Bacterial and Archaeal Assemblages from Two Size Fractions in Submarine Groundwater Near an Industrial Zone


Abstract
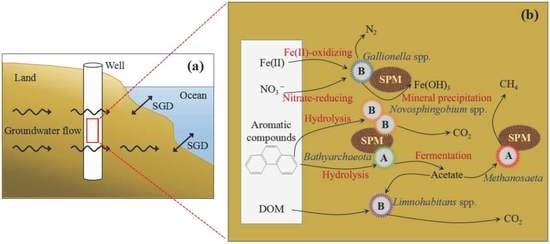
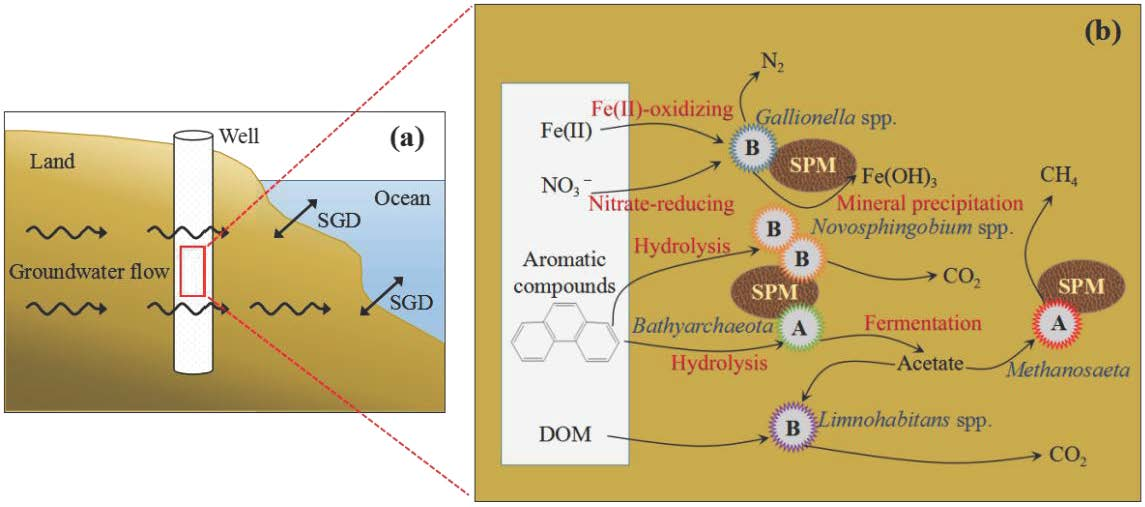
:
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Measurements of Chemical Parameters
2.2. DNA Extraction, PCR Amplification and Illumina MiSeq Sequencing
2.3. Sequence Data Processing, OTU Clustering, and Taxonomic Assignment
2.4. Phylogenetic Analyses
2.5. Statistical Analyses
2.6. Predictive Functional Profiling
3. Results
3.1. Site Description and Environmental Characteristics
3.2. Bacterial and Archaeal Diversity
3.3. Bacterial and Archaeal Distribution
3.4. Phylogenetic Analyses
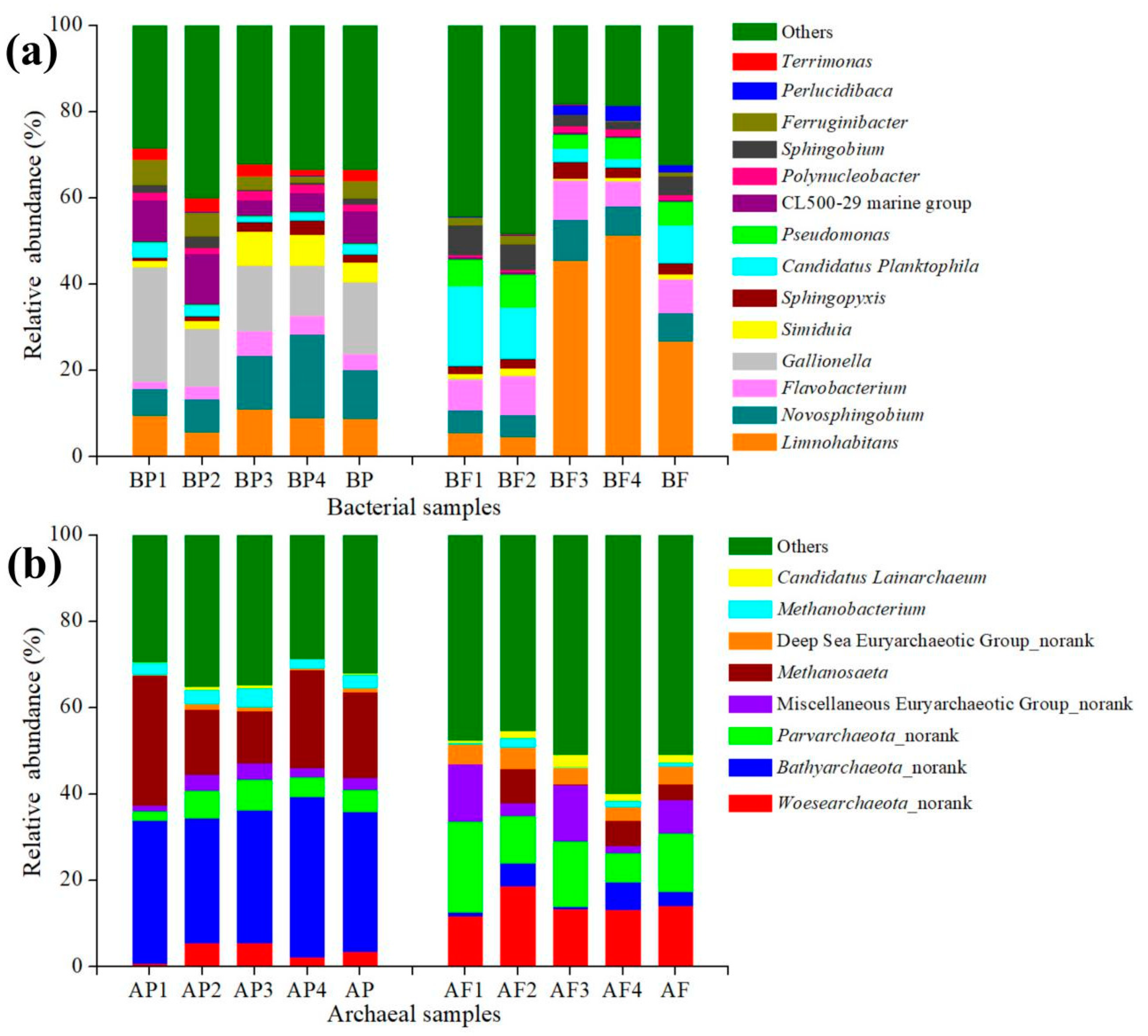
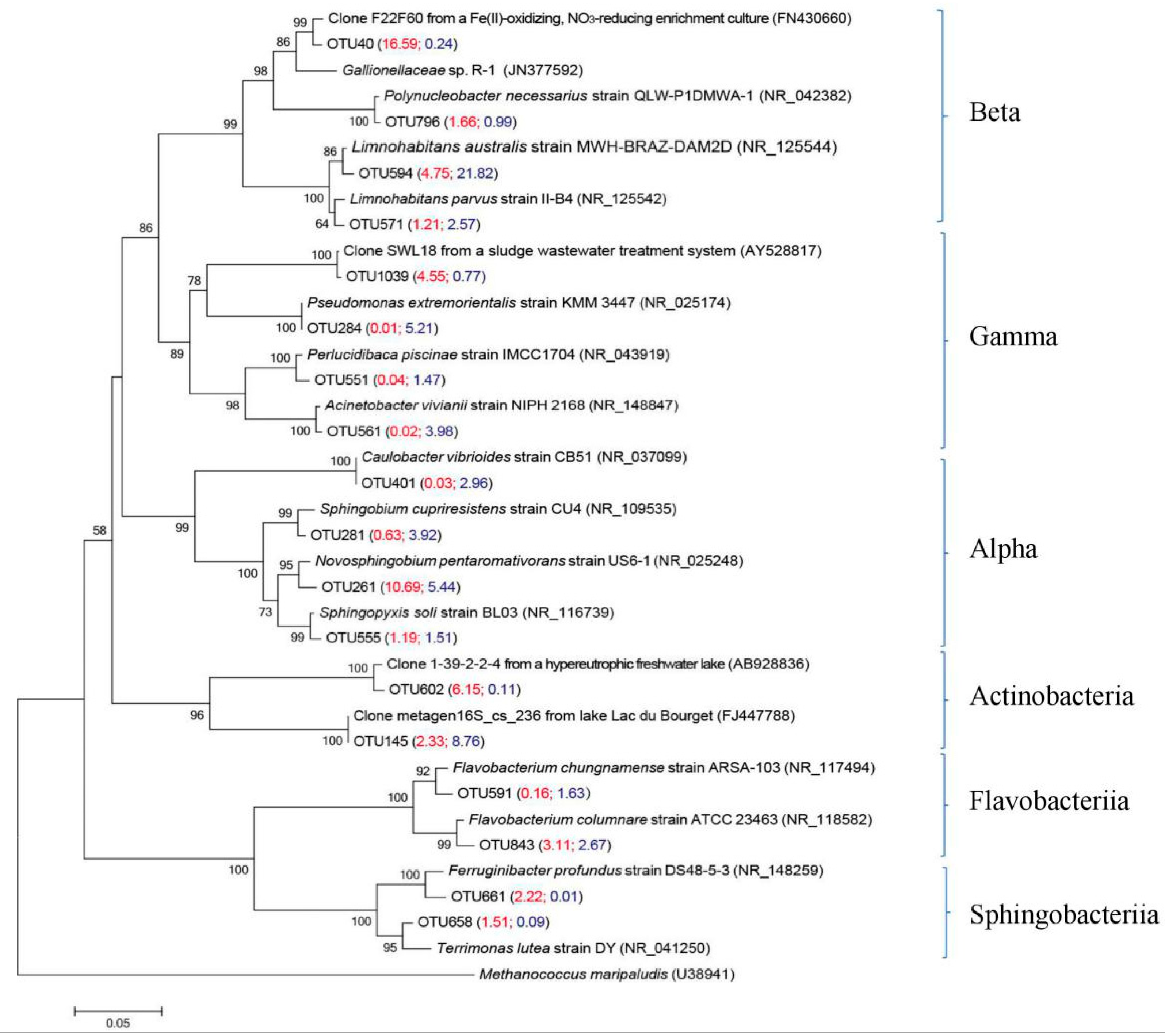
3.4.1. Main Bacterial Groups in Two Size Fractions
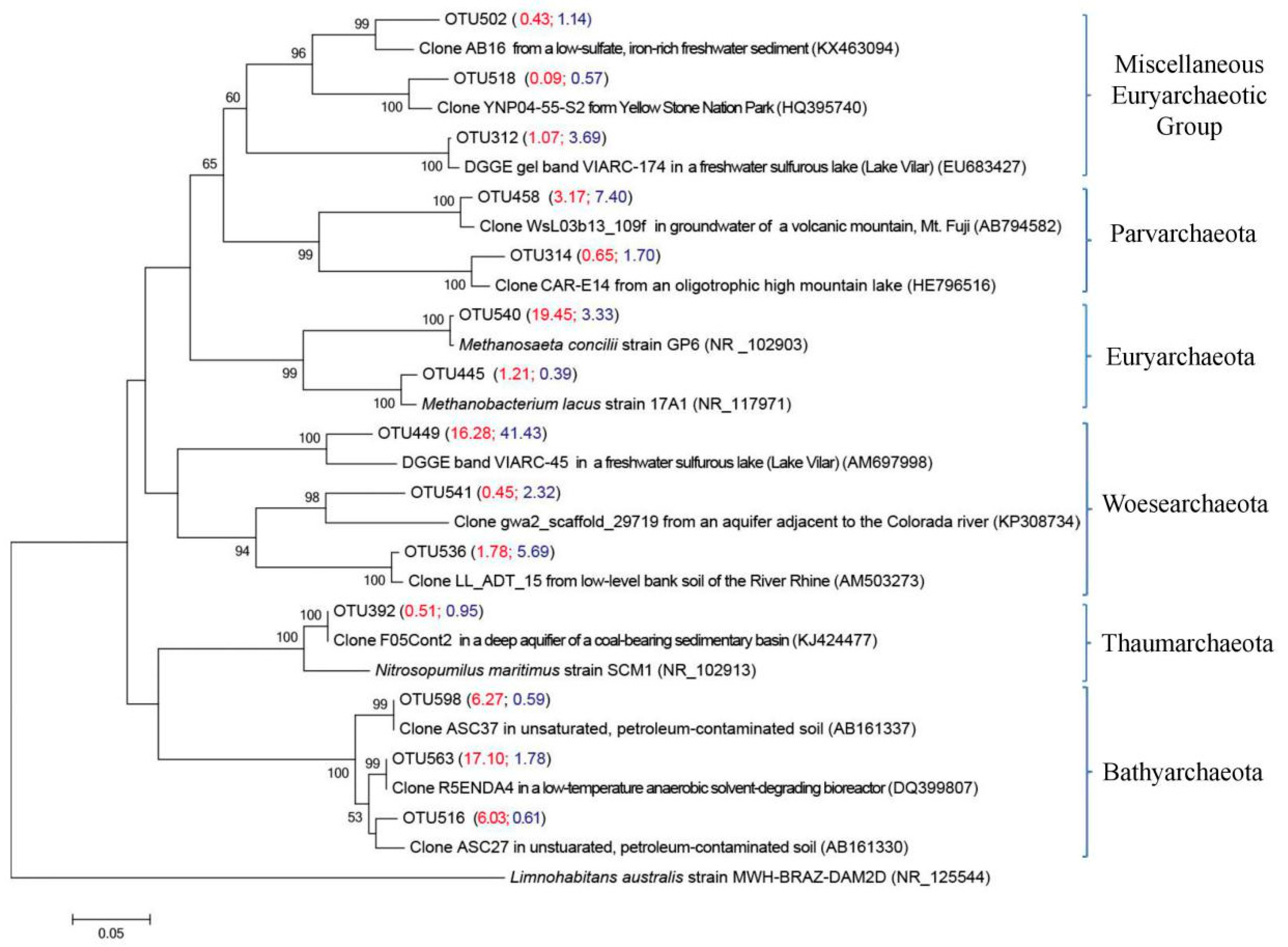
3.4.2. Main Archaeal Groups in Two Size Fractions
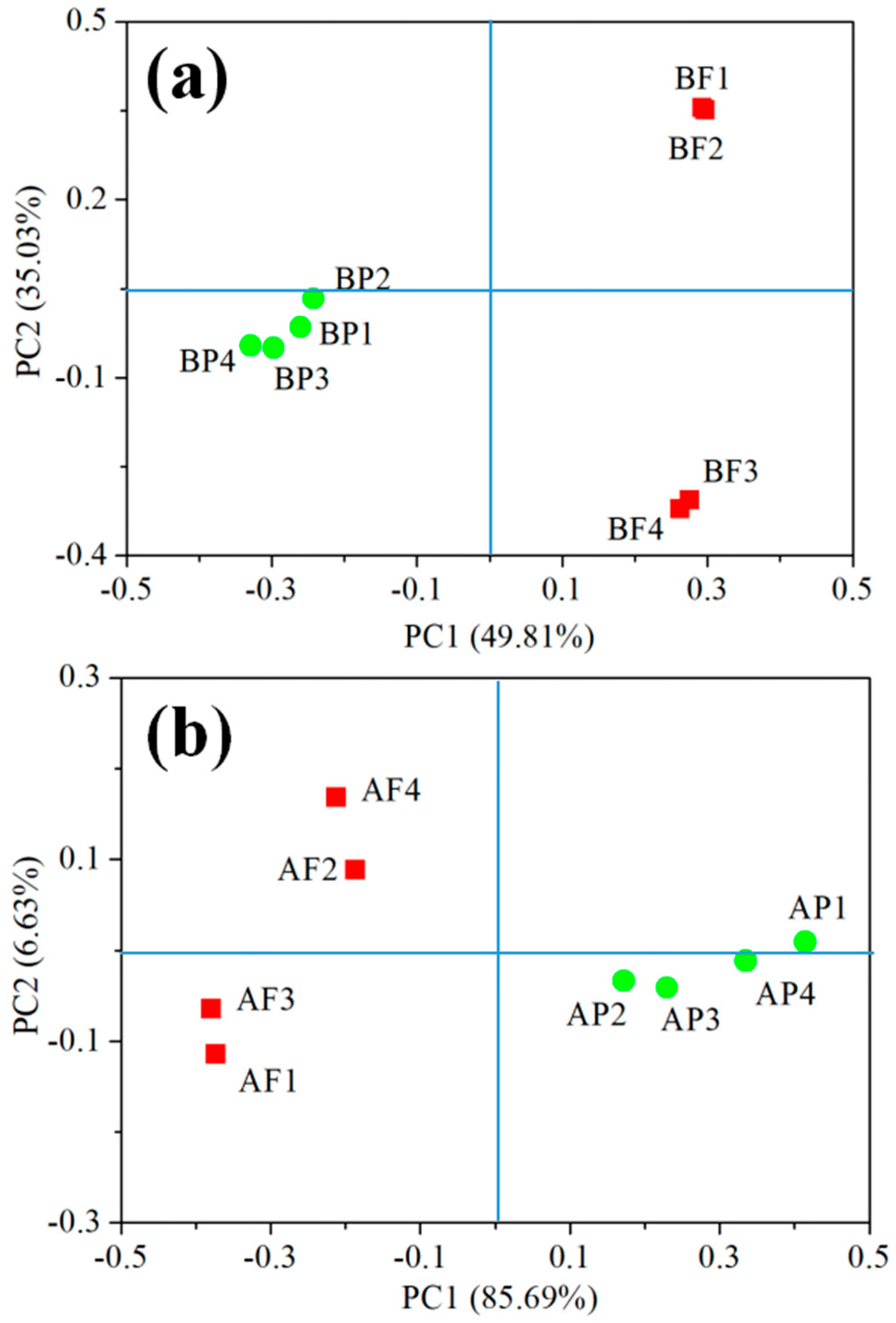
3.5. Distinct Bacterial and Archaeal Communities in Two Size Fractions
4. Discussion
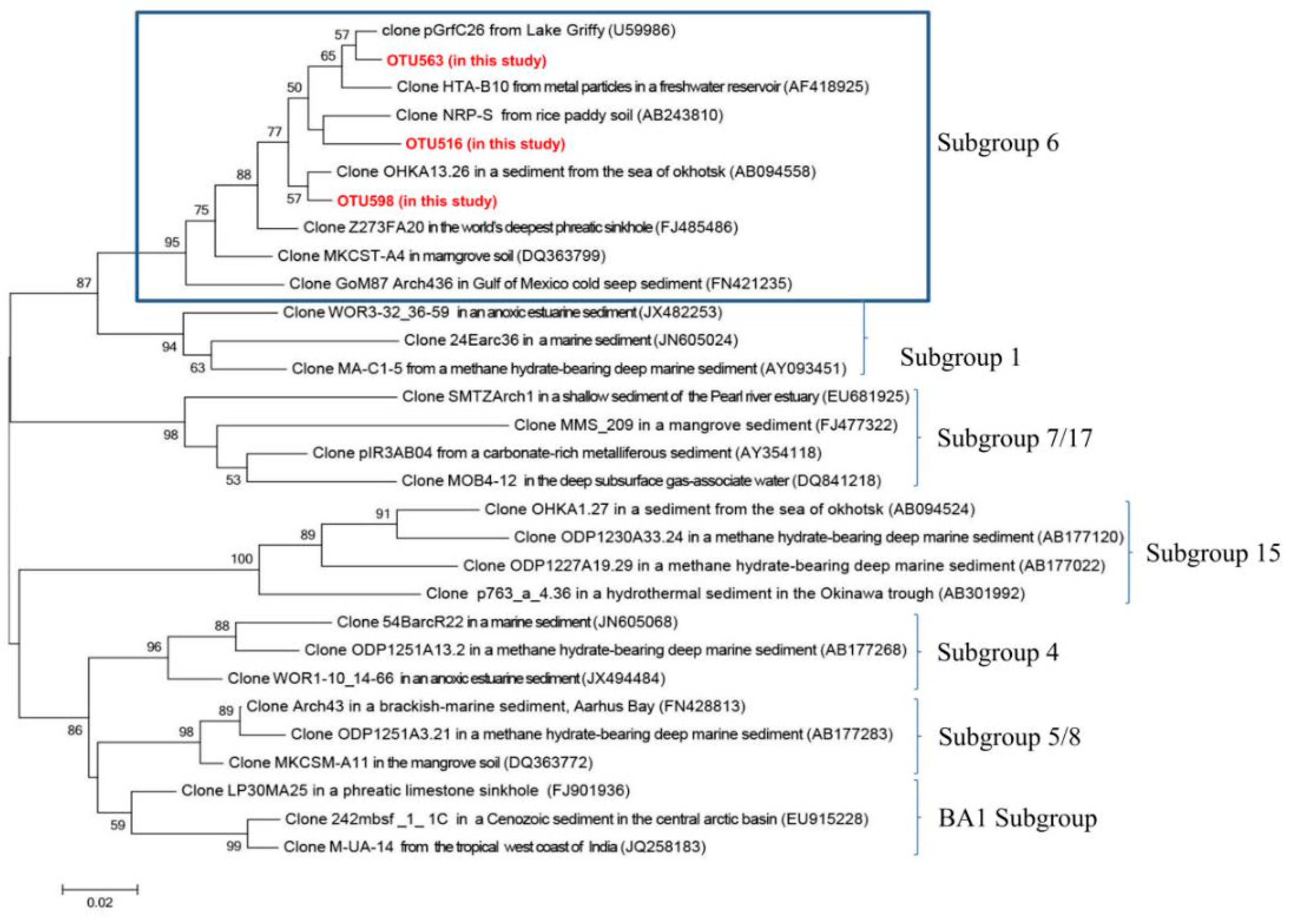
4.1. The Ecological Niches of Bathyarchaeota in Submarine Groundwater Systems
4.2. Microbial Candidate for In Situ Bioremediation in SGD
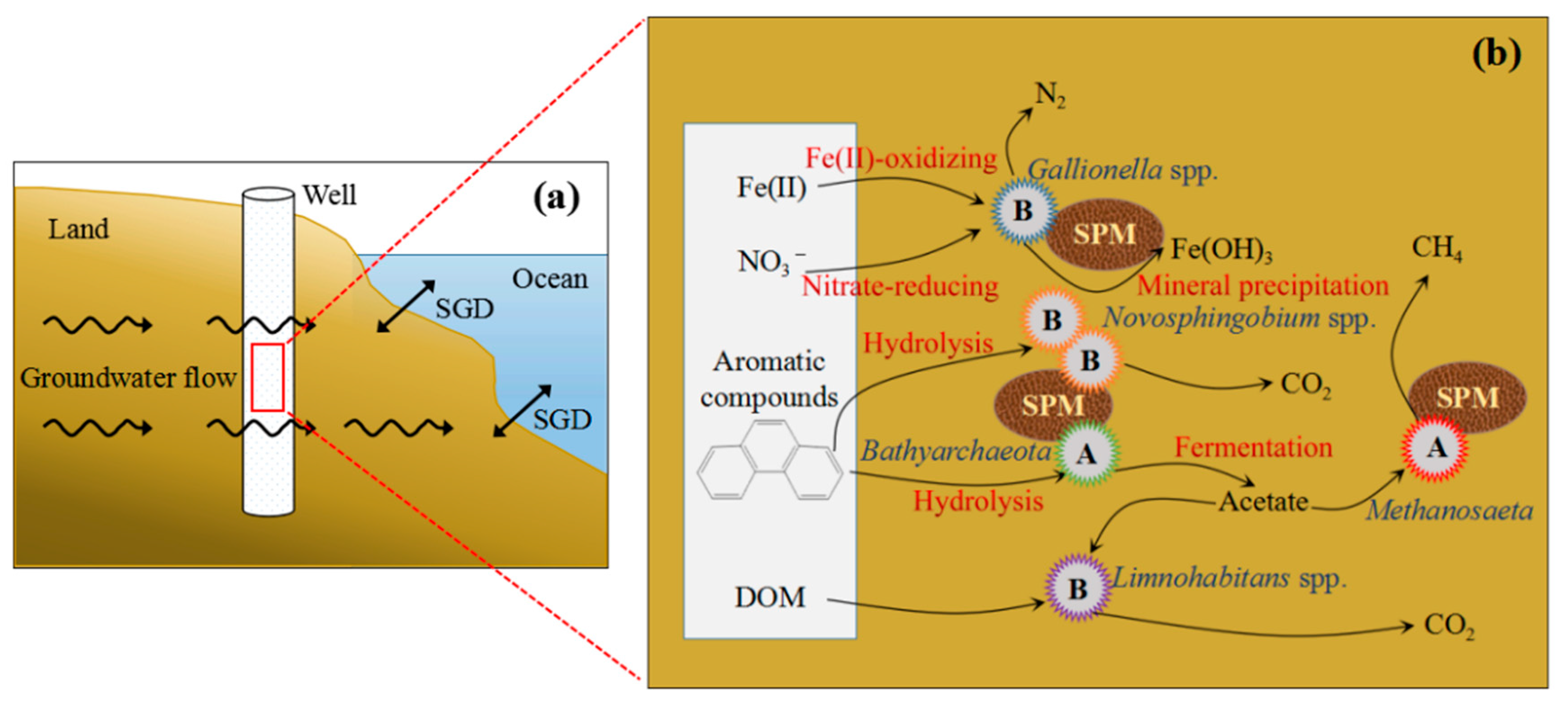
4.3. Influence of Key Microbes on SGD in Qinzhou Bay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burnett, W.C.; Bokuniewicz, H.; Huettel, M.; Moore, W.S.; Taniguchi, M. Groundwater and pore water inputs to the coastal zone. Biogeochemistry 2003, 66, 3–33. [Google Scholar] [CrossRef]
- Moore, W.S. The subterranean estuary: A reaction zone of ground water and sea water. Mar. Chem. 1999, 65, 111–125. [Google Scholar] [CrossRef]
- Moore, W.S. The effect of submarine groundwater discharge on the ocean. Annu. Rev. Mar. Sci. 2010, 2, 59–88. [Google Scholar] [CrossRef] [PubMed]
- Pavlidou, A.; Papadopoulos, V.P.; Hatzianestis, I.; Simboura, N.; Patiris, D.; Tsabaris, C. Chemical inputs from a karstic submarine groundwater discharge (SGD) into an oligotrophic Mediterranean coastal area. Sci. Total Environ. 2014, 488, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, J.; Cukrov, N.; Du, J. Porewater-derived nutrient fluxes in a coastal aquifer (Shengsi Island, China) and its implication. Estuar. Coast. Shelf Sci. 2019, 218, 204–211. [Google Scholar] [CrossRef]
- Luo, X.; Jiao, J.J. Submarine groundwater discharge and nutrient loadings in Tolo Harbor, Hong Kong using multiple geotracer-based models, and their implications of red tide outbreaks. Water Res. 2016, 102, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.; Lee, Y.W.; Kim, G. Large submarine groundwater discharge and benthic eutrophication in Bangdu Bay on volcanic Jeju Island, Korea. Limnol. Oceanogr. 2005, 50, 1393–1403. [Google Scholar] [CrossRef]
- McCoy, C.; Viso, R.; Peterson, R.N.; Libes, S.; Lewis, B.; Ledoux, J.; Voulgaris, G.; Smith, E.; Sanger, D. Radon as an indicator of limited cross-shelf mixing of submarine groundwater discharge along an open ocean beach in the South Atlantic Bight during observed hypoxia. Cont. Shelf Res. 2011, 31, 1306–1317. [Google Scholar] [CrossRef]
- Slomp, C.P.; Van Cappellen, P. Nutrient inputs to the coastal ocean through submarine groundwater discharge: Controls and potential impact. J. Hydrol. 2004, 295, 64–86. [Google Scholar] [CrossRef]
