
Tuning the Catalytic Water Oxidation Activity through Structural Modifications of High-Nuclearity Mn-oxo Clusters [Mn18M] (M = Sr2+, Mn2+)


Abstract
:1. Introduction
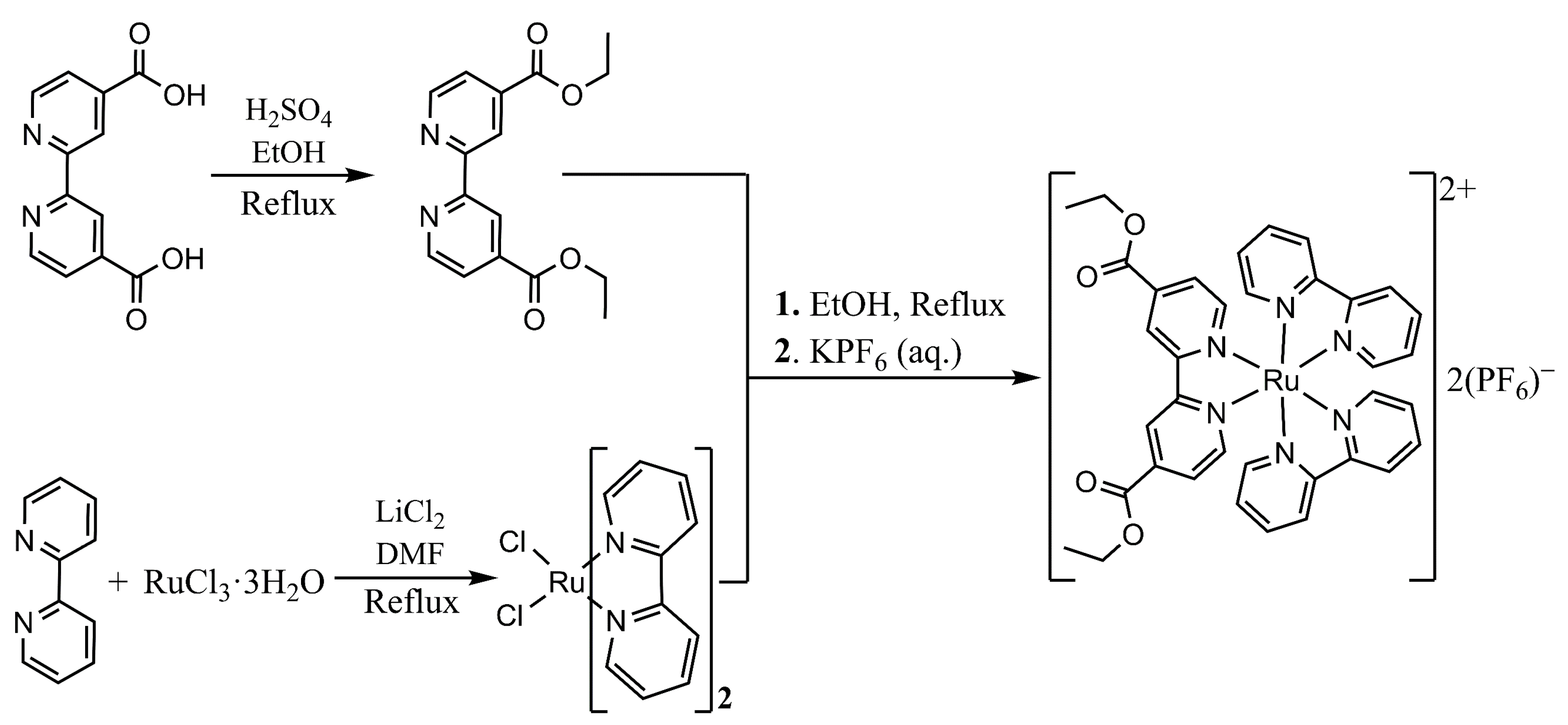
2. Materials and Methods
