, shows that the extent of anthropogenic effects on cloud formation is currently the largest source of uncertainty for predictive understanding of global anthropogenic radiative forcing [
2]. Both direct and indirect effects are heavily influenced by the composition of aerosols on a per-particle level [
4,
5,
6]. To better understand and predict the influence of industrialization, one aspect of particular interest is the effect that anthropogenic emissions have on the per-particle composition of aerosols and their impacts on local and global climate [
2,
7].
One underlying reason for this uncertainty is the complex manner in which aerosol composition changes over time and distance through coagulation, condensation, and chemical reaction [
8]. Because aerosol radiative forcing and cloud formation depend on the individual particle composition, it is important to know how atmospheric components are mixed within a population of aerosols. How these components are mixed plays a large role in determining the manner and extent to which radiative forcing is affected. For example, the coating of soot by organics can change the direct radiative forcing of those aerosols by as much as a factor of 2.4 over pure soot [
5,
9,
10,
11]. Hence, in this case, it is important to know whether soot and organics coexist in the same aerosol particle. How components are mixed in an aerosol sample is referred to as its mixing state. This mixing state can range anywhere from an internal mixture where each component is evenly distributed throughout all particles, to an external mixture where each component occupies its own population of particles. Many atmospheric models assume one of these extremes throughout their simulation [
12,
13,
14]. Some models include a specific aspect of aerosol mixing such as the mixing state of black carbon [
4,
15], while other, nascent, models will account for a more complete mixing state [
16]. Mixing state values for coated black carbon (BC) have been determined using a single-particle soot photometer (SP2) based on the time delay between light scattering and soot incandescence but thermodynamic properties of organic coatings must be assumed to infer coating thicknesses, making the technique qualitative [
17,
18]. This approach also becomes less applicable if inorganic dominant or non-soot containing particles are of interest. A real-time method for determining aerosol mixing state index has been achieved by using single particle mass spectrometry [
19], although this technique is blind to detailed aerosol morphology.
Recently, more nuanced metrics were developed to quantify the mixing state of a population of aerosols [
20]. Here, Riemer and West utilize an information theoretic approach to determine specific mixing states in populations of aerosols. Particle-specific mass fractions are used to calculate both bulk and individual particle diversity parameters. The mixing state of a population is then calculated from the ratio of individual and bulk diversities. This method of mixing state determination necessitates a mass quantitative method of determining per-particle composition. Spectromicroscopy techniques are uniquely suited to analyze both the morphology and the comprehensive mixing state of a population of aerosols. Here, these quantitative mixing state metrics are applied to microscopy images of particle samples collected in the central Amazon basin.
In this study, we determine the mass fractions of 14 elements on the exact same set of particles using the complementary techniques of Scanning Transmission X-ray Microscopy with Near-Edge X-ray Absorption Fine Structure spectroscopy (STXM/NEXAFS) and Scanning Electron Microscopy coupled with Computer Controlled Energy Dispersive X-ray spectroscopy (SEM/EDX). Each technique is limited in which elements it can investigate. STXM/NEXAFS is limited by the energy range of the synchrotron insertion device as well as the beamtime available for sample analysis. STXM/NEXAFS has the advantage of providing quantitative measurements of light, low Z (atomic number) elements (C, N, and O), as well as some heavier elements with L-shell absorption edges in the same energy range (e.g., K and Ca) [
21,
22]. Although SEM/EDX provides a faster method of per-particle spectromicroscopy, it is only considered quantitative for higher Z elements (Z > 11, Na) [
7,
23,
24]. These two techniques are inherently complementary, with each technique providing mass information on elements that the other cannot adequately probe and both providing this information on an individual particle level. Both techniques have been used in tandem on microscopy samples previously [
25]. In that study, O’Brien et al. [
25] used STXM/NEXAFS and SEM/EDX to characterize individual particles from northern California. From this, mixing state parameters were calculated; however, because STXM and SEM were conducted on different particles within a given sample, separate mixing states were calculated from each technique. The current work combines STXM and SEM data at the single particle level in a similar way to Piens et al., 2016 [
26], where both techniques were used together to determine hygroscopicity of individual particles. The per-particle elemental mass fractions determined herein are used to calculate an elemental mixing state for particles collected at three sampling sites.
Aerosol production in the Amazon basin plays an important role in global climate due to the large scale of biogenic emissions from the tropical forest often mixed with pollutants from vegetation fires (mostly related to deforestation and pasture burning) [
27,
28,
29,
30]. South America contributes significantly to the global aerosol carbon budget; ~17% of global soot emissions are produced in Central and South America combined [
31]. Aerosols are also subject to long range transport and thus are of importance to global models [
32]. This environmentally important region of Central Amazonia contains Manaus, a city with over two million people. Manaus is a large industrial manufacturing city as a consequence of its free trade status since the 1960s. The juxtaposition of pristine rainforest with a large anthropogenic center presents a unique circumstance for studying how native biogenic aerosols are affected by emissions from an industrial city [
33]. To take advantage of this unique location, the Observations and Modeling of the Green Ocean Amazon (GoAmazon2014/5) field campaign was conducted from January 2014 through December 2015 [
34,
35]. The GoAmazon campaign was developed with multiple scientific objectives, two of which involve the biogenic and anthropogenic interactions studied here.