- UNESCO. Submarine Groundwater Discharge: Management Implications, Measurements and Effects; IHP-VI, Series on Groundwater No. 5, IOC Manuals and Guides No. 44; UNESCO: Paris, France, 2004. [Google Scholar]
- Chen, X.; Lao, Y.; Wang, J.; Du, J.; Liang, M.; Yang, B. Submarine Groundwater-Borne Nutrients in a Tropical Bay (Maowei Sea, China) and Their Impacts on the Oyster Aquaculture. Geochem. Geophys. Geosyst. 2018, 19, 932–951. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, F.; Lao, Y.; Wang, X.; Du, J.; Santos, I.R. Submarine groundwater discharge-derived carbon fluxes in mangroves: An important component of blue carbon budgets? J. Geophys. Res. Oceans 2018, 123, 6962–6979. [Google Scholar] [CrossRef]
- Zhang, J.L.; Li, Y.Y.; Wang, Y.H.; Zhang, Y.Y.; Zhang, D.; Zhang, R.J.; Li, Y.; Zhang, G. Spatial distribution and ecological risk of polychlorinated biphenyls in sediments from Qinzhou Bay, Beibu Gulf of South China. Mar. Pollut. Bull. 2014, 80, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.G.; Lin, Q.; Yu, Z.L.; Wang, X.N.; Ke, C.L.; Ning, J.J. Speciation and risk of heavy metals in sediments and human health implications of heavy metals in edible nekton in Beibu Gulf, China: A case study of Qinzhou Bay. Mar. Pollut. Bull. 2015, 101, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.; Millward, G.E. Suspended particles: Their role in estuarine biogeochemical cycles. Estuar. Coast. Shelf Sci. 2002, 55, 857–883. [Google Scholar] [CrossRef]
- Suzumura, M.; Kokubun, H.; Arata, N. Distribution and characteristics of suspended particulate matter in a heavily eutrophic estuary, Tokyo Bay, Japan. Mar. Pollut. Bull. 2004, 49, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Rosa, F.; Bloesch, J.; Rathke, D.E. Sampling the settling and suspended particulate matter (SPM). In Handbook of Techniques for Aquatic Sediments Sampling; Murdoch, A., MacKnight, S.D., Eds.; CRC Press: Boca Raton, FL, USA, 1994; pp. 97–129. [Google Scholar]
- Uncles, R.J.; Joint, I.; Stephens, J.A. Transport and retention of suspended particulate matter and bacteria in the Humber-Ouse Estuary, United Kingdom, and their relationship to hypoxia and anoxia. Estuaries 1998, 21, 597–612. [Google Scholar] [CrossRef]
- Kravchishina, M.D.; Mitzkevich, I.N.; Veslopolova, E.F.; Shevchenko, V.P.; Lisitzin, A.P. Relationship between the suspended particulate matter and microorganisms in the White Sea waters. Oceanology 2008, 48, 837–854. [Google Scholar] [CrossRef]
- Perkins, T.L.; Perrow, K.; Rajko-Nenow, P.; Jago, C.F.; Jones, D.L.; Malham, S.K.; McDonald, J.E. Decay rates of faecal indicator bacteria from sewage and ovine faeces in brackish and freshwater microcosms with contrasting suspended particulate matter concentrations. Sci. Total Environ. 2016, 572, 1645–1652. [Google Scholar] [CrossRef] [Green Version]
- Boehm, A.B.; Shellenbarger, G.G.; Paytan, A. Groundwater discharge: Potential association with fecal indicator bacteria in the surf zone. Environ. Sci. Technol. 2004, 38, 3558–3566. [Google Scholar] [CrossRef]
- Ye, Q.; Liu, J.A.; Du, J.Z.; Zhang, J. Bacterial diversity in submarine groundwater along the coasts of the yellow sea. Front. Microbiol. 2016, 6, 1519. [Google Scholar] [CrossRef]
