Quantifying Impacts of Aerosol Mixing State on Nucleation-Scavenging of Black Carbon Aerosol Particles



Abstract
:1. Introduction
2. Methodology
2.1. PartMC-MOSAIC: A Particle-Resolved Approach to Simulated Aerosol Dynamics and Chemistry
2.2. Setup of Idealized Urban Plume Scenarios
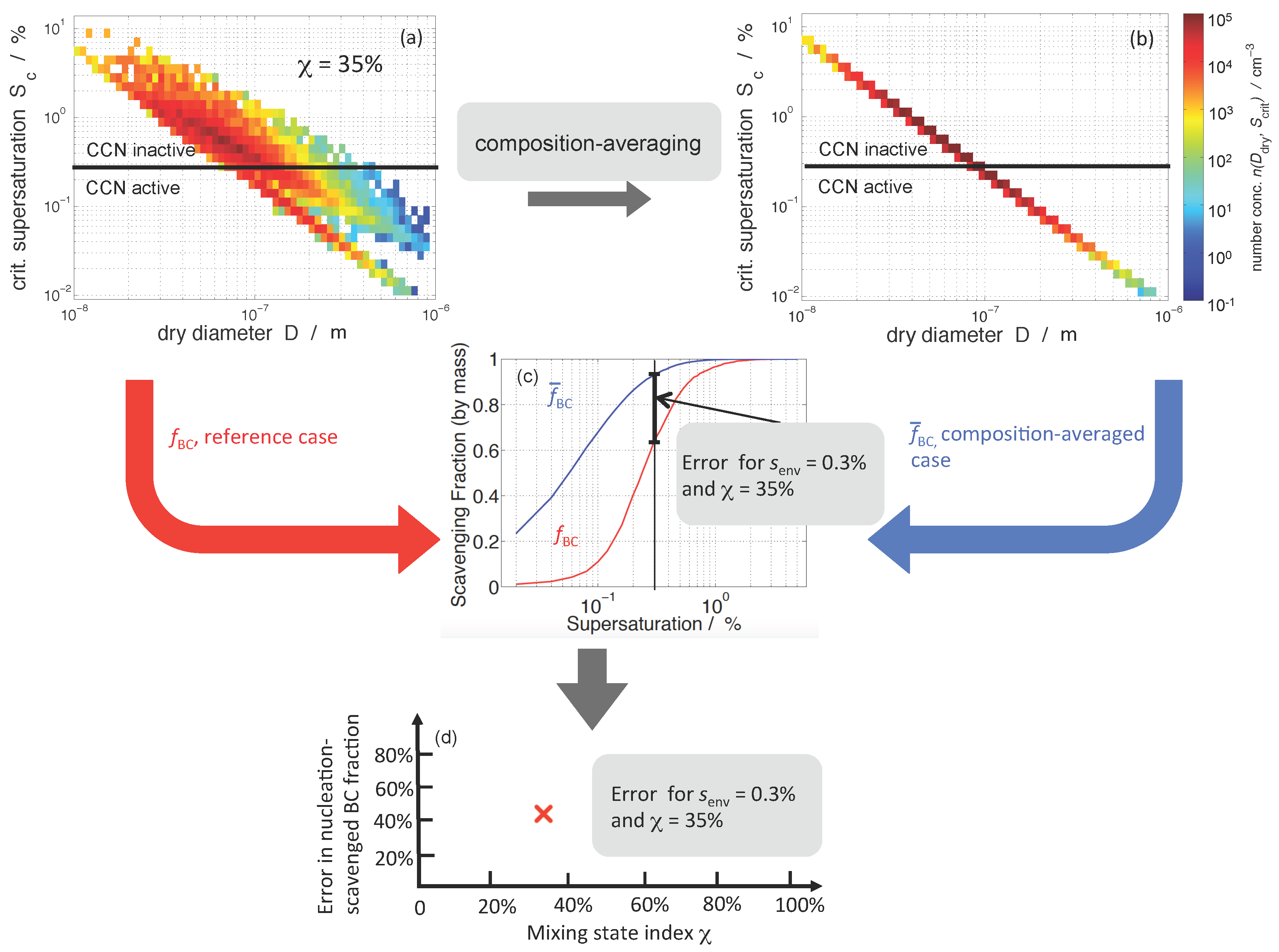
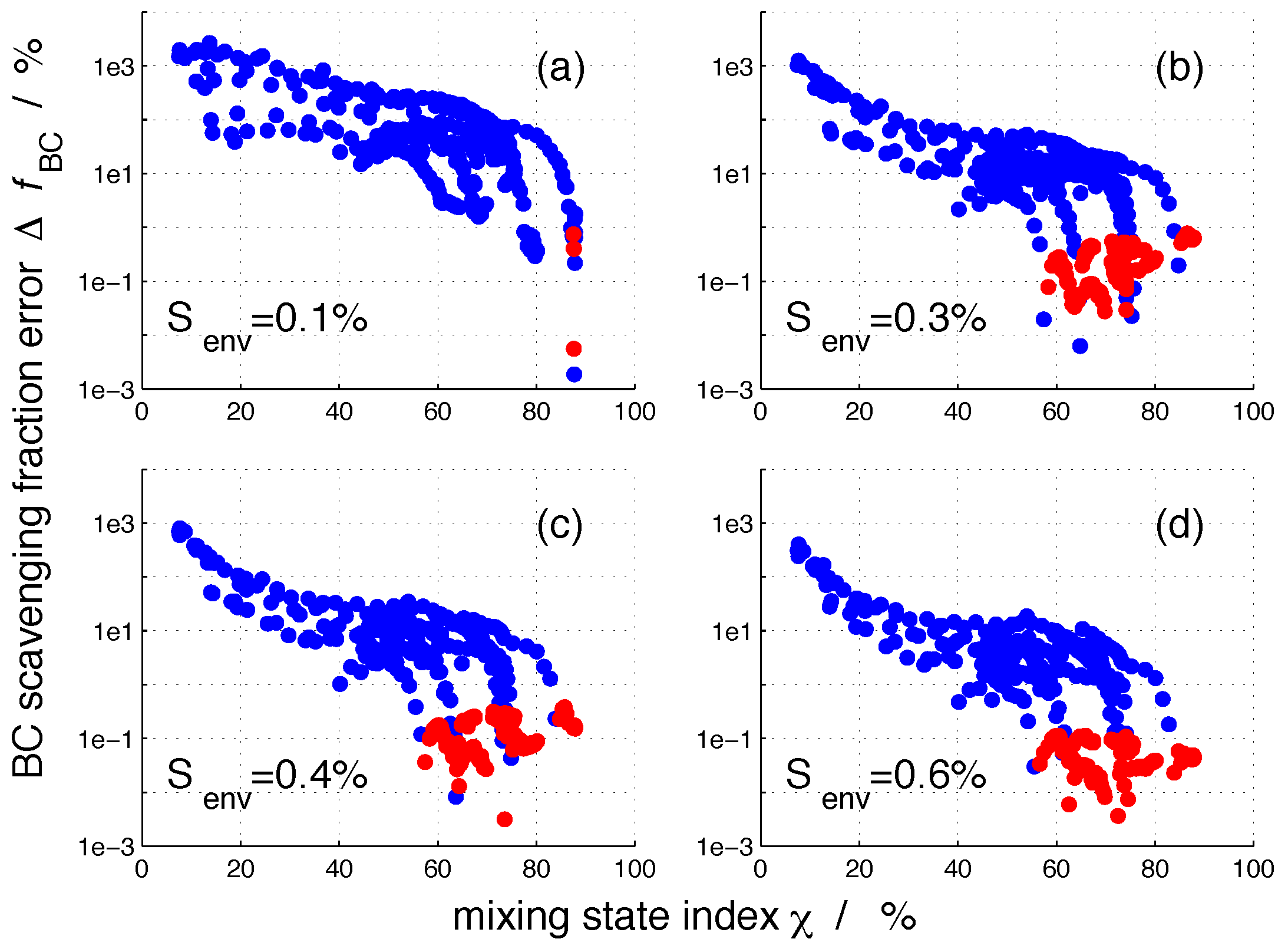
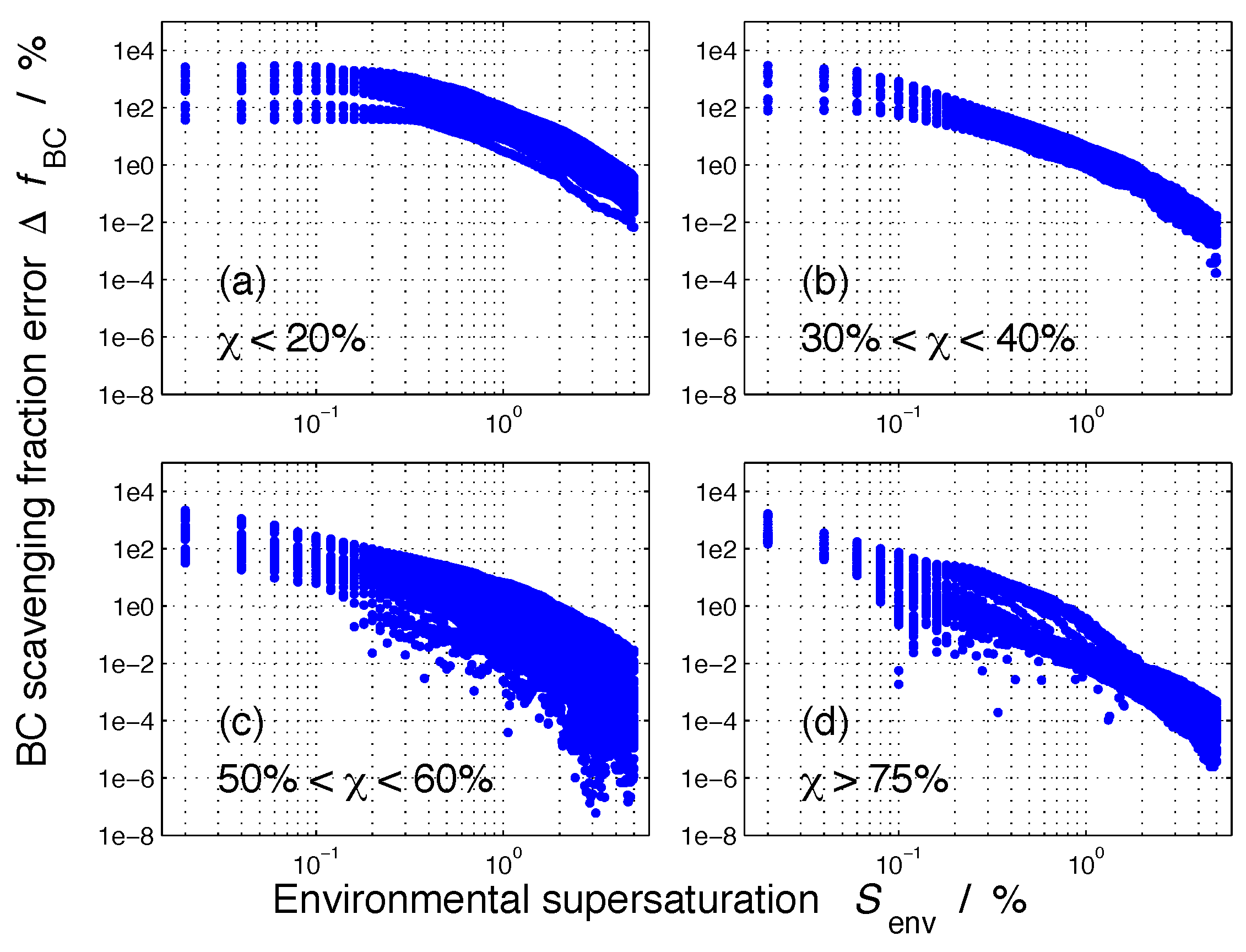
2.3. Framework of Error Quantification
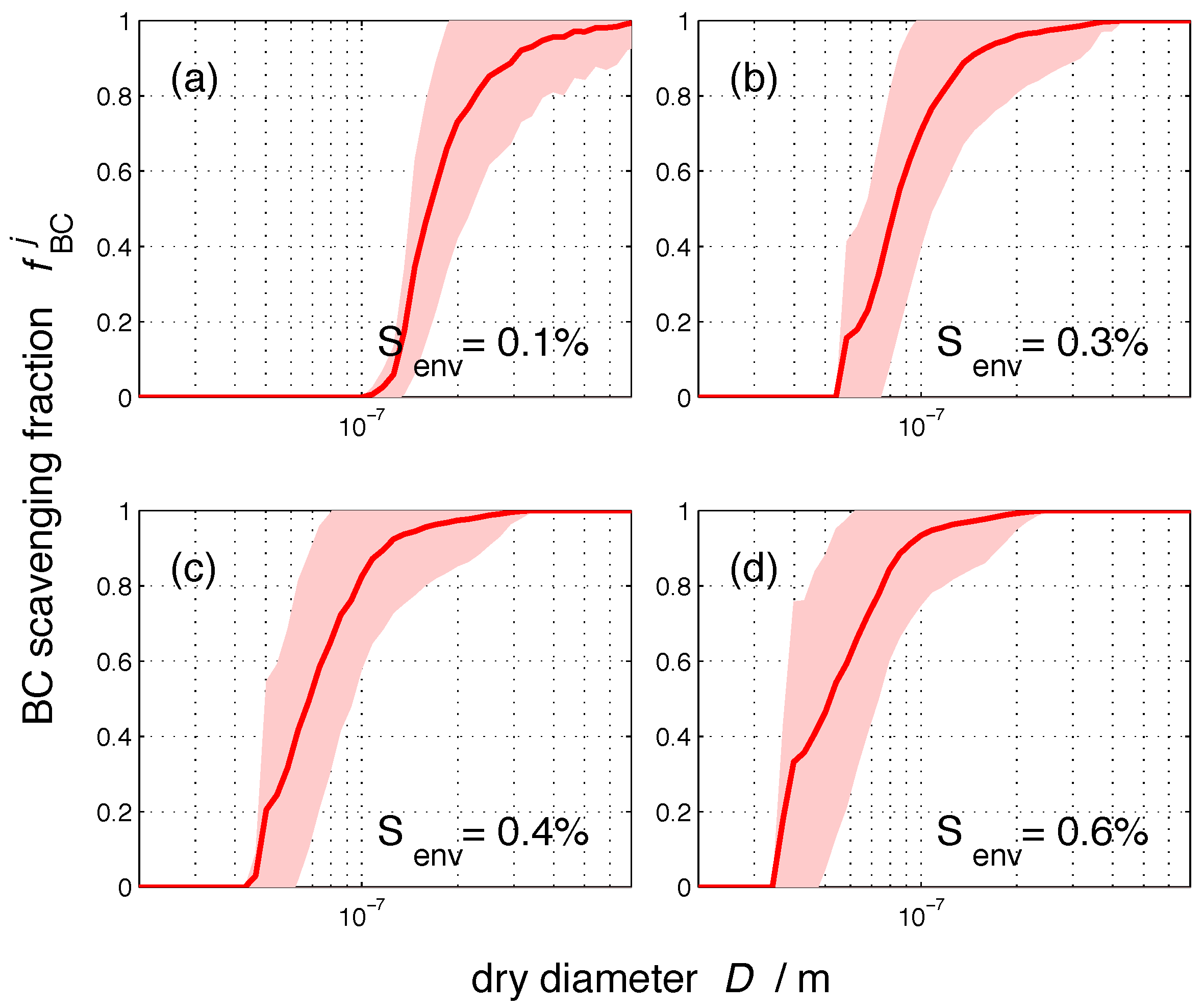
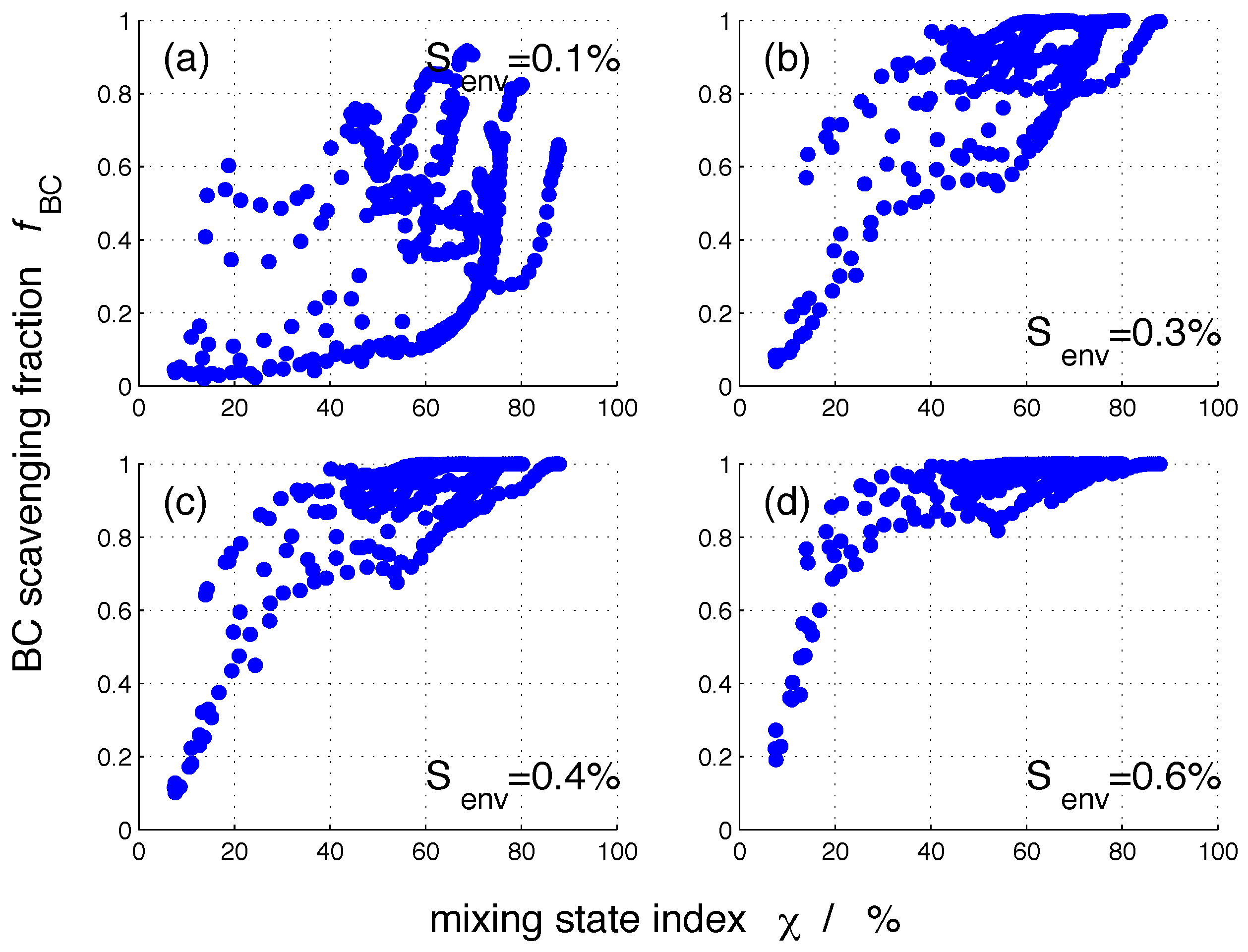
3. Results
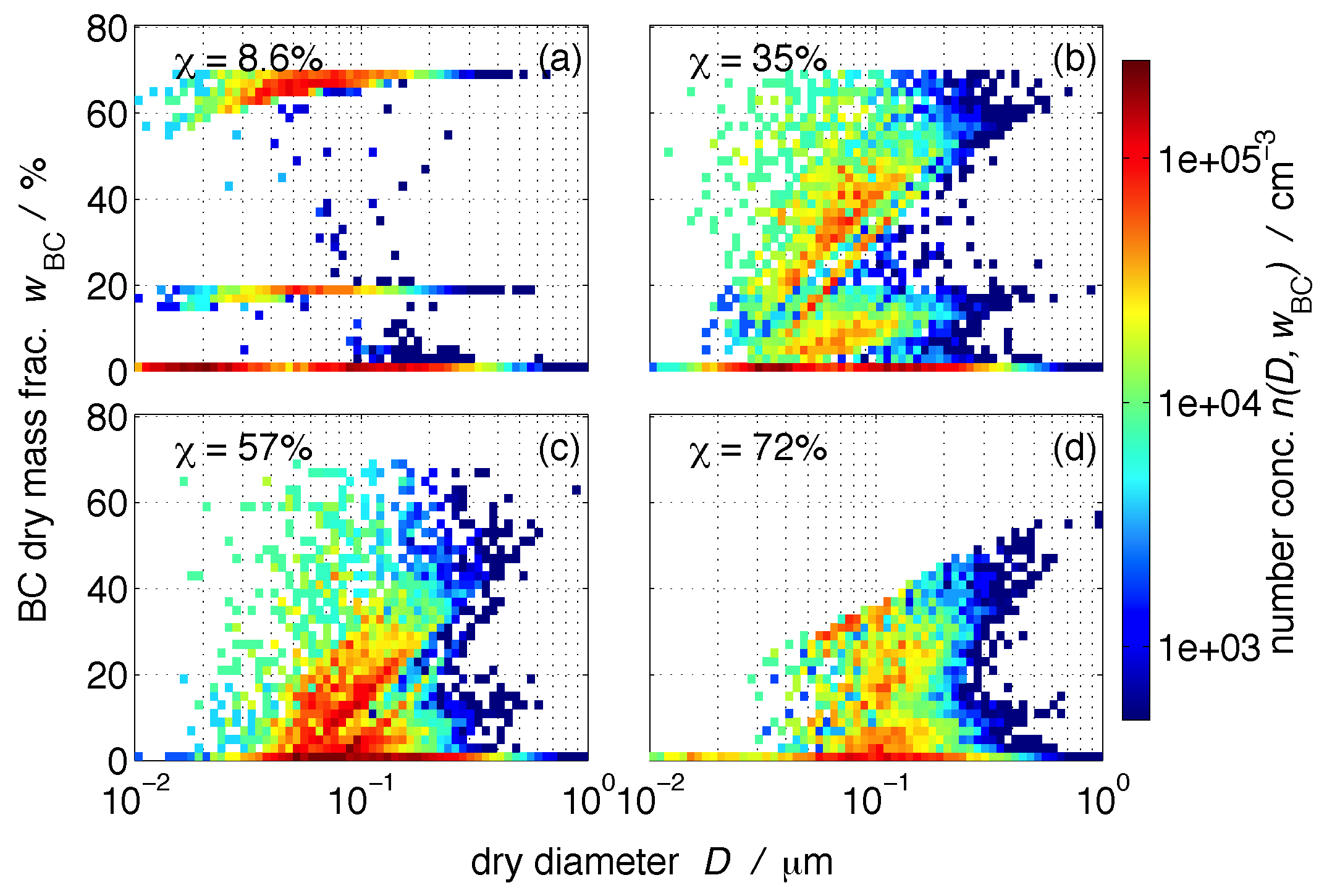
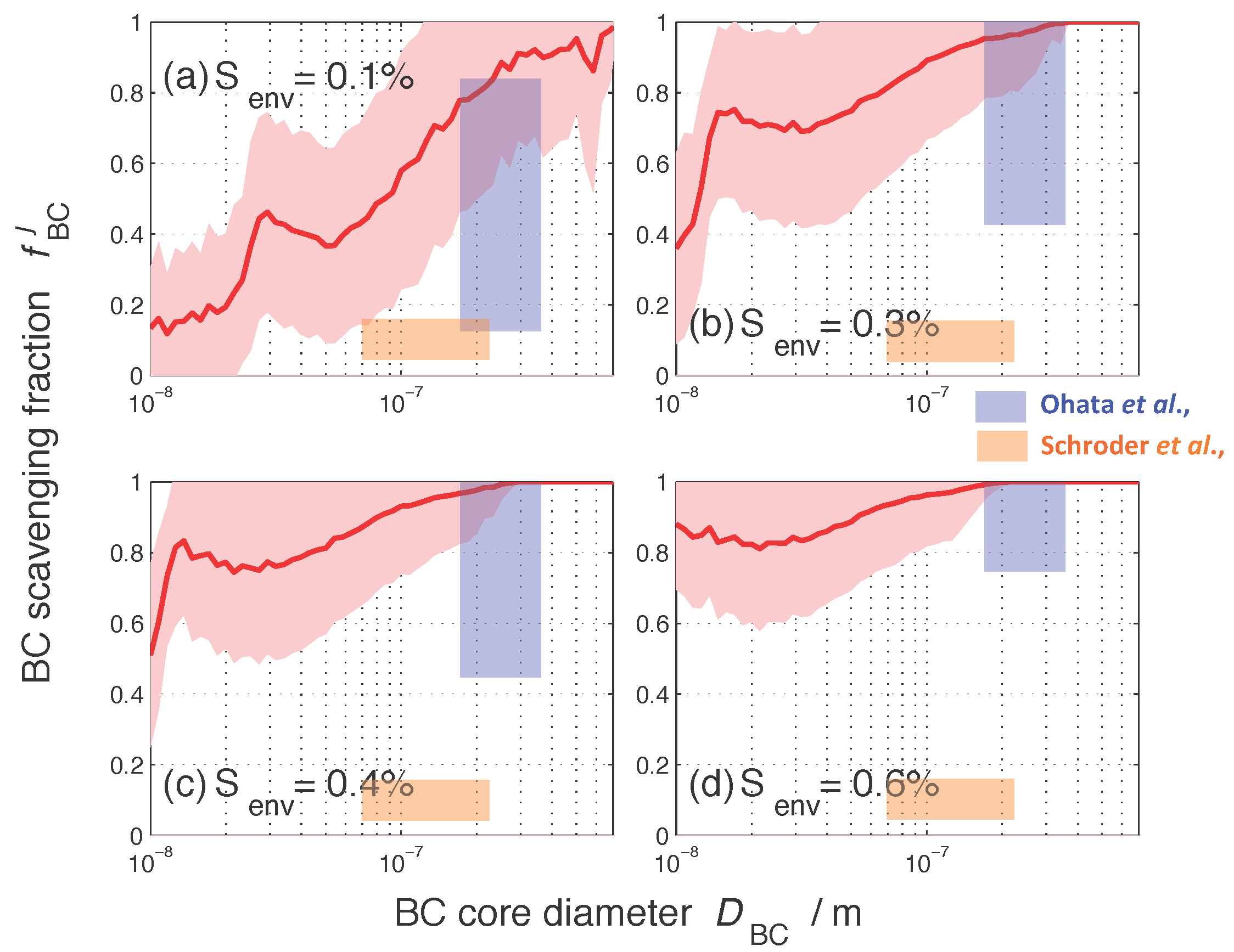
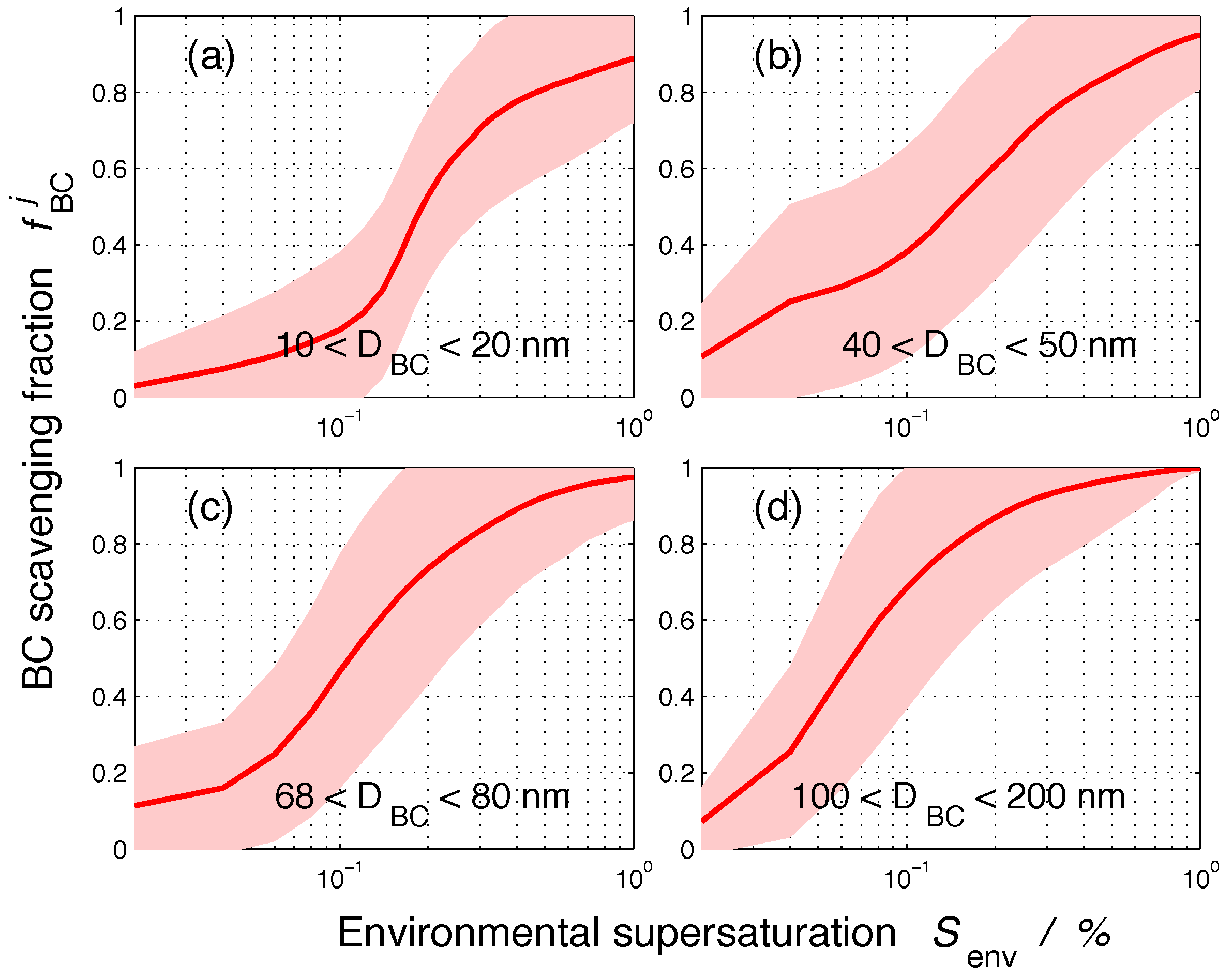
3.1. Size-Resolved Nucleation-Scavenged BC Mass Fraction
3.2. Nucleation-Scavenged BC Mass Fraction and Aerosol Mixing State
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Riemer, N.; West, M. Quantifying aerosol mixing state with entropy and diversity measures. Atmos. Chem. Phys. 2013, 13, 11423–11439. [Google Scholar] [CrossRef]
- Pöschl, U. Atmospheric Aerosols: Ccomposition, Transformation, Climate and Health Effects. Angew. Chem. Int. Ed. 2005, 44, 7520–7540. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.D.; Bodhaine, B.A.; Dutton, E.G.; Schnell, R.C. Aerosol black carbon measurement at the South Pole: Initial results 1986–1987. Geophys. Res. Lett. 1988, 15, 1193–1196. [Google Scholar] [CrossRef]