2. Experiments
2.1. Sampling Site Description
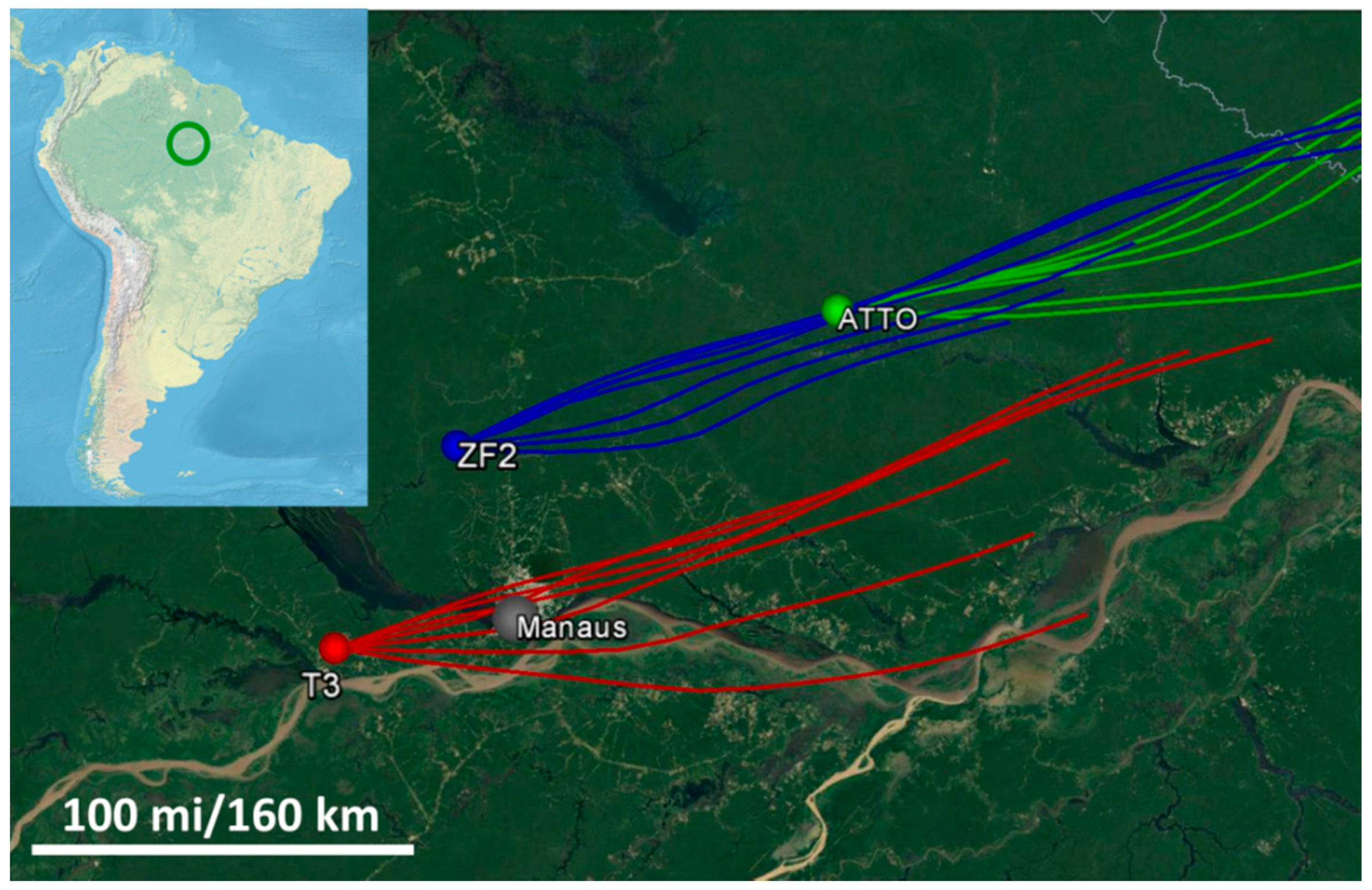
As part of the GoAmazon field campaign, two Intensive Operating Periods (IOPs) were conducted during 2014, with IOP2 taking place during the dry season from the 15 August through the 15 October 2014 [
34]. This campaign was conducted over central Amazonia with multiple sampling sites around the city of Manaus (
Figure 1). Northeasterly trade winds in this region dictate the general wind direction over the area and so the sampling sites were located with this in mind. These trade winds carry marine aerosols from the ocean inland and, during the wet season, can also carry supermicron mineral dust from the Sahara [
36]. For the wet season, secondary organic aerosols (pure liquid or with a soot/inorganic core) dominate the submicrometer size range [
37,
38,
39]. During the dry season, however, a large fraction of the aerosol population can be attributed to large scale biomass burning [
30].
For this study, particle samples from three sampling sites were studied: the Amazon Tall Tower Observatory (ATTO; T0a), the Terrestrial Ecosystem Science site (ZF2; T0t), and the Atmospheric Radiation Measurement (ARM) site located near Manacapuru (T3). The ATTO site is located approximately 150 km upwind of Manaus and serves as a background site. During the wet season, near-pristine conditions can be observed here; but, because the dry season is dominated by biomass burning particles, the ATTO site will serve as a regional background rather than a background of pure biogenic particles as might be expected [
40]. The ZF2 site is located about 140 km directly downwind of the ATTO site. The final site, T3, is 70 km downwind of Manaus and often experiences the pollution plume from Manaus [
34,
35]. Site locations and characteristics are presented in Martin et al. 2016 [
34]. For additional background information about the sampling sites, please see Andreae et al., 2015, Artaxo et al., 2013, and Martin et al., 2016 [
28,
34,
40].
2.2. Sample Collection
At the three sampling sites, atmospheric particle samples were collected on silicon nitride (Si
3N
4) membranes overlaid on a 5 × 5 mm silicon chip frame with a central 0.5 × 0.5 mm window (100 nm thick membrane, Silson Inc., Southam, England). Samples were collected using a Micro-Orifice Uniform Deposit Impactor (MOUDI, MSP MOUDI-110, Shoreview, MN, USA) on the dates and times shown in
Table 1. HYSPLIT back trajectories were examined for each sampling period to confirm the wind patterns seen in
Figure 1. These samples were then analyzed sequentially with the two spectromicroscopy techniques discussed in the following sections.
2.3. STXM Data Collection and Image Processing
Samples were first imaged at the STXM beamline 5.3.2.2 (ALS, Berkeley, CA, USA) at the Advanced Light Source (ALS) [
43]. The energy range of this STXM (200–600 eV) end station enables the quantitative study of carbon, nitrogen, and oxygen. Energy selected soft X-rays were focused down to a ~30 nm spot size and directed onto the sample surface. After a suitable 15 × 15 μm region was located, the sample stage was then raster scanned, with 40 nm steps, using piezo-electric stages to capture an image at a specific energy. This process was then repeated at multiple photon energies to produce a stack of images with an absorption spectrum associated with each 40 × 40 nm pixel. For each element, photon energies were chosen before and after the k-shell absorption edge: 278 and 320 eV for carbon, 400 and 430 eV for nitrogen, and 525 and 550 eV for oxygen [
44]. Additional images were also taken near the carbon edge at 285.4 and 288.5 eV, for the RC = CR and RCOOR C1s→π* transitions, respectively, in order to partly characterize the molecular speciation of carbon [
45].
Any displacement between images within a stack is corrected by a routine based on Guizar-Sicarios’ image registration algorithm [
46]. Regions within a given stack were then identified as particles or substrate using Otsu’s method on that stack’s average intensity image over all 8 energies [
47]. Background subtraction of a given element’s pre-edge intensity image from its post-edge image is then performed to account for any absorbing species not attributed to that element.
The recorded intensity at each pixel determined to be a particle was converted to optical density using:
where
OD is optical density,
I is intensity of the pixel, and
I0 is the background intensity. This is followed by a conversion to mass with the following formula:
where
m is the mass of a specific element at that pixel,
A is the area of that pixel, and
µpre and
µpost refer to the mass absorption coefficients for that specific element before and after the absorption edge, respectively. Mass absorption coefficients have been both empirically and theoretically determined for a variety of elements as tabulated in Henke et al., 1993 [
44].
Previously developed algorithms for determining the speciation of carbon using 278, 285.4, 288.5, and 320 eV were applied to each Field Of View (FOV) as well. This mapping technique uses a series of thresholds to identify inorganics, soot, and organic carbon. Total carbon is taken to be
OD320 −
OD278, pixels with an
OD278/
OD320 ratio 0.5 or greater are rich in inorganics, and pixels with an elevated (0.35) ratio of sp
2 bonding compared to total carbon (
OD288.5 −
OD278)/(
OD320 −
OD278) are indicative of soot [
45].