- Halliday, E.; Gast, R.J. Bacteria in beach sands: An emerging challenge in protecting coastal water quality and bather health. Environ. Sci. Technol. 2010, 45, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.M.; Hong, G.H.; Zhang, J.; Ye, X.W.; Jiang, X.L. Nutrient budgets for large Chinese estuaries. Biogeosciences 2009, 6, 2245–2263. [Google Scholar] [CrossRef] [Green Version]
- Cerdà-Domènech, M.; Rodellas, V.; Folch, A.; Garcia-Orellana, J. Constraining the temporal variations of Ra isotopes and Rn in the groundwater end-member: Implications for derived SGD estimates. Sci. Total Environ. 2017, 595, 849–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, J.; Liu, Y.; Lin, X.; Zhang, H.; Zeng, J.; Hou, J.; Yang, Y.; Yao, T.; Knight, R.; Chu, H. Geographic distance and pH drive bacterial distribution in alkaline lake sediments across Tibetan Plateau. Environ. Microbiol. 2012, 14, 2457–2466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, A.C.C.; Cleary, D.F.R.; Almeida, A.; Cunha, A.; Dealtry, S.; Mendonça-Hagler, L.C.S.; Smalla, K.; Gomes, N.C. Denaturing gradient gel electrophoresis and barcoded pyrosequencing reveal unprecedented archaeal diversity in mangrove sediment and rhizosphere samples. Appl. Environ. Microbiol. 2012, 78, 5520–5528. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Li, Y.F.; Chen, Y.R.; Yang, J.L.; Bao, W.Y.; Guo, X.; Liang, X.; Shi, Z.Y.; Li, J.-L.; Ding, D.-W. Effects of substratum type on bacterial community structure in biofilms in relation to settlement of plantigrades of the mussel Mytilus coruscus. Int. Biodeter. Biodegr. 2014, 96, 41–49. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platformindependent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.R.; Thurber, R.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814. [Google Scholar] [CrossRef]
- Cho, H.M.; Kim, G. Determining groundwater Ra end-member values for the estimation of the magnitude of submarine groundwater discharge using Ra isotope tracers. Geophys. Res. Lett. 2016, 43, 3865–3871. [Google Scholar] [CrossRef] [Green Version]
- Fabisch, M.; Beulig, F.; Akob, D.M.; Küsel, K. Surprising abundance of Gallionella-related iron oxidizers in creek sediments at pH 4.4 or at high heavy metal concentrations. Front. Microbiol. 2013, 4, 390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, M.W.; Kasalický, V.; Jezbera, J.; Brandt, U.; Šimek, K. Limnohabitans australis sp. nov., isolated from a freshwater pond, and emended description of the genus Limnohabitans. Int. J. Syst. Evol. Microbiol. 2010, 60, 2946–2950. [Google Scholar] [CrossRef] [PubMed]
- Kasalický, V.; Jezbera, J.; Šimek, K.; Hahn, M.W. Limnohabitans planktonicus sp. nov. and Limnohabitans parvus sp. nov., planktonic betaproteobacteria isolated from a freshwater reservoir, and emended description of the genus Limnohabitans. Int. J. Syst. Evol. Microbiol. 2010, 60, 2710–2714. [Google Scholar] [CrossRef]
- Blöthe, M.; Roden, E.E. Composition and activity of an autotrophic Fe (II)-oxidizing, nitrate-reducing enrichment culture. Appl. Environ. Microbiol. 2009, 75, 6937–6940. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.N.; Frigon, D.; Raskin, L. Populations related to Alkanindiges, a novel genus containing obligate alkane degraders, are implicated in biological foaming in activated sludge systems. Environ. Microbiol. 2007, 9, 1898–1912. [Google Scholar] [CrossRef]