- Hansen, J.; Nazarenko, L. Soot climate forcing via snow and ice albedos. PNAS 2004, 101, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Bond, T.C.; Doherty, S.J.; Fahey, D.W.; Forester, P.M.; Berntsen, T.; DeAngelo, B.J.; Flanner, M.G.; Ghan, S.; Kärcher, B.; Koch, D.; et al. Bounding the role of black carbon in the climate system: A scientific assessment. J. Geophys. Res. 2013, 118, 5380–5552. [Google Scholar] [Green Version]
- Hansen, J.; Sato, M.; Ruedy, R. Radiative forcing and climate response. J. Geophys. Res. Atmos. 1997, 102, 6831–6864. [Google Scholar] [CrossRef]
- Ackerman, A.S.; Toon, O.B.; Stevens, D.E.; Heymsfield, A.J.; Ramanathan, V.; Welton, E.J. Reduction of tropical cloudiness by soot. Science 2000, 288, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Koch, D.; Schulz, M.; Kinne, S.; McNaughton, C.; Spackman, J.R.; Balkanski, Y.; Bauer, S.; Berntsen, T.; Bond, T.C.; Boucher, O.; et al. Evaluation of black carbon estimations in global aerosol models. Atmos. Chem. Phys. 2009, 9, 9001–9026. [Google Scholar] [CrossRef]
- Koch, D.; Genio, A.D.D. Black carbon semi-direct effects on cloud cover: Review and synthesis. Atmos. Chem. Phys. 2010, 10, 7685–7696. [Google Scholar] [CrossRef] [Green Version]
- Chylek, P.; Hallett, J. Enhanced absorption of solar radiation by cloud droplets containing soot particles in their surface. Q. J. R. Meteorol. Soc. 1992, 118, 167–172. [Google Scholar] [CrossRef]
- Wendisch, M.; Mertens, S.; Ruggaber, A.; Nakajima, T. Vertical profiles of aerosol and radiation and the influence of a temperature inversion: Measurements and radiative transfer calculations. J. Appl. Meteorol. 1996, 35, 1703–1715. [Google Scholar] [CrossRef]
- Flanner, M.G.; Zender, C.S.; Randerson, J.T.; Rasch, P.J. Present day climate forcing and response from black carbon in snow. J. Geophys. Res. 2007, 112. [Google Scholar] [CrossRef]