2.4. SEM/EDX Data Collection
The same sample windows previously imaged with STXM were imaged again with a computer controlled scanning electron microscope (FEI, Quanta 3D FEG, Hillsboro, AL, USA) coupled with energy dispersive X-ray spectroscopy (CCSEM/EDX, PNNL, Richland, CA, USA). The SEM utilized a field emission tip to produce an electron beam which was directed and focused onto a sample with an accelerating voltage of 20 kV which can cause core shell atomic electrons to be ejected from the sample. Higher shell electrons then relax into the newly created orbital hole, releasing an elementally characteristic photon recorded by an energy dispersive X-ray detector (EDAX PV7761/54 ME with Si(Li) detector, Mahwah, NJ, USA). As the electron beam was scanned over the sample, the transmitted electron image was used to identify the exact same FOVs from the previous STXM images. Once a FOV previously analyzed with STXM is located, a 10,000× image (30 nm/pixel resolution) was captured. This image combines both transmitted and backscattered electron images to improve particle detection [
23]. A threshold contrast level was then set to identify which areas of the collected image counted as particles using the “Genesis” software from EDAX, Inc. A software filter was then applied which discounts particles that are too small (e.g., noise spikes) or too large (e.g., multiple nearby particles counted as a single large particle). The electron beam was then directed towards each identified particle in sequence and an EDX spectrum was collected. Afterwards, software was used to fit the peaks of eleven relevant elements selected for this study: Na, Mg, P, S, Cl, K, Ca, Mn, Fe, Ni, and Zn. Some elements of interest have been included in the spectral fit, but omitted from quantitation, including Al, Si, and Cu due to background sources of these elements: (1) the STXM sample holder where the Si
3N
4 windows sat was made of Al and was inserted into the SEM as well, (2) the mounting stage that holds samples inside the microscope was fabricated from beryllium-copper alloy, (3) the EDX data was collected using a Si(Li) detector with a 10 mm
2 active area. Each of these circumstances could contribute background signal for the elements in question.
After data has been collected from both SEM and STXM, individual particle mass information is contained in two sets of images: one from STXM and one from SEM. Due to differing contrast mechanisms, image resolution, and other factors, particles do not necessarily appear the same between images taken with the two techniques. The manual matching of particles was performed using pattern recognition to ensure proper alignment of the image sets from both techniques.
2.5. Quantifying Higher Z Elements
Using the aforementioned methods, STXM yields quantitative, absolute mass information on a sub-particle basis. SEM/EDX is more limited in this aspect, being quantitative for elements with Z > 11 (Na) but only semi-quantitative for C, N, and O [
23]. Due to the EDAX software used for EDX data collection and analysis, there is an additional caveat to the quantitation of Z > 11 elements: the software reports only the relative mass percentages compared to the elements chosen during data processing. In order to properly quantify the mixing state, the absolute mass of each element in each particle is necessary. To determine these absolute masses, a system of equations was set up using the following equation types:
For each pixel, ODi is the optical density taken at energy i, is the density and t is the thickness of the sample (at that pixel), fa is the mass fraction of element a, and µa,i is the mass absorption coefficient of element a at energy i. Equation (4) is a general relationship, which equates the ratio of two absolute mass fractions (fx and fy) with the ratio of relative mass percentages (rel.%x and rel.%y) produced by the EDAX software. Equation (3) utilizes the quantitative nature of STXM whereas the relative mass percent of elements with Z > 11 were used in Equation (4) to combine the quantitative abilities of SEM/EDX. This system was then solved for the 14 absolute mass fractions (fa) of each element chosen in this study.
Equation (5) is an assumption that is valid when the 14 elements analyzed comprise close to 100% of the particle’s composition. Here, systematic error in the calculated mass fractions of specific particles can be introduced in particles where elements not considered represent a significant portion of that particle’s mass (e.g., mineral dust and Si or Al). During the Amazonian dry season, Al and Si represent 0.3% and 0.4% of the average fine mode particle mass [
48]. This mass fraction error becomes negligible, however, when the ensemble diversity values or mixing state index is considered due to the overwhelming mass of C, N, and O in each particle.
After both sets of images are matched and the corresponding light and heavy element information has been processed quantitatively, mass information for each FOV is contained in sets of maps, one for each element analyzed.
2.6. Mixing State Parameterization
The method of parameterizing mixing state used here is based on calculating mass fractions for different groupings of the individual components defined and is reproduced from Riemer and West [
20]. The absolute mass of a given component
, within
a given particle
, is labeled as
where
= 1, ...,
A (and
A is the total number of components) and
= 1, ...,
N (the total number of particles). From this, the following relationships are established:
Mass fractions are then established from these relationships with:
where
is the mass fraction of a particle within a sample,
is the mass fraction of component a within a sample, and
is the mass fraction of component a within particle
i.
These mass fractions are used to calculate the Shannon entropy (also called information entropy) for each particle, each component, and for the bulk using Equations (10)–(12), respectively.
Each type of mass fraction can be thought of as a probability, and thus the collection of mass fractions defines a probability distribution. The Shannon entropy of a probability distribution quantifies how uniform the distribution is. Shannon entropy is maximized if every element in the distribution is equally probable, and the entropy decreases the more likely any individual element becomes [
20]. With this information entropy, diversity values are defined with the following equations:
The diversity values contain the same type of information, but represent it in another way. Each diversity value represents the effective number of species (weighted by mass) within a given population (i.e.,
Di represents the number of species within a specific particle,
Dα is the average number of species within any given particle, and
Dγ represents the number of species within the entire sample). From these diversity values, the mixing state index is defined as
This definition compares how many species exist, on average, within individual particles, with the total number of species identified in the sample. χ is at a minimum of 0 when Dα is 1, corresponding to each particle being comprised of exactly one species. A mixing state index of 1 occurs when Dα and Dγ are equal, meaning that each particle has the same composition as the bulk sample.
2.7. Error in Mixing State Index, χ
The measurement uncertainty of χ due to STXM, EDX, or the system of equations was found to be insignificant compared to the statistical uncertainty of χ within each cluster and thus only the statistical uncertainty is considered here. To determine this uncertainty, the statistical uncertainty in Dα, and Dγ were found separately.
Determining statistical uncertainty in
Dγ starts with
fa from Equation (9). From Riemer and West [
20],
fa is a ratio of the total mass of the
ath component and the total mass of the sample, however this is equivalent to the ratio of the mean mass of the
ath component and the mean mass of particles within the sample:
where
is the mean mass of the
ath component and
is the mean mass of particles within the sample. From this, the standard error (for a 95% confidence level) can be determined for
and
which is then propagated through Equations (9)–(13).
The statistical uncertainty in
Dα was found by first rearranging and combining Equations (11) and (13):
and, because this takes the form of an expected value
, the error in
Hα can be found with Equation (15) and then propagated with Equation (16) to determine the error in
Dα.
2.8. k-Means Clustering
All analyzed particles were combined and a k-means clustering algorithm was then used to group particles into clusters [
49]. A vector of 18 variables were used for k-means clustering: the quantitative elemental mass fractions composition of the 14 elements chosen, the Circular Equivalent Diameter (CED) [
1],
Di, the mass fraction of carbon attributed to soot, and the area fraction of the particle dominated by inorganics. In this way, particles were clustered based on size, elemental composition, as well as on how carbon speciation was distributed. The square root of these parameters was used in the clustering algorithm to enhance trace elements in accordance with Rebotier et al. [
50]. CED is used here as the descriptor of particle size due to it being readily calculable from STXM data. While aerodynamic equivalent diameter is the physical parameter determining MOUDI sampling, it is difficult to retrieve from microscopy data.
The correct number of clusters was initially chosen based on a combination of two common methods: the elbow method, and the silhouette method [
51]. Using these two methods, 12 clusters were identified.
3. Results
As a general trend, during the dry season, the whole Amazon Basin experiences a significantly higher aerosol number concentration and CO
(g) concentration compared to the wet season, largely due to in-Basin fires [
30,
40]. Furthermore, in addition to biomass burning, emissions from Manaus are often observed at the T3 site (downwind of Manaus), sporadically at the ZF2 site (upwind but near the city) and rarely at ATTO (upwind and ~150 km away).
Table 2 outlines some supporting measurements made at the three sites.