- Sohn, J.H.; Kwon, K.K.; Kang, J.H.; Jung, H.B.; Kim, S.J. Novosphingobium pentaromativorans sp. nov., a high-molecular-mass polycyclic aromatic hydrocarbon-degrading bacterium isolated from estuarine sediment. Int. J. Syst. Evol. Microbiol. 2004, 54, 1483–1487. [Google Scholar] [CrossRef]
- Choi, J.H.; Kim, M.S.; Jung, M.J.; Roh, S.W.; Shin, K.S.; Bae, J.W. Sphingopyxis soli sp. nov., isolated from landfill soil. Int. J. Syst. Evol. Microbiol. 2010, 60, 1682–1686. [Google Scholar] [CrossRef]
- Lee, S.; Weon, H.Y.; Kim, S.J.; Ahn, T.Y. Flavobacterium koreense sp. nov., Flavobacterium chungnamense sp. nov., and Flavobacterium cheonanense sp. nov., isolated from a freshwater reservoir. J. Microbiol. 2011, 49, 387–392. [Google Scholar] [CrossRef]
- LaFrentz, B.R.; Waldbieser, G.C.; Welch, T.J.; Shoemaker, C.A. Intragenomic heterogeneity in the 16S rRNA genes of Flavobacterium columnare and standard protocol for genomovar assignment. J. Fish Dis. 2014, 37, 657–669. [Google Scholar] [CrossRef]
- Enright, A.M.; McGrath, V.; Gill, D.; Collins, G.; O’Flaherty, V. Effect of seed sludge and operation conditions on performance and archaeal community structure of low-temperature anaerobic solvent-degrading bioreactors. Syst. Appl. Microbiol. 2009, 32, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Kasai, Y.; Takahata, Y.; Hoaki, T.; Watanabe, K. Physiological and molecular characterization of a microbial community established in unsaturated, petroleum-contaminated soil. Environ. Microbiol. 2005, 7, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Barber, R.D.; Zhang, L.; Harnack, M.; Olson, M.V.; Kaul, R.; Ingram-Smith, C.; Smith, K.S. Complete genome sequence of Methanosaeta concilii, a specialist in aceticlastic methanogenesis. J. Bacteriol. 2011, 193, 3668–3669. [Google Scholar] [CrossRef] [PubMed]
- Llirós, M.; Casamayor, E.O.; Borrego, C. High archaeal richness in the water column of a freshwater sulfurous karstic lake along an interannual study. FEMS Microbiol. Ecol. 2008, 66, 331–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, R.; Klose, M.; Noll, M.; Kemnitz, D.; Bodelier, P.L. Soil type links microbial colonization of rice roots to methane emission. Glob. Chang. Biol. 2008, 14, 657–669. [Google Scholar] [CrossRef]
- Castelle, C.J.; Wrighton, K.C.; Thomas, B.C.; Hug, L.A.; Brown, C.T.; Wilkins, M.J.; Frischkorn, K.R.; Tringe, S.G.; Singh, A.; Markillie, L.M.; et al. Genomic expansion of domain archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr. Biol. 2015, 25, 690–701. [Google Scholar] [CrossRef]
- Kubo, K.; Lloyd, K.G.; Biddle, J.F.; Amann, R.; Teske, A.; Knittel, K. Archaea of the Miscellaneous Crenarchaeotal Group are abundant, diverse and widespread in marine sediments. ISME J. 2012, 6, 1949–1965. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, K.G.; Schreiber, L.; Petersen, D.G.; Kjeldsen, K.U.; Lever, M.A.; Steen, A.D.; Stepanauskas, R.; Richter, M.; Kleindienst, S.; Lenk, S.; et al. Predominant archaea in marine sediments degrade detrital proteins. Nature 2013, 496, 215–218. [Google Scholar] [CrossRef]
- Evans, P.N.; Parks, D.H.; Chadwick, G.L.; Robbins, S.J.; Orphan, V.J.; Golding, S.D.; Tyson, G.W. Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science 2015, 350, 434–438. [Google Scholar] [CrossRef] [Green Version]