- Samset, B.H.; Myhre, G.; Herber, A.; Kondo, Y.; Li, S.M.; Moteki, N.; Koike, M.; Oshima, N.; Schwarz, J.P.; Balkanski, Y.; et al. Modelled black carbon radiative forcing and atmospheric lifetime in AeroCom Phase II constrained by aircraft observations. Atmos. Chem. Phys. 2014, 14, 12465–12477. [Google Scholar] [CrossRef]
- Wilcox, E.M.; Thomas, R.M.; Praveen, P.S.; Pistone, K.; Bender, F.A.M.; Ramanathan, V. Black carbon solar absorption suppresses turbulence in the atmospheric boundary layer. Proc. Natl. Acad. Sci. USA 2015, 113, 11794–11799. [Google Scholar] [CrossRef] [PubMed]
- Ding, A.; Huang, X.; Nie, W.; Sun, J.; Kerminen, V.M.; Petäjä, T.; Su, H.; Cheng, Y.; Yang, X.Q.; Wang, M.; et al. Enhanced haze pollution by black carbon in megacities in China. Geophys. Res. Lett. 2016, 43, 2873–2879. [Google Scholar] [CrossRef]
- Chen, W.; Lee, Y.H.; Adams, P.J.; Nenes, A.; Seinfeld, J.H. Will black carbon mitigation dampen aerosol indirect forcing? Geophys. Res. Lett. 2010, 37, L09801. [Google Scholar] [CrossRef]
- Bond, T.C.; Sun, H. Can reducing black carbon emissions counteract global warming? Environ. Sci. Technol. 2005, 39, 5921–5926. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.Z. Short-term effects of controlling fossil-fuel soot, biofuel soot and gases, and methane on climate, Arctic ice, and air pollution health. J. Geophys. Res. 2010, 115. [Google Scholar] [CrossRef]
- Ohata, S.; Moteki, N.; Mori, T.; Koike, M.; Kondo, Y. A key process controlling the wet removal of aerosols: New observational evidence. Sci. Rep. 2016, 6, 34113. [Google Scholar] [CrossRef] [PubMed]