Most values listed for the 12 September and 13 September sample at T3 are consistent with their sample-period monthly averages, even considering time of day each sample was collected. The data from 14 September, however, shows a marked increase in particle concentration, nitrate, organic, and CO concentration, along with a small increase in BC. This is indicative of a heavy pollution plume which, in this case, had recently passed over the T3 sampling site (see
Figure S2). AMS and particle concentration data for the T3 sites show a reasonable agreement with either background or polluted conditions previously reported, as do ozone measurements [
52,
53]. The monthly average values for 13 September and 14 September are often similar due to the similar (though not identical) sampling time from 8:00 to 12:00 and from 9:00 to 12:00, respectively. The similarity between monthly average particle concentrations for 12 September and 13 September are purely coincidental.
Particle concentration and AMS/ACSM data were not available for the ZF2 site during this study. Concentrations of CO, ozone, and BC values agreed well with their sample-period monthly averages with the lone exception of ozone levels for the 3–6 October sample period. This increase is also reflected, albeit to a much lesser degree, in an increase of CO and BC levels. From
Figure S3, it appears that sample collection began in the middle of a period of higher than average pollution levels. Temporary enhancements in BC due to emissions from Manaus have been observed previously at ZF2 [
54]. Overall the levels of CO, ozone, and BC at ZF2 are smaller with respect to T3 values as is expected. ACSM data collected for past studies at site ZF2 fits well with the trends seen in the limited data presented here [
28]. Concentrations of aerosol components can fluctuate depending on the day but September (2012) averages for ammonium, chloride, nitrate, organics, and sulfate have been reported as 0.46, 0.01, 0.22, 13.9, 0.37 µg m
−3, respectively by Artaxo et al., 2013 [
28].
Unsurprisingly, the ATTO site shows the lowest levels of almost all presented aerosol and gas components. The sample collected on 15 October also appears to be fairly average with respect to the sample-period monthly average for this collection time. The low particle concentration suggests a sample with low pollution levels which makes this sample ideal for our purposes. The values presented for the ATTO site are also in fair agreement with previously published data [
40].
The supporting data tabulated here has been collected from different instruments at the three sites and so a direct comparison could be suspect, especially in the case of BC measurements [
55]. However, considering the agreement with published literature and qualitative use of
Table 2 in the current work, we believe any associated error is acceptable.
Figure 2 shows an example FOV and the type of data calculated for all three sites. Each particle has an OD map (which is proportional to mass, refer to Equation (2) for C, N, and O as well as a C speciation map. In
Figure 2, the STXM grayscale image shown is the average intensity map over the four C edge images. There is a correlation between the brightest spots and the identification of soot in the C speciation map. This speciated image is possible due to the sub-particle spatial resolution achievable with STXM mapping, which is highlighted in the C, N, and O maps. Potential inter-site differences can be seen in this figure: the ATTO sample shows large inorganic inclusions coated by organics along with Na, Mg, and Cl representing the bulk of the higher Z elements. The particles present at ATTO also often look like either inorganic aerosols from biomass burning events or small biogenic K salt particles (due to the KCl or NaCl inorganic cores), or secondary organics, with a few particles appearing to be sea spray [
56,
57,
58]. The ZF2 sample has a consistent circular morphology with appreciable mixing between the three carbon species. ZF2 particles often look amorphous with some particles appearing to be sulfate-based aerosols [
59]. Lastly, the T3 sample is the most varied in terms of morphology and in elemental composition with S, P, and K all present in many of the particles sampled. Unsurprisingly, soot inclusions are much more common in the T3 sample. Particles from this site often look like biomass burning particles with a few fractal soot particles as well [
60]. It is important to keep in mind that the particle morphologies presented have possibly changed from their original state when collected. This change could be due to the impaction of particles during sampling, or the changes in relative humidity experienced as these particles are collected, stored, transported, and placed in vacuum before STXM or SEM images can be obtained. Liquid particles can spread upon impact making them appear larger on microscopy substrates. Particles with high water content can effloresce at lower relative humidity leaving solid phase inorganic components. Loss of highly volatile organic carbon or volatile inorganics like ammonium nitrate is also possible, making concentrations detected here a lower limit for inorganic and organic species.
The SEM grayscale image shows the slightly different views presented by the two techniques, with particle shapes appearing different between them along with a higher spatial resolution image (10 nm vs. 40 nm with STXM). Soot inclusions identified in the C speciation map are also seen as bright spots in the SEM grayscale image in addition to many of the inorganic inclusions [
23]. From the EDX data collection, mass fraction maps for each element (on a per-particle basis) were used to calculate individual particle diversity (
Di) values for each particle. Another aspect of the maps is the varying background level between SEM images, seen especially in the high background of the ZF2 image. This is a consequence of the brightness and contrast levels being set before EDX acquisition and was performed to ensure that the maximum number of particles were detected by the CCSEM particle detection software.
3.1. Clustering and Source Attribution
For each of the 12 clusters, determined by the k-means algorithm, a random representative sample of 40 particles (taken from any sample or sampling site) was selected for the images shown in
Figure 3. The average elemental composition of each cluster is shown in
Figure 4 along with the fraction of each cluster collected at the three sampling sites. Finally,
Table 3 outlines the assigned colors and labels, as well as some relevant descriptive statistics for each cluster. As can be seen in the average particle diversity column in
Table 3, most clusters have a
Dα value near either 2.4 or 3.6 (with a single exception). These two values define the “low” or “high” diversity referred to in the cluster names and are discussed in more detail in
Section 3.5. A similar source apportionment was discussed in a previous SEM based study; however, it was conducted during the wet season when biogenic aerosols dominate [
37]. During the dry season, these biogenic particles are still present but are overwhelmed by aerosols derived from biomass burning.
One notable aspect of
Figure 4 is the ratio of elemental Cl to S in each of the clusters shown. From the EDX spectroscopy data presented here, the mass fraction of Cl is often greater or at least similar to that of S. This is apparently contradicted by
Table 2, where the concentrations of chloride are an order of magnitude less than the concentrations of sulfate. There are, however, a few extenuating circumstances for this comparison. Firstly, the chloride level in
Table 2 is that of non-refractory material owing to the AMS’s method of volatilizing particles at ~600 °C. This is well below the vaporization temperature for NaCl and KCl, two major sources of Cl (and inorganics in general). Hence,
Table 2 AMS data underestimates Cl mass fractions. Another requirement to allow direct comparison is to change concentration of sulfate to S by multiplying by the ratio of molar masses (~32/96), reducing the concentrations seen in
Table 2 to about one third. The third circumstance is the potential for beam damage using the two sequential microscopy techniques here. Some of the inorganic inclusions/cores detected using STXM/NEXAFS spectroscopy may be particularly sensitive to electron beam damage. These sensitive inorganics (particularly ammonium sulfate) could have been volatilized during the scanning/locating phase of SEM and therefore would not be well characterized with subsequent EDX spectroscopy. This carries two consequences: a possible underestimation in the mass fraction of S, and the identification of inorganic regions with STXM without the detection of many inorganic elements by EDX to explain the inclusions. This issue of S quantification is further highlighted when the S/Cl ratios in
Figure 4 are compared with previous Particle Induced X-ray Emission (PIXE) measurements which report aerosol S concentrations an order of magnitude greater than Cl concentrations [
61,
62]. In addition to PIXE, Artaxo et al., 1994 used factor analysis to determine broad particle classes including soil dust, biogenic, marine, and biomass burning classes. One finding of relevance is the high degree of correlation between biogenic particles and S concentration [
62]. For the current work, this could suggest an underestimation in the number of clusters hypothesized to contain biogenic particles.