- Lazar, C.S.; Baker, B.J.; Seitz, K.; Hyde, A.S.; Dick, G.J.; Hinrichs, K.U.; Teske, A.P. Genomic evidence for distinct carbon substrate preferences and ecological niches of Bathyarchaeota in estuarine sediments. Environ. Microbiol. 2016, 18, 1200–1211. [Google Scholar] [CrossRef]
- Fry, J.C.; Parkes, R.J.; Cragg, B.A.; Weightman, A.J.; Webster, G. Prokaryotic biodiversity and activity in the deep subseafloor biosphere. FEMS Microbiol. Ecol. 2008, 66, 181–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, J.; Xu, J.; Qin, D.; He, Y.; Xiao, X.; Wang, F. Genetic and functional properties of uncultivated MCG archaea assessed by metagenome and gene expression analyses. ISME J. 2014, 8, 650–659. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Li, M.; Perumal, V.; Feng, X.; Fang, J.; Xie, J.; Sievert, S.M.; Wang, F. Genomic and enzymatic evidence for acetogenesis among multiple lineages of the archaeal phylum Bathyarchaeota widespread in marine sediments. Nat. Microbiol. 2016, 1, 16035. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Wang, R.; Wang, H.; Gong, L.; Man, B.; Xu, Y. Distribution of Bathyarchaeota Communities Across Different Terrestrial Settings and Their Potential Ecological Functions. Sci. Rep. 2017, 7, 45028. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Liu, X.; Dong, X. Methanosaeta harundinacea sp. nov., a novel acetate-scavenging methanogen isolated from a UASB reactor. Int. J. Syst. Evol. Microbiol. 2006, 56, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; He, M.; Yang, Z.; Lin, C.; Quan, X.; Wang, H. Distribution of polycyclic aromatic hydrocarbons in water, suspended particulate matter and sediment from Daliao River watershed, China. Chemosphere 2007, 68, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.; Wu, W.; Gentry, T.J.; Carley, J.; Corbin, G.A.; Carroll, S.L.; Watson, D.B.; Jardine, P.M.; Zhou, J.; Criddle, C.S.; et al. Bacterial community succession during in situ uranium bioremediation: Spatial similarities along controlled flow paths. ISME J. 2009, 3, 47–64. [Google Scholar] [CrossRef]
- Gan, H.M.; Hudson, A.O.; Rahman, A.Y.A.; Chan, K.G.; Savka, M.A. Comparative genomic analysis of six bacteria belonging to the genus Novosphingobium: Insights into marine adaptation, cell-cell signaling and bioremediation. BMC Genom. 2013, 14, 431. [Google Scholar] [CrossRef]
- Yabuuchi, E.; Yano, I.; Oyaizu, H.; Hashimoto, Y.; Ezaki, T.; Yamamoto, H. Proposals of Sphingomonas paucimobilis gen. nov. and comb. nov., Sphingomonas parapaucimobilis sp. nov., Sphingomonas yanoikuyae sp. nov., Sphingomonas adhaesiva sp. nov., Sphingomonas capsulata comb. nov., and two genospecies of the genus Sphingomonas. Microbiol. Immunol. 1990, 34, 99–119. [Google Scholar] [CrossRef]
- Tiirola, M.A.; Männistö, M.K.; Puhakka, J.A.; Kulomaa, M.S. Isolation and characterization of Novosphingobium sp. strain MT1, a dominant polychlorophenol-degrading strain in a groundwater bioremediation system. Appl. Environ. Microbiol. 2002, 68, 173–180. [Google Scholar] [CrossRef]