- Schroder, J.C.; Hanna, S.J.; Modini, R.L.; Corrigan, A.L.; Kreidenwies, S.M.; Macdonald, A.M.; Noone, K.J.; Russell, L.M.; Leaitch, W.R.; Bertram, A.K. Size-resolved observations of refractory black carbon particles in cloud droplets at a marine boundary layer site. Atmos. Chem. Phys. 2015, 15, 1367–1383. [Google Scholar] [CrossRef]
- Weingartner, E.; Burtscher, H.; Baltensperger, H. Hygroscopic properties of carbon and diesel soot particles. Atmos. Environ. 1997, 31, 2311–2327. [Google Scholar] [CrossRef]
- Hitzenberger, R.; Berner, A.; Giebl, H.; Drobesch, K.; Kasper-Giebl, A.; Loeflund, M.; Urban, H.; Puxbaum, H. Black carbon (BC) in alpine aerosols and cloud water—Concentrations and scavenging efficiencies. Atmos. Environ. 2001, 35, 5135–5141. [Google Scholar] [CrossRef]
- Cozic, J.; Verheggen, B.; Mertes, S.; Connolly, P.; Bower, K.; Petzold, A.; Baltensperger, U.; Weingartner, E. Scavenging of black carbon in mixed phase clouds at the high alpine site Jungfraujoch. Atmos. Chem. Phys. 2007, 7, 1797–1807. [Google Scholar] [CrossRef]
- Hallberg, A.; Ogren, J.; Noone, K.; Heintzenberg, J.; Berner, A.; Solly, I.; Kruisz, C.; Reischl, G.; Fuzzi, S.; Facchini, M.; et al. Phase partitioning for different aerosol species in fog. Tellus B 1992, 44, 545–555. [Google Scholar] [CrossRef]
- Hallberg, A.; Noone, K.; Ogren, J.; Svenningsson, I.; Flossmann, A.; Wiedensohler, A.; Hansson, H.C.; Heintzenberg, J.; Anderson, T.; Arends, B.; et al. Phase partitioning of aerosol particles in clouds at Kleiner Feldberg. J. Atmos. Chem. 1994, 19, 107–127. [Google Scholar] [CrossRef]