3.1.1. Soot Clusters (LDS1, LDS2, HDS)
The HDS cluster is characterized by a thick organic coating around a soot core. The high levels of organics and K suggests that this cluster mostly originated from biomass burning, but may also contain urban emissions [
63]. This, combined with the overwhelming majority of HDS particles being from site T3, suggests that they are predominantly anthropogenic in nature. This cluster’s enhanced mass fraction of Na, Cl, Mg, and S indicates a contribution from marine aerosols. Together with the appreciable amount of P and K, this results in the higher particle diversity seen in this cluster. Further supporting this cluster’s identity is that enhancement of these elements have been observed previously during biomass burning events in the presence of a background rich in marine aerosols [
64,
65].
In addition to LDS1 lacking the P, K, and Ca that HDS has, LDS1 also has a smaller amount of Na, Mg, S, and Cl which results in the lower average particle diversity. The decreased abundance of K and Cl may indicate urban combustion sources such as diesel engines as opposed to biomass burning [
56]. The coatings of organics around the soot cores seen in this cluster are much thinner compared to other soot containing clusters which suggests that particles in this cluster are less aged [
66]. The vast majority of particles from this cluster type are from site T3; given that T3 is downwind of Manaus, this suggests fresh urban soot emissions as this cluster’s source [
35].
The large and multiple soot inclusions and the presence of fractal soot are indications that LDS2 is comprised of particles with a contribution from combustion [
67,
68]. Particles in LDS2 are found mostly at site T3 which points towards Manaus being the source of these aerosols. The sometimes substantial organic coating on many of these particles is most likely due to condensation during travel of fresh aerosols from Manaus travel to the T3 sampling site. With the exception of the one night time sample, all T3 samples were collected during the mid to late morning (~9:00 to 12:00) which has been seen in other urban areas to correspond to an increased in aged soot over fresh soot owing to the increase in photochemistry [
5].
3.1.2. Organic Clusters (LDOrg, HDOrg)
HDOrg is comprised of small particles with their carbon being entirely organic dominant. This cluster has a substantial amount of the heavier elements (Z = 11 (Na) and above) driving the diversity up. The presence of P specifically is important as these elements, coupled with the carbon speciation, suggest that the particles from this cluster are biological in origin [
61]. This cluster also contains the highest number of particles with collocated Na and S, which has been previously shown to suggest biogenic particles [
69]. The HDOrg cluster also contains an appreciable amount of K and, given that one sample was taken at night, could include biological particles derived from fungal spores [
58].
LDOrg is similar in carbon speciation, morphology, and size but lacks the heavier elements contained in HDOrg. The almost entirely C, N, and O composition, small size, and the organic carbon speciation suggest that particles in this cluster are secondary organic aerosols. This is supported by a slight majority of particles in this cluster coming from the general direction of the ATTO site, where a dominant appearance of biogenic secondary organic aerosols and a smaller influence from anthropogenic emissions is expected. The high O/C ratio, along with the dearth of inorganics, may make this cluster comparable to aged ambient oxygenated aerosols [
70].
As discussed further on in
Section 3.2, both organic clusters are unique in that they make up a sizeable fraction of particles at all sampling sites.
3.1.3. Inorganic Clusters (LDI, HDI1, HDI2, VHDI)
Other than C, N, O, and trace levels of Mg, no other elements are observed in the LDI cluster. Carbon speciation, however, shows a clear inorganic core with an organic coating. This leaves only a few options for the identity of the inorganic cores seen here. One possibility is that the inorganic core is composed of elements not analyzed here, such as Si or Al. Due to the Al mounting plate and the Si(Li) detector used, we are not able to quantitatively detect Al and Si. However, a more likely possibility is that, as mentioned above, the inorganic cores that were initially detected with STXM were particularly sensitive to electron beam damage leading to these sensitive inorganics (possibly ammonium sulfate) being poorly characterized by EDX.
HDI1 is characterized by large, vaguely cubic, inorganic inclusions coated with organics. This cluster has a fairly high fraction of O, Na, and Mg while containing the highest amount of Cl of any cluster. The HD1 cluster is also unique in that particles in it were collected almost exclusively at the ATTO site. Because of this, we suspect these particles represent marine aerosols [
71,
72,
73]. The organic coating here is substantial and is likely due to aging as aerosols are transported inland to the ATTO site. The transport of particles inland over a large distance is also reflected in the O/C ratio, where the particles (specifically the organic coatings) may have oxidized more than other clusters.
HDI2 is characterized by many small inorganic inclusions speckled throughout the particles which are not as localized as with the HDI1 cluster. There are small soot inclusions and an increased presence of P, K, and S as compared to HDI1. These particles are mainly seen at the ZF2 site with a smaller portion present at ATTO. Thus, it is possible that this cluster is associated with spore rupturing but further investigation is needed to apportion this cluster [
74].
The VHDI cluster is unique in that it possesses the highest
Dα value of any of the clusters at 4.83, well above both the nominal “high diversity” value of 3.6 and the second highest
Dα value of 3.83. This cluster also has a large statistical error of 1.92 (at a 95% confidence level), which could indicate multiple disparate groups are present in this cluster. This cluster is comprised mostly of particles from ZF2, but ATTO and T3 particles contribute substantially as well. The VHDI cluster’s elemental composition is similar to that of HDI2, but with a decreased C and O mass fraction and an enhancement of the other elements, especially K (often seen in inorganic salt grains from biomass burning) [
30,
75]. Inorganics are seen both as large localized inclusions, and as many small inclusions speckled throughout the particle. This cluster’s high diversity and larger statistical spread may also be indicative of the varied biomass burning fuels and burning conditions present.
3.1.4. Mixed Clusters (LDM, HDM)
The LDM cluster is characterized by all three carbon speciation types being present in many of the particles. The presence of inorganic inclusions along with the lack of heavier elements suggests ammonium nitrate (and possibly ammonium sulfate) as the identity of the inorganics. This cluster is seen almost entirely at the ZF2 site which, along with its low diversity and few elemental constituents, may indicate a local aerosol source near site ZF2. In which case, particles would have little time or distance to age and scavenge new elements. The presence of soot in the LDM cluster might suggest these aerosols come from the same source as the HDM cluster.
The species of carbon found in the HDM cluster’s particles are well mixed with soot, non-carbonaceous inorganic, and organic carbon found in varying ratios. The large soot inclusions, high diversity, and substantial presence of higher Z elements may point to an industrial or automotive origin. Although the sizeable representation of the HDM cluster at T3 supports this, a slightly larger representation is seen at site ZF2. This raises the possibility that some emissions from service vehicles driving to or past the ZF2 site, or nearby generators may be collected at the ZF2 site. Emissions from Manaus are not uncommon either and could account for this cluster’s presence at ZF2 [
38].
3.1.5 Miscellaneous (Misc.) Clusters
Particles placed in this cluster were most likely grouped due to their supermicron size rather than their composition or diversity. This cluster is comprised of some large rectangular crystals, particles which did not fit well into the other clusters, as well as cases of particles with multiple large inclusions (inorganic or soot) each encased within individual lobes. Since we could not include Al and Si in our analysis, this cluster may also contain mineral dust particles (e.g., quartz and kaolinite) coated with organic material.
This last particle type may contain adjacent particles being erroneously deemed a single particle by our detection algorithm because of overlap of the organic coating upon impaction. This grouping of multiple individual particles into agglomerations much larger than expected for the given MOUDI stage could have caused them to be placed in the Misc. cluster.