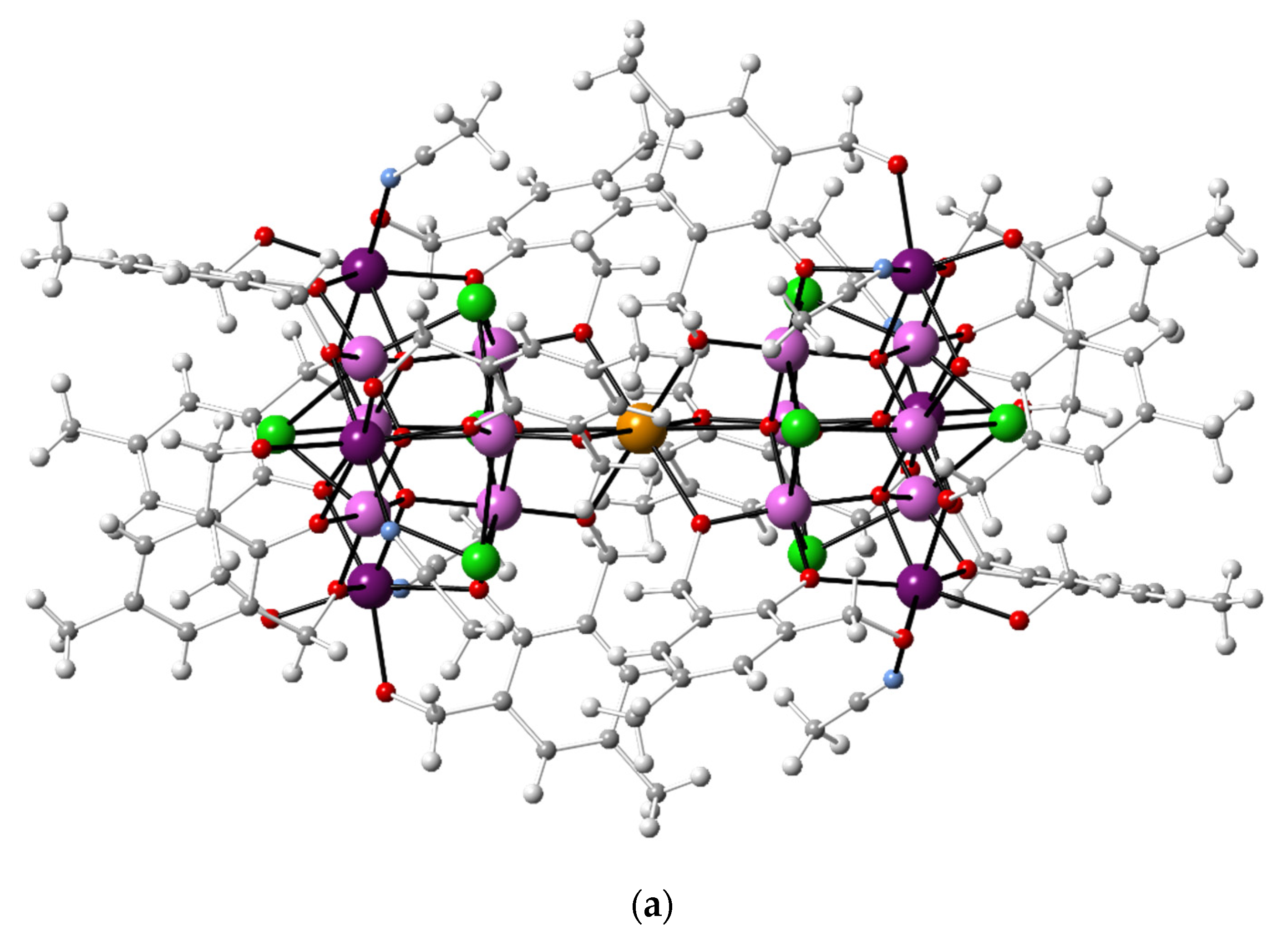
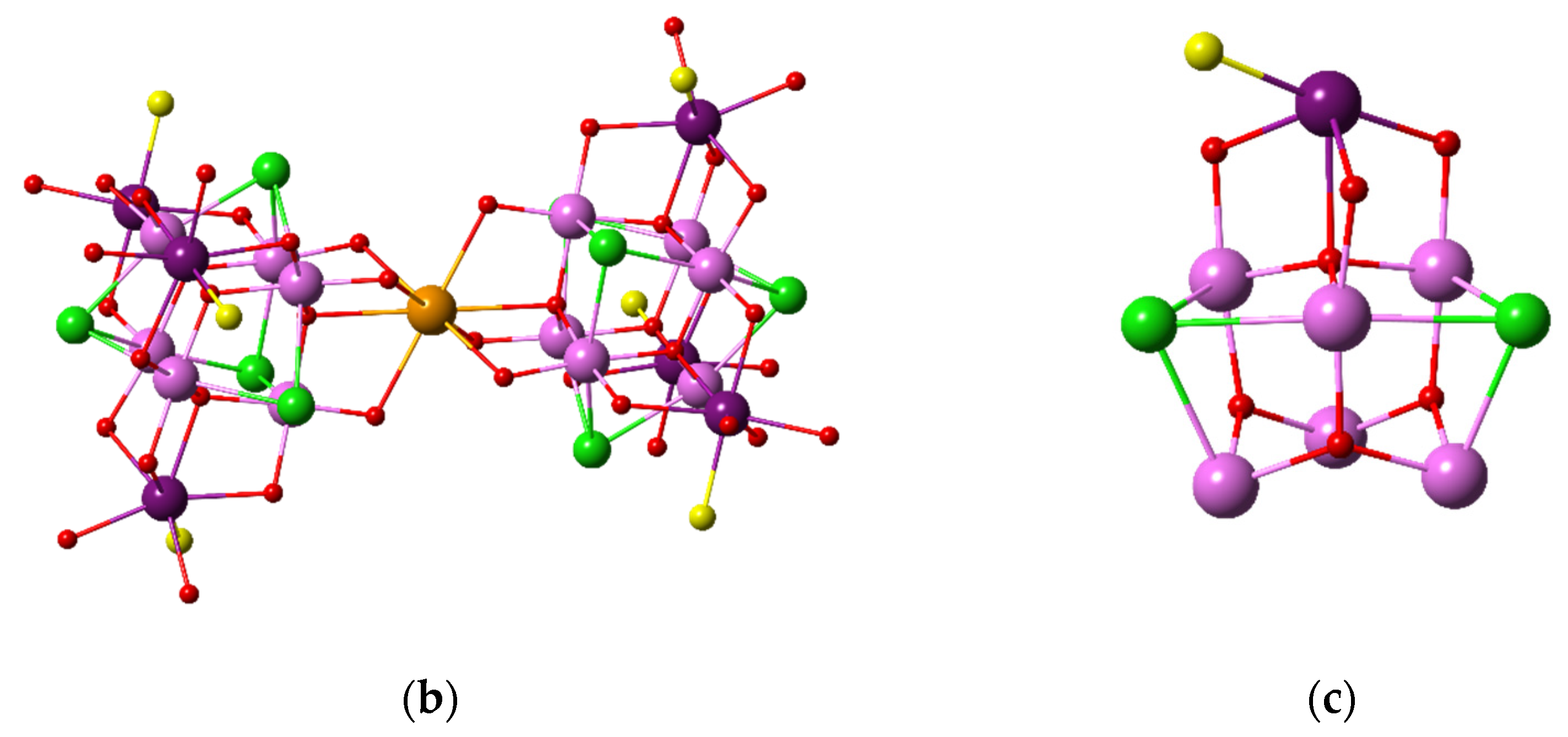
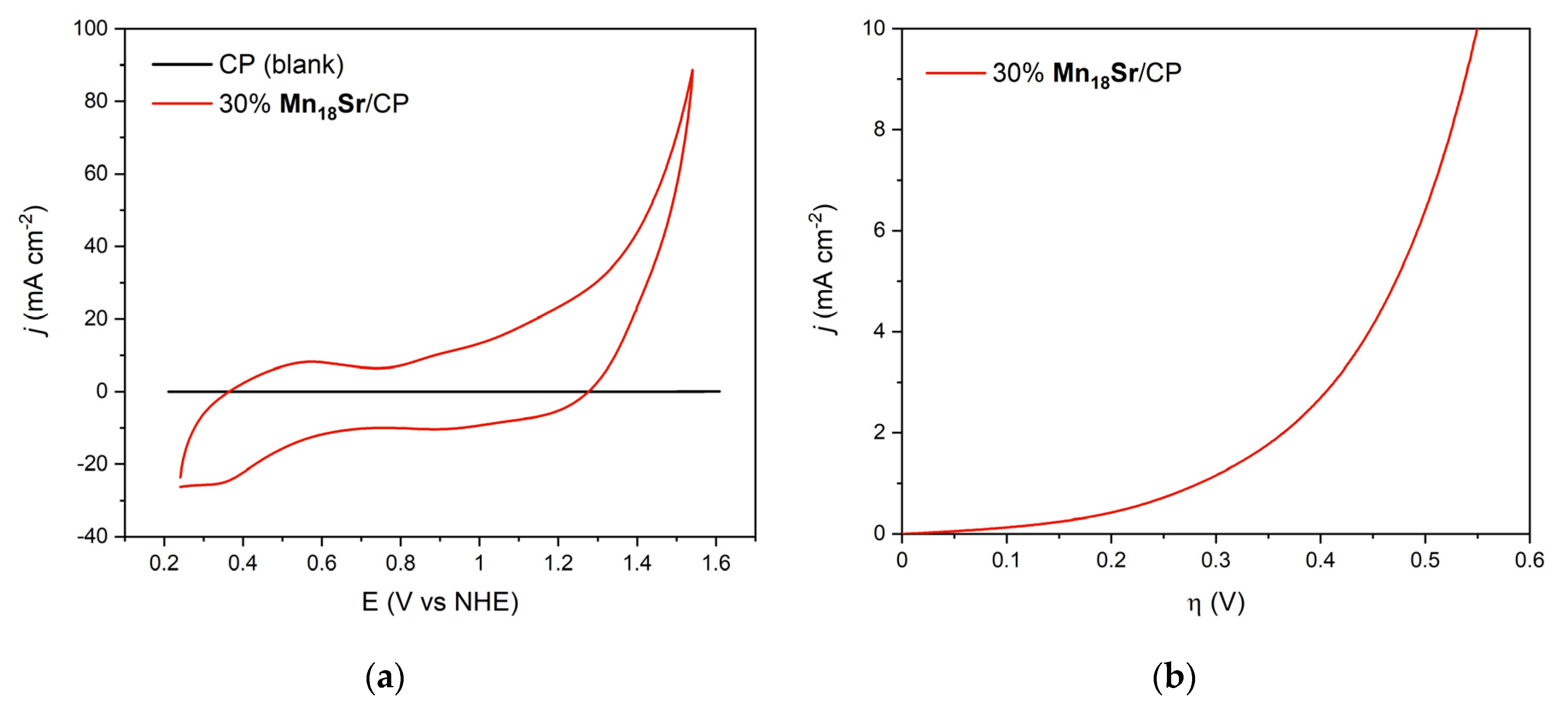