- Sellegri, K.; Laj, P.; Dupuy, R.; Legrand, M.; Preunkert, S.; Putaud, J.P. Size-dependent scavenging efficiencies of multicomponent atmospheric aerosols in clouds. J. Geophys. Res. Atmos. 2003, 108. [Google Scholar] [CrossRef]
- Kasper-Giebl, A.; Koch, A.; Hitzenberger, R.; Puxbaum, H. Scavenging efficiency of ‘aerosol carbon’ and sulfate in supercooled clouds at Mt. Sonnblick (3106 m asl, Austria). J. Atmos. Chem. 2000, 35, 33–46. [Google Scholar] [CrossRef]
- Gieray, R.; Wieser, P.; Engelhardt, T.; Swietlicki, E.; Hansson, H.C.; Mentes, B.; Orsini, D.; Martinsson, B.; Svenningsson, B.; Noone, K.; et al. Phase partitioning of aerosol constituents in cloud based on single-particle and bulk analysis. Atmos. Environ. 1997, 31, 2491–2502. [Google Scholar] [CrossRef]
- Hitzenberger, R.; Berner, A.; Kromp, R.; Kasper-Giebl, A.; Limbeck, A.; Tscherwenka, W.; Puxbaum, H. Black carbon and other species at a high-elevation European site (Mount Sonnblick, 3106 m, Austria): Concentrations and scavenging efficiencies. J. Geophys. Res. Atmos. 2000, 105, 24637–24645. [Google Scholar] [CrossRef]
- Heintzenberg, J.; Leck, C. Seasonal variation of the atmospheric aerosol near the top of the marine boundary layer over Spitsbergen related to the Arctic sulphur cycle. Tellus B Chem. Phys. Meteorol. 1994, 46, 52–67. [Google Scholar] [CrossRef]
- Winkler, P. The growth of atmosphierc aerosol particles as a function of the relative humidity—II. an improved concept of mixed nuclei. Aerosol Sci. 1973, 4, 373–387. [Google Scholar] [CrossRef]