One notable particle type seen in this cluster is a collection of particles with a rectangular inorganic core with a small patch of organic carbon in the center. Some of these inorganic cores wrap around the carbon center while some others have a side missing but they all retain the same basic shape. The elemental composition of the inorganic portion contains small amounts of Na and Mg, a relatively large amount of S along with most of the particle’s N and O mass fraction. These particles are observed on the only night time sample that was collected. This, along with the particles being found mainly at site T3 could suggest an industrial process whose emissions become easier to identify at night when other sources of aerosols (automotive) experience a decrease. Fragments of ruptured biological particles also may be a possibility based on their elemental composition [
74].
3.2. Cluster Type Dependence on Sampling Site
The cluster contributions at each sampling site are shown in
Figure 5 separated by stage. Although particles from all cluster types were seen at each location, some particle types were predominantly associated with a particular sampling site. The clusters labeled LDOrg, HDOrg, HDI1, and HDI2 account for ~86% of the particles seen at the ATTO site. To account for a similar share of particles at site ZF2 one must consider the clusters labeled: HDI2, VHDI, LDOrg, HDOrg, LDM, and HDM. Site T3 requires LDS1, LDS2, HDS, LDOrg, HDOrg, and HDM to be accounted for to define a similar portion of particles.
As the ATTO sampling site is less polluted and representative of biogenic aerosols, the presence of both organic clusters as well as two inorganic clusters with possible biogenic origins is expected. Conversely, the relative absence of soot clusters or the mixed clusters further highlights the ATTO site’s remoteness from regional anthropogenic (urban) influences. However, even this site is far from being pristine, as shown by the presence of significant amounts of BC, presumably from long-range transport.
While the ZF2 site contains many of the same clusters present at the ATTO site, there are some notable differences. The presence of the HDI1 cluster is diminished (~1% as compared to ATTO’s 26%), and both mixed clusters are seen in substantial amounts. The largest difference between the two stages is the enhancement of the LDM cluster in stage 7 data and the minor increase in all three soot clusters in stage 8 particles.
Site T3 shows the presence of many clusters, with all three soot clusters present in substantial amounts. This is expected, as automotive exhaust or energy production through fuel oil burning will produce soot particles that travel to site T3 [
76]. Both organic clusters are present with a slight enhancement in stage 8 particles. Because both organic clusters are seen in reasonable amounts at each sampling site, these particles may be part of the aerosol background inherent to sampling in a heavily forested region. Stage 8 particles are also devoid of LDI, HDI1, and Misc. clusters, but few of these were seen in stage 7 and so this absence may be due to insufficient sampling.
Another aspect of
Figure 5 is how many clusters make up most of each site’s aerosol population, for which we use the following metric. Each site’s cluster contribution is sorted in descending order and an effective number of clusters is found using
, where r is the rank of each cluster’s contribution to that site’s population (with 1 assigned to the cluster with the largest contribution),
fr is the fraction of that site’s population that cluster r accounts for, and
E(
r) is the effective number of clusters. This metric will vary, in this case from 1 to 12, where the lower the effective number of clusters, the better a given site is characterized by fewer clusters. The values calculated from this metric are listed in
Table 4. This metric highlights the increased diversity of sites T3 and ZF2 with respect to the ATTO site. Site ZF2’s cluster composition is more varied. This is possibly due to specific events occurring during sampling, or by virtue of being closer to Manaus and therefore more susceptible to anthropogenic emissions. Site ZF2 samples were also collected over multiple days meaning some of the cluster variability may be due to the inclusion of both day and nighttime aerosols. This higher cluster variety could also be attributed to a local aerosol source as mentioned previously. Site T3 stage 7 shows the highest cluster variability due to T3’s proximity to (and location downwind of) the anthropogenic center of Manaus. Stage 8 of site T3, in contrast, shows the lowest cluster variability. This may be the influence of fresh emissions coming from Manaus, which tend to be smaller in size and similar in composition (placing many of them in the same cluster).
3.3. Cluster Size Dependence
Although relatively few supermicron particles were collected, most clusters included some fraction of both sub- and supermicron particles. Only one cluster (Misc) was exclusively supermicron in size, whereas three clusters (LDS1, LDI, LDOrg) included exclusively submicron particles. Referencing
Figure 6, the only clusters observed in the supermicron size range were those labeled: Misc (located around 2 µm with a very small percentage), LDM, HDM, HDI2, LDS2, and HDI1. Supermicron particles in the clusters HDS, HDOrg, and VHDI particles were also observed, but in very small numbers. Many clusters that make up the supermicron range represent more aged species.
The submicron range is composed of many more clusters relative to the supermicron range. Many clusters in the submicron range were often labeled as less aged than the ones found in the supermicron range. This qualitative observation is supported by
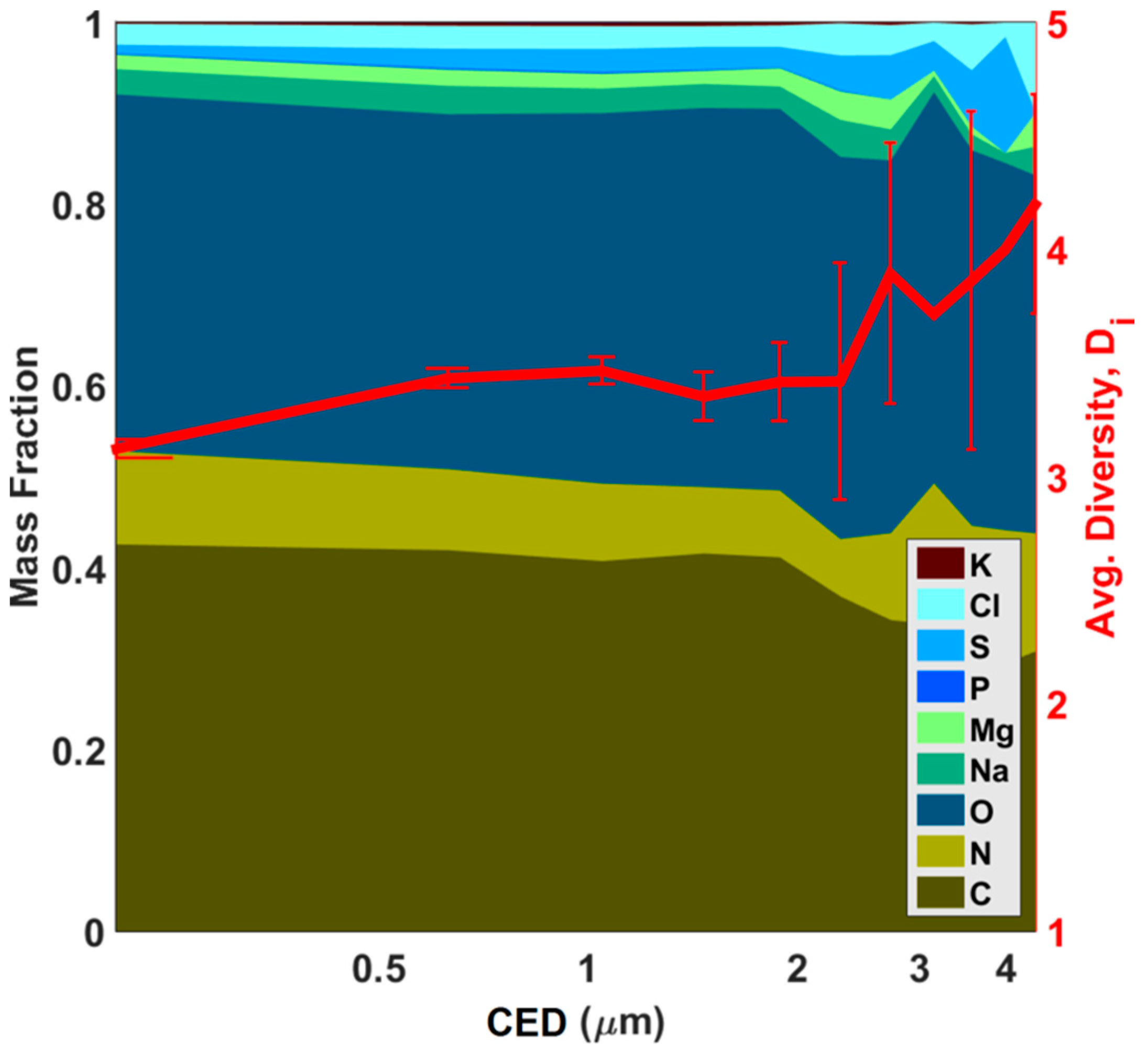
Figure 7 where there is an increasing trend in individual particle diversity (
Dα) with increasing particle size and the notion that
Dα is correlated with the extent of particle aging.