- Yan, Q.X.; Hong, Q.; Han, P.; Dong, X.J.; Shen, Y.J.; Li, S.P. Isolation and characterization of a carbofuran-degrading strain Novosphingobium sp. FND-3. FEMS Microbiol. Lett. 2007, 271, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Lai, Q.; Zheng, T.; Shao, Z. Novosphingobium indicum sp. nov., a polycyclic aromatic hydrocarbon-degrading bacterium isolated from a deep-sea environment. Int. J. Syst. Evol. Microbiol. 2009, 59, 2084–2088. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Onda, K.; Morita, T.; Luxmy, B.S.; Tada, K.; Miya, A.; Murakami, T. Contribution of the estrogen-degrading bacterium Novosphingobium sp. strain JEM-1 to estrogen removal in wastewater treatment. J. Environ. Eng. 2010, 136, 890–896. [Google Scholar] [CrossRef]
- Kappler, A.; Straub, K.L. Geomicrobiological cycling of iron. Rev. Mineral. Geochem. 2005, 59, 85–108. [Google Scholar] [CrossRef]
- Konhauser, K.O.; Amskold, L.; Lalonde, S.V.; Posth, N.R.; Kappler, A.; Anbar, A. Decoupling photochemical Fe (II) oxidation from shallow-water BIF deposition. Earth Planet. Sci. Lett. 2007, 258, 87–100. [Google Scholar] [CrossRef]
- Pérez, M.T.; Sommaruga, R. Differential effect of algal-and soil-derived dissolved organic matter on alpine lake bacterial community composition and activity. Limnol. Oceanogr. 2006, 51, 2527–2537. [Google Scholar] [CrossRef]
- Šimek, K.; Kasalický, V.; Zapomělová, E.; Horňák, K. Alga-derived substrates select for distinct betaproteobacterial lineages and contribute to niche separation in Limnohabitans strains. Appl. Environ. Microbiol. 2011, 77, 7307–7315. [Google Scholar] [CrossRef]
- Hörtnagl, P.; Pérez, M.T.; Zeder, M.; Sommaruga, R. The bacterial community composition of the surface microlayer in a high mountain lake. FEMS Microbiol. Ecol. 2010, 73, 458–467. [Google Scholar] [CrossRef]
- Glaeser, S.P.; Grossart, H.P.; Glaeser, J. Singlet oxygen, a neglected but important environmental factor: Short-term and long-term effects on bacterioplankton composition in a humic lake. Environ. Microbiol. 2010, 12, 3124–3136. [Google Scholar] [CrossRef]
- Kasalický, V.; Jezbera, J.; Hahn, M.W.; Šimek, K. The diversity of the Limnohabitans genus, an important group of freshwater bacterioplankton, by characterization of 35 isolated strains. PLoS ONE 2013, 8, e58209. [Google Scholar] [CrossRef]
- Chapelle, F. Ground-Water Microbiology and Geochemistry; John Wiley & Sons: New York, NY, USA, 2001. [Google Scholar]
- Chapelle, F.H.; Lovley, D.R. Competitive exclusion of sulfate reduction by Fe (lll)-reducing bacteria: A mechanism for producing discrete zones of high-iron ground water. Groundwater 1992, 30, 29–36. [Google Scholar] [CrossRef]
- Appelo, C.A.J.; Postma, D. Geochemistry, Groundwater and Pollution; Balkema: Amsterdam, The Netherlands, 1994. [Google Scholar]
- Postma, D.; Boesen, C.; Kristiansen, H.; Larsen, F. Nitrate reduction in an unconfined sandy aquifer: Water chemistry, reduction processes, and geochemical modeling. Water Resour. Res. 1991, 27, 2027–2045. [Google Scholar] [CrossRef]
- Engesgaard, P.; Kipp, K.L. A geochemical transport model for redox-controlled movement of mineral fronts in groundwater flow systems: A case of nitrate removal by oxidation of pyrite. Water Resour. Res. 1992, 28, 2829–2843. [Google Scholar] [CrossRef]
- Hartog, N.; Griffioen, J.; van der Weijden, C.H. Distribution and reactivity of O2-reducing components in sediments from a layered aquifer. Environ. Sci. Technol. 2002, 36, 2338–2344. [Google Scholar] [CrossRef] [PubMed]