- Ching, J.; Riemer, N.; West, M. Impacts of black carbon mixing state on black carbon nucleation scavenging: Insights from a particle-resolved model. J. Geophys. Res. Atmos. 2012, 117. [Google Scholar] [CrossRef]
- Ching, J.; Riemer, N.; West, M. Impacts of black carbon particles mixing state on cloud microphysical properties: Sensitivity to environmental conditions. J. Geophys. Res. Atmos. 2016, 121, 5990–6013. [Google Scholar] [CrossRef]
- Ching, J.; Fast, J.; West, M.; Riemer, N. Metrics to quantify the importance of mixing state for CCN activity. Atmos. Chem. Phys. 2017, 17, 7445–7458. [Google Scholar] [CrossRef]
- Riemer, N.; West, M.; Zaveri, R.; Easter, R. Simulating the evolution of soot mixing state with a particle-resolved aerosol model. J. Geophys. Res. Atmos. 2009, 114, D09202. [Google Scholar] [CrossRef]
- Zaveri, R.A.; Easter, R.C.; Fast, J.D.; Peters, L.K. Model for Simulating Aerosol Interactions and Chemistry (MOSAIC). J. Geophys. Res. Atmos. 2008, 113, D13204. [Google Scholar] [CrossRef]
- Zaveri, R.A.; Peters, L.K. A new lumped structure photochemical mechanism for large-scale applications. J. Geophys. Res. Atmos. 1999, 104, 30387–30415. [Google Scholar] [CrossRef]
- Schell, B.; Ackermann, I.J.; Binkowski, F.S.; Ebel, A. Modeling the formation of secondary organic aerosol within a comprehensive air quality model system. J. Geophys. Res. 2001, 106, 28275–28293. [Google Scholar] [CrossRef]