3.4. Composition and Diversity Size Dependence
Submicron particles, as seen in
Figure 7, have a high fraction of C, N, and O. With
Dα values, calculated for both sub- and supermicron particles being 3.3 and 3.4, respectively, submicron particles appear to be the least diverse. However, the error analysis described below, renders this merely suggestive rather than conclusive. As particle size increases, two things are observed: (1) average particle diversity increases slightly and (2) the fraction of inorganics increases. Because of the ubiquity of C, N, and O in aerosol particles, the average particle diversity will almost always be slightly above 3. Given the relatively constant ratios of C, N, and O (with O/C ≈ 0.91 and N/C ≈ 0.22), individual particle diversity is not dependent on these elements; with the exception of soot. Rather, it is mainly the presence of heavier elements which are responsible for any increase in diversity. These larger particles, often represented by more aged clusters like LDS2, HDM, or HDI1, have had sufficient time and travel distance to acquire additional elements during the aging process. A similar conclusion was observed during the Carbonaceous Aerosol and Radiative Effects Study (CARES) conducted in 2010 in California, where heavier elements appreciably affected the mixing state of particles and increased with size, while C, N, and O remained constant [
25,
37].
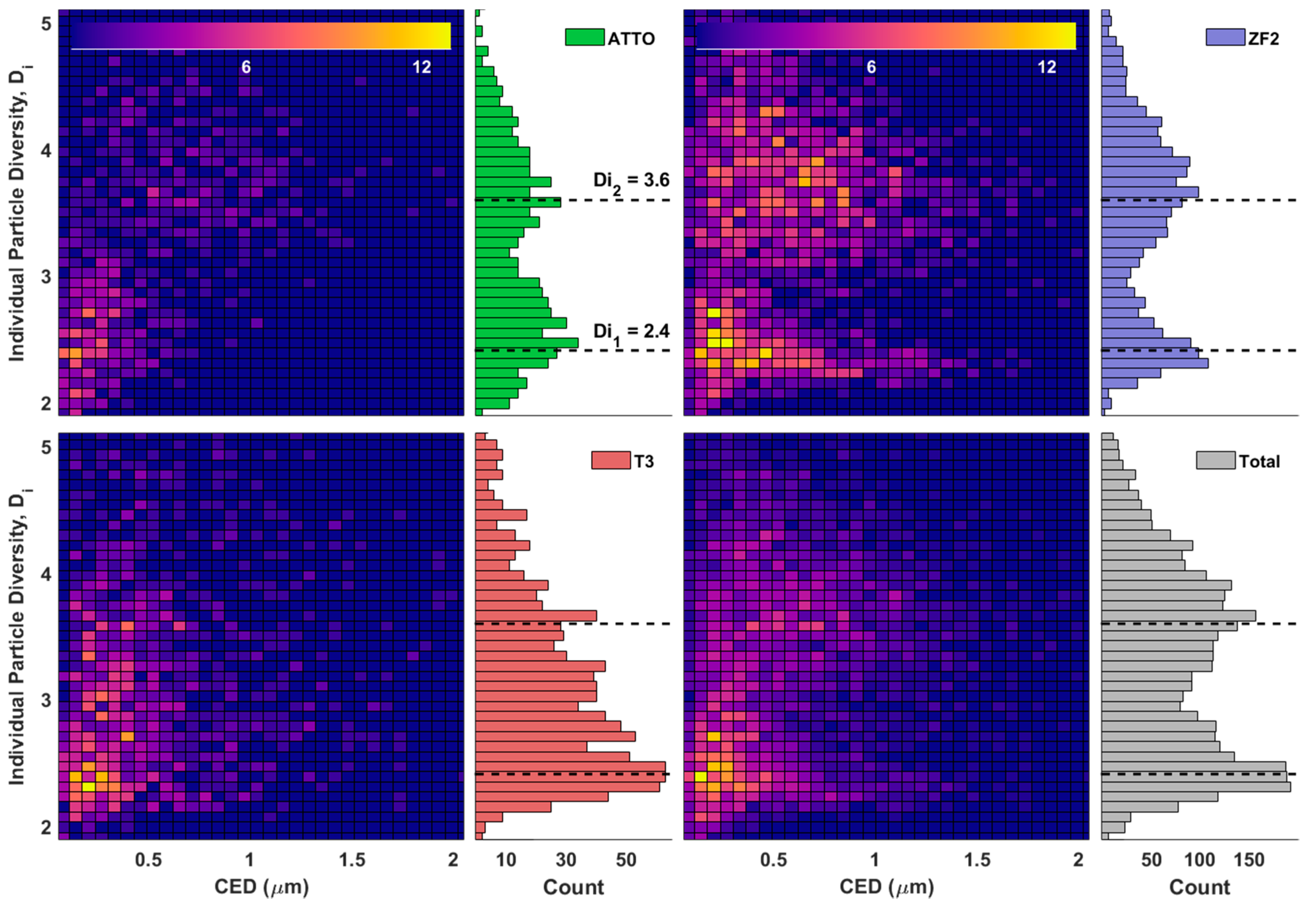
After clustering, most clusters were assigned so that their average particle diversity (
Dα) was close to one of the two modes present in
Figure 8. This clear distinction between the two diversity modes is what the high and low diversity cluster names are referring to.
The bimodality seen here may represent the separation between fresh and more aged aerosol particles. The diversity values of the lower and upper mode of the combined data set were 2.4 and 3.6, respectively. Considering that the three elements C, N, and O dominate the mass fractions of most particles, it is fitting for one mode to be below and one mode to be above 3. Particles in the lower diversity group are mostly C, N, and O with very little presence of other elements. The differing mass fraction between each of these elements causes the diversity to drop below 3.
This bimodality is absent in the T3 samples, having only the less diverse mode. The production of soot from transportation or fuel combustion is most likely the cause of this enhancement of lower diversity particles because of soot’s relative elemental purity.
The two dimensional histograms between
Di and CED in
Figure 8 serve to reinforce the idea that smaller particles tend to be less diverse. These smaller, less diverse particles are also less spread out whereas the more diverse particles show a wider spread in both diversity and size.