- Straub, K.L.; Benz, M.; Schink, B.; Widdel, F. Anaerobic, nitrate-dependent microbial oxidation of ferrous iron. Appl. Environ. Microbiol. 1996, 62, 1458–1460. [Google Scholar] [PubMed]
- Cho, H.M.; Kim, G.; Kwon, E.Y.; Moosdorf, N.; Garcia-Orellana, J.; Santos, I.R. Radium tracing nutrient inputs through submarine groundwater discharge in the global ocean. Sci. Rep. 2018, 8, 2439. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site Descriptions | Environmental Chemical Parameters | ||
|---|---|---|---|
| Latitude | 21°43′39.92″ N | Temperature (°C) | 17.2 |
| Longitude | 108°37′30.43″ E | Salinity | 0.5 |
| Location | Qinzhou City, Guangxi Province | 222Rn (Bq m−3) | 4140 |
| Well depth (m) | ~ 4 | DIC (mmol L−1) | 1.3 |
| Characteristics of sampling site | In the Gaoshatou Village; near an industrial zone; 800 meters from the coast; garbage found | DOC (mmol L−1) | 1.0 |
| NO3− (μmol L−1) | 5.2 | ||
| NO2− (μmol L−1) | 0.42 | ||
| Characteristics of submarine groundwater | Turbid water; smelly water | NH4+ (μmol L−1) | 4.8 |
| PO43− (μmol L−1) | 0.31 | ||
| SiO32− (μmol L−1) | 166 | ||
| Method | Distance | Bacteria | Archaea | ||
|---|---|---|---|---|---|
| R2 | p Value | R2 | p Value | ||
| PERMANOVA | Bray-Curtis | 0.51 | 0.02 | 0.78 | 0.02 |
| OTU | Proportion of OTUs (%) | Closest Relatives | Similarity | |
|---|---|---|---|---|
| BP | BF | |||
| OTU 594 | 4.7 | 21.8 | Limnohabitans australis strain MWH-BRAZ-DAM2D [35] | NR_125544 (99.2%) |
| OTU 571 | 1.2 | 2.6 | Limnohabitans parvus strain II-B4 [36] | NR_125542 (98.9%) |
| OTU 40 | 16.6 | 0.2 | Clone F22F60 from a Fe(II)-oxidizing, nitrate-reducing enrichment culture [37] | FN430660 (98.9%) |
| OTU 1039 | 4.5 | 0.8 | Gammaproteobacterial clone SWL18 from a sludge wastewater treatment system [38] | AY528817 (99.7%) |
| OTU 261 | 10.7 | 5.4 | Novosphingobium pentaromativorans strain US6-1 [39] | NR_025248 (98.4%) |
| OTU 555 | 1.2 | 1.5 | Sphingopyxis soli strain BL03 [40] | NR_116739 (99.2%) |
| OTU 843 | 3.1 | 2.7 | Flavobacterium chungnamense strain ARSA-103 [41] | NR_117494 (98.7%) |
| OTU 591 | 0.1 | 1.6 | Flavobacterium columnare strain ATCC 23463 [42] | NR_118582 (98.4%) |
| OTU | Proportion of OTUs (%) | Closest Relatives | Similarity | |
|---|---|---|---|---|
| AP | AF | |||
| OTU 563 | 29.4 (OTU 563, 516 and 598) | 3.0 (OTU 563, 516 and 598) | Clone R5ENDA4 from low-temperature anaerobic solvent-degrading bioreactors [43] | DQ399807 (100%) |
| OTU 516 | Clone ASC27 in unsaturated, petroleum-contaminated soil [44] | AB161330 (100%) | ||
| OTU 598 | Clone ASC37 in unsaturated, petroleum-contaminated soil [44] | AB161337 (100%) | ||
| OTU 540 | 19.5 | 3.3 | Acetoclastic methanoarchaeon Methanosaeta concilii strain GP6 [45] | NR_102903 (99.6%) |
| OTU 312 | 1.1 | 3.7 | DGGE gel band VIARC-174 from the water column of Lake Vilar [46] | EU683427 (99.2%) |
| OTU 458 | 3.2 | 7.4 | Clone WsL03b13_109f from groundwater of a volcanic mountain, Mt. Fuji (Unpublished) | AB794582 (98.4%) |
| OTU 536 | 1.8 | 5.7 | Clone LL_ADT_15 from low-level bank soil of the River Rhine [47] | AM503273 (98.8%) |
| OTU 541 | 0.5 | 2.3 | Clone gwa2_scaffold_29719 from an aquifer adjacent to Colorado River [48] | KP308734 (87.0%) |
| OTU 449 | 16.3 | 41.4 | VIARC-45 retrieved from the water column of Lake Vilar [46] | AM697998 (92.5%) |