- Zaveri, R.; Barnard, J.; Easter, R.; Riemer, N.; West, M. Particle-resolved simulation of aerosol size, composition, mixing state, and the associated optical and cloud condensation nuclei activation properties in an evolving urban plume. J. Geophys. Res. Atmos. 2010, 115, D17210. [Google Scholar] [CrossRef]
- Kaiser, J.; Hendricks, J.; Righi, M.; Riemer, N.; Zaveri, R.A.; Metzger, S.; Aquila, V. The MESSy aerosol submodel MADE3 (v2. 0b): Description and a box model test. Geosci. Model Dev. 2014, 7, 1137–1157. [Google Scholar] [CrossRef] [Green Version]
- Fierce, L.; Bond, T.C.; Bauer, S.E.; Mena, F.; Riemer, N. Black carbon absorption at the global scale is affected by particle-scale diversity in composition. Nat. Commun. 2016, 7, 12361. [Google Scholar] [CrossRef] [PubMed]
- Fierce, L.; Riemer, N.; Bond, T.C. Toward reduced representation of mixing state for simulating aerosol effects on climate. Bull. Am. Meteorol. Soc. 2017, 98, 971–980. [Google Scholar] [CrossRef]
- Tian, J.; Brem, B.; West, M.; Bond, T.; Rood, M.; Riemer, N. Simulating aerosol chamber experiments with the particle-resolved aerosol model PartMC. Aerosol Sci. Technol. 2017, 51, 856–867. [Google Scholar] [CrossRef]
- Riemer, N.; West, M.; Zaveri, R.; Easter, R. Estimating black carbon aging time-scales with a particle-resolved aerosol model. J. Aerosol Sci. 2010, 41, 143–158. [Google Scholar] [CrossRef]
- Fierce, L.; Riemer, N.; Bond, T.C. Explaining variance in black carbon’s aging timescale. Atmos. Chem. Phys. 2015, 15, 3173–3191. [Google Scholar] [CrossRef]
- United States Environmental Protection Agency. Report to Congress on Black Carbon; Technical Report EPA-450/R-12-001; United States Environmental Protection Agency: Washington, DC, USA, 2012.