The increased spread seen in the aged aerosol group may be due to the variety of ways that aerosols can age and differences in distances traveled from the aerosols origin. Because the same variability isn’t seen in the smaller, less diverse, fresh aerosol group we suspect these particles have sources closer to where they were sampled. By sampling particles with nearby sources, the elemental composition and, by extension, particle diversity will be determined by the method of production and therefore be much less variable.
3.5. Mixing State of Particles at Different Sampling Sites
Entropy metrics were used to quantify mixing state for each sample analyzed here.
Figure 9 shows the mixing state index (
χ) corresponding to particles in each sample. In this case, the variation in mixing state index is small, with all samples having a χ bounded between 0.8 and 0.9. This is a result of
Dα and
Dγ consistently being around 3.4 and 3.9, respectively.
In the previous study by O’Brien et al. [
25], similar sets of STXM and SEM/EDX data were collected for the CARES field campaign. In that study, the same diversity and mixing state parameterization was used except that STXM data (elements C, N, and O) and SEM/EDX (elements Na, Mg, S, P, Cl, K, Fe, Zn, Al, Si, Mn, and Ca) data were analyzed independently. They found that mixing state index values for heavier elements usually ranged from 0.4–0.6 with some values as high as 0.9. The values of χ for C, N, and O generally ranged from 0.75 to 0.9. The mixing state indices retrieved exclusively from STXM data closely matches the values determined in this paper. This suggests that χ is almost entirely determined by C, N, and O due to these three elements dominating the mass of the individual particles and the sample as a whole.
A point of note is the small spread of Dγ values within a given sampling site. With Dγ representing bulk diversity, these values serve to compare the average elemental composition (for the 14 elements chosen) of all aerosols, condensed down into one number. For a given site, samples analyzed were taken during the same season of the same year with similar wind trajectories, sampling times and sampling duration. It is expected then, that there will be a consistency in how much of any given element is present in the aerosol population, based on how much of each element is emitted and included in the particulate material. For Dγ to vary wildly from one day to another, or from one sampling period to another, would require an event or aerosol source producing substantially more of one element than usual.
The spread in
Dα values among samples within a sampling site is much wider than that of
Dγ. A large spread in
Dα is expected when a singular diversity value is calculated from samples containing the variety of distinct particle types seen in
Figure 3. This value is more susceptible to change from one sampling date to another compared to
Dγ and depends on how much of each aerosol type is collected during a given sampling time.
The increase in the average particle diversity (Dα) with respect to increasing particle size is hinted at here, albeit in less certain terms. Focusing on samples collected where both stage 7 and 8 data were analyzed, average Dα values appear to be larger for stage 7 particles.
Samples collected at site T3 were expected to have a lower mixing state than either the ATTO or the ZF2 site. This hypothesis was borne from the quantity of fresh emissions in Manaus, specifically soot production from combustion, which would serve to drive the mixing state downwards towards total external mixing. However, the end result of the error calculations in
Section 2.7 is that the values of χ for each point in
Figure 8a become statistically unresolvable, as can be seen from the large error bars. This is also not an issue that would be solved with any reasonable amount of extra data collection but is instead mainly the consequence of the intrinsic spread in
Dα.
4. Conclusions
Presented here is a quantitative combination between two complementary per-particle spectromicroscopy techniques, STXM/NEXAFS and SEM/EDX, on the exact same data set. STXM/NEXAFS data was collected at the C, N, and O K-edges on a sub-particle level. This allowed not only the quantitative determination of C, N, and O absolute masses, but also carbon speciation and morphology. SEM/EDX allowed the approximate composition of the inorganic fraction to be determined and then quantified along with the STXM data. The combination of these two techniques enables almost all atmospherically relevant elements to be quantitatively probed on a per-particle basis. The potential issue with S detection discussed above could be mitigated entirely in future measurements by conducting STXM measurements at the S L-edge to obtain S mass fractions. This combined technique could be especially useful for identifying aerosol sources using elemental tracers or unique elemental compositions.
Using particle-specific elemental composition, size, carbon speciation, and individual particle diversity (
Di), k-means clustering was used to separate particles into 12 clusters. The cluster average of these same parameters allowed for potential sources to be assigned. It was found that the stage 7 of the T3 site had a more varied population of particles (as defined by the effective cluster number) and contained more soot-containing clusters than either the ATTO or ZF2 site. Clusters also exhibited size dependence, with a large portion of supermicron particles assigned to high diversity clusters which have been hypothesized to represent more aged particles. This approach could be used for even larger data sets, especially those located at long-standing measurement facilities. From this, diurnal, seasonal, or yearly changes in the aerosol population could be monitored directly. Application of this combined technique would be especially fruitful near large pollution sources, as these anthropogenic sources are difficult to model without the size-resolved composition presented here [
77]. The clustering presented here offers an opportunity not only to classify particles but also to identify sources, which can be invaluable in determining the effects of trade or environmental protection policies. The largest detriments to the utility of this composite technique are the long analysis times needed and the requirement for two separate instruments as well as beam time at a synchrotron light source.
Utilizing the composite data set to determine a quantitative mixing state index revealed that particles at site T3 were more externally mixed than at the other two sites. Error analysis, however, shows a fairly large uncertainty in the elemental mixing state for all samples, with statistical errors in
χ ranging from 0.3 to 0.8. These error estimates do show that when calculating mixing state by using the 14 elements listed here, mixing state values are close together and show most samples to be highly (between 80 and 90%) internally mixed. Size-dependent trends were also observed in individual particle diversity, with larger particles being slightly more diverse (3.3 and 3.4 for sub and supermicron particles, respectively). This size-dependent trend in diversity was seen even more drastically within the fine mode, with particles <0.5 µm having an average
Di value of around 2.4 and particles >0.5 µm having about a 3.6 diversity value, with a much larger spread of diversity values for larger particles. This difference may identify a separation between fresh and aged aerosols in terms of diversity. This result and the experimental method could be useful for climate models, allowing an experimental mixing state and size-resolved particle composition to be used rather than assumed to improve model performance [
78,
79,
80]. Even though this type of individual particle microscopy study is time-consuming, regions that are important to global climate models (such as the Amazon) may benefit from the improved accuracy of an experimentally determined mixing state.
The quantitative mixing state index presented is a useful tool, but its utility can be readily expanded. Two of the advantages that this combined spectromicroscopy technique has are the ability to identify morphology both of the particles as a whole and of the constituents within the particle. Due to the general nature of the mixing state parameterization, the mixing state index and its interpretation is heavily dependent on what components were used. In this study, 14 elements were used, however the omission or addition of just a few elements (especially abundant elements such as carbon) could drastically alter the value of
χ. Because of this, specifics about which parameters were used and how relevant they are to the samples being studied must be examined before interpreting the value of
χ. How well mixed individual elements are also may have limited usefulness to modelers or in general. With the exception of elemental carbon, mass fractions of specific elements (like nitrogen) are less chemically relevant than the molecules or ions they may be found in (nitrate vs. ammonium for example). Future work will build upon this composite technique to instead determine masses and a molecular mixing state for chemically and atmospherically relevant species such as nitrate, carbonate, sulfate, and etc. Modification in this way could allow our current combined technique to determine an aerosol population’s radiative forcing contribution due to both direct and indirect effects. Specific aspects about the indirect effect like hygroscopicity or the number of effective Cloud Condensation Nuclei (CCN) within a population of aerosols could also be gleaned from this method [
81]. This type of modification would bolster the usefulness of this technique as well as the usefulness of
χ for climate modelers.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}