| Pathway | Relative Abundance | Definition | |
|---|---|---|---|
| >0.45 μm Size Fraction | 0.2–0.45 μm Fraction | ||
| ko00351 | 0.00 | 0.00 | 1,1,1-Trichloro-2,2-bis(4-chlorophenyl)ethane (DDT) degradation |
| ko00361 | 0.22 | 0.21 | Chlorocyclohexane and chlorobenzene degradation |
| ko00362 | 1.31 | 1.55 | Benzoate degradation |
| ko00363 | 0.19 | 0.23 | Bisphenol degradation |
| ko00364 | 0.13 | 0.12 | Fluorobenzoate degradation |
| ko00621 | 0.21 | 0.18 | Dioxin degradation |
| ko00622 | 0.20 | 0.15 | Xylene degradation |
| ko00623 | 0.34 | 0.34 | Toluene degradation |
| ko00624 | 0.27 | 0.29 | Polycyclic aromatic hydrocarbon degradation |
| ko00625 | 0.50 | 0.52 | Chloroalkane and chloroalkene degradation |
| ko00626 | 0.48 | 0.54 | Naphthalene degradation |
| ko00627 | 0.88 | 1.09 | Aminobenzoate degradation |
| ko00633 | 0.16 | 0.13 | Nitrotoluene degradation |
| ko00642 | 0.22 | 0.18 | Ethylbenzene degradation |
| ko00643 | 0.20 | 0.20 | Styrene degradation |
| ko00791 | 0.10 | 0.10 | Atrazine degradation |
| ko00903 | 0.76 | 0.92 | Limonene and pinene degradation |
| ko00930 | 0.56 | 0.70 | Caprolactam degradation |
| Total | 6.73 | 7.46 | |
| Pathway | Relative Abundance | Definition | |
|---|---|---|---|
| >0.45 μm Size Fraction | 0.2–0.45 μm Fraction | ||
| ko00281 | 0.19 | 0.25 | Geraniol degradation |
| ko00361 | 0.01 | 0.03 | Chlorocyclohexane and chlorobenzene degradation |
| ko00362 | 0.49 | 0.53 | Benzoate degradation |
| ko00363 | 0.01 | 0.04 | Bisphenol degradation |
| ko00364 | 0.00 | 0.01 | Fluorobenzoate degradation |
| ko00621 | 0.01 | 0.03 | Dioxin degradation |
| ko00622 | 0.00 | 0.01 | Xylene degradation |
| ko00623 | 0.33 | 0.29 | Toluene degradation |
| ko00624 | 0.11 | 0.12 | Polycyclic aromatic hydrocarbon degradation |
| ko00625 | 0.17 | 0.23 | Chloroalkane and chloroalkene degradation |
| ko00626 | 0.05 | 0.11 | Naphthalene degradation |
| ko00627 | 0.23 | 0.29 | Aminobenzoate degradation |
| ko00630 | 1.03 | 1.05 | Glyoxylate and dicarboxylate metabolism |
| ko00633 | 0.87 | 0.77 | Nitrotoluene degradation |
| ko00642 | 0.01 | 0.02 | Ethylbenzene degradation |
| ko00643 | 0.01 | 0.03 | Styrene degradation |
| ko00791 | 0.00 | 0.02 | Atrazine degradation |
| ko00903 | 0.27 | 0.29 | Limonene and pinene degradation |
| ko00930 | 0.18 | 0.20 | Caprolactam degradation |
| Total | 3.97 | 4.31 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Ye, Q.; Du, J.; Zhang, J. Bacterial and Archaeal Assemblages from Two Size Fractions in Submarine Groundwater Near an Industrial Zone. Water 2019, 11, 1261. https://doi.org/10.3390/w11061261
Chen X, Ye Q, Du J, Zhang J. Bacterial and Archaeal Assemblages from Two Size Fractions in Submarine Groundwater Near an Industrial Zone. Water. 2019; 11(6):1261. https://doi.org/10.3390/w11061261
Chicago/Turabian StyleChen, Xiaogang, Qi Ye, Jinzhou Du, and Jing Zhang. 2019. "Bacterial and Archaeal Assemblages from Two Size Fractions in Submarine Groundwater Near an Industrial Zone" Water 11, no. 6: 1261. https://doi.org/10.3390/w11061261
APA StyleChen, X., Ye, Q., Du, J., & Zhang, J. (2019). Bacterial and Archaeal Assemblages from Two Size Fractions in Submarine Groundwater Near an Industrial Zone. Water, 11(6), 1261. https://doi.org/10.3390/w11061261