- Petters, M.D.; Kreidenweis, S.M. A single parameter representation of hygroscopic growth and cloud condensation nucleus activity. Atmos. Chem. Phys. 2007, 7, 1961–1971. [Google Scholar] [CrossRef]
- Curtis, J.H.; Riemer, N.; West, M. A single-column particle-resolved model for simulating the vertical distribution of aerosol mixing state: WRF-PartMC-MOSAIC-SCM v1.0. Geosci. Model Dev. 2017, 10, 4057–4079. [Google Scholar] [CrossRef]
- Oshima, N.; Kondo, Y.; Moteki, N.; Takegawa, N.; Koike, M.; Kita, K.; Matsui, H.; Kajino, M.; Nakamura, H.; Jung, J.; et al. Wet removal of black carbon in Asian outflow: Aerosol Radiative Forcing in East Asia (A-FORCE) aircraft campaign. J. Geophys. Res. 2012, 117. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Site | Citations | Environment | Average Scavenged BC Mass Fraction |
|---|---|---|---|
| Po Valley, Italy | [24] | Urban | 0.06 |
| Kleiner Feldberg, Germany | [25] | Rural | 0.15 |
| Puy de Dome, France | [26] | Mid altitude (1465 m) | 0.33 |
| Mt. Sonnblick, Austria | [27] | Mid altitude (3106 m) | 0.45 |
| Rax, Austria | [22] | Mid altitude (1644 m) | 0.54 |
| Great Dun Fell, UK | [28] | Rural-Coastal | 0.57 |
| Jungfraujoch, Switzerland | [23] | High altitude (3850 m) | 0.61 |
| Mt. Sonnblick, Austria | [29] | High altitude (3106 m) | 0.74 |
| Spitzbergen, Norway | [30] | Arctic | 0.80 |
| Mt. Soledad, La Jolla, USA | [20] | Marine-Coastal | 0.01–0.1 |
| Tokyo, Japan | [19] | Urban | 0.1–1.0 |
| Emission | Emission Strength () | Mean Diameter (m) | Geometric Standard Deviation | Composition by Mass |
|---|---|---|---|---|
| Meat cooking | 0.0864 | 1.9 | POA | |
| Diesel vehicles | 0.05 | 1.7 | POA, BC | |
| Gasoline vehicles | 0.05 | 1.7 | POA, BC |
| Initial/Background | Number Concentration () | Mean Diameter (m) | Geometric Standard Deviation | Composition by Mass |
|---|---|---|---|---|
| Aitken mode | 0.02 | 1.45 | ||
| SOA | ||||
| BC | ||||
| Accumulation mode | 0.116 | 1.65 | ||
| SOA | ||||
| BC |
| Chemical Species | Mixing Ratio (ppbv) | Emission Flux () |
|---|---|---|
| Nitrogen oxide | 0.1 | 15.9 |
| Nitrogen dioxide | 1.0 | 0.84 |
| Nitric acid | 1.0 | - |
| Ozone | 50.0 | - |
| Hydrogen peroxide | 1.1 | - |
| Carbon monoxide | 80 | 291.3 |
| Sulfur dioxide | 0.8 | 2.51 |
| Ammonia | 0.5 | 6.11 |
| Hydrogen chloride | 0.7 | - |
| Methane | 2200 | - |
| Ethane | 1.0 | - |
| Formaldehyde | 1.2 | 1.68 |
| Methanol | 0.12 | 0.28 |
| Methyl hydrogen peroxide | 0.5 | - |
| Acetaldehyde | 1.0 | 0.68 |
| Paraffin carbon | 2.0 | 96.0 |
| Acetone | 1.0 | 1.23 |
| Ethene | 0.2 | 7.28 |
| Terminal olefin carbons | 2.43 | |
| Internal olefin carbons | 2.43 | |
| Toluene | 0.1 | 4.04 |
| Xylene | 0.1 | 2.41 |
| Lumped organic nitrate | 0.1 | - |
| Peroxyacetyl nitrate | 0.8 | - |
| Higher organic acid | 0.2 | - |
| Higher organic peroxide | - | |
| Isoprene | 0.5 | 0.23 |
| Alcohols | - | 3.45 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ching, J.; West, M.; Riemer, N. Quantifying Impacts of Aerosol Mixing State on Nucleation-Scavenging of Black Carbon Aerosol Particles. Atmosphere 2018, 9, 17. https://doi.org/10.3390/atmos9010017
Ching J, West M, Riemer N. Quantifying Impacts of Aerosol Mixing State on Nucleation-Scavenging of Black Carbon Aerosol Particles. Atmosphere. 2018; 9(1):17. https://doi.org/10.3390/atmos9010017
Chicago/Turabian StyleChing, Joseph, Matthew West, and Nicole Riemer. 2018. "Quantifying Impacts of Aerosol Mixing State on Nucleation-Scavenging of Black Carbon Aerosol Particles" Atmosphere 9, no. 1: 17. https://doi.org/10.3390/atmos9010017
APA StyleChing, J., West, M., & Riemer, N. (2018). Quantifying Impacts of Aerosol Mixing State on Nucleation-Scavenging of Black Carbon Aerosol Particles. Atmosphere, 9(1), 17. https://doi.org/10.3390/atmos9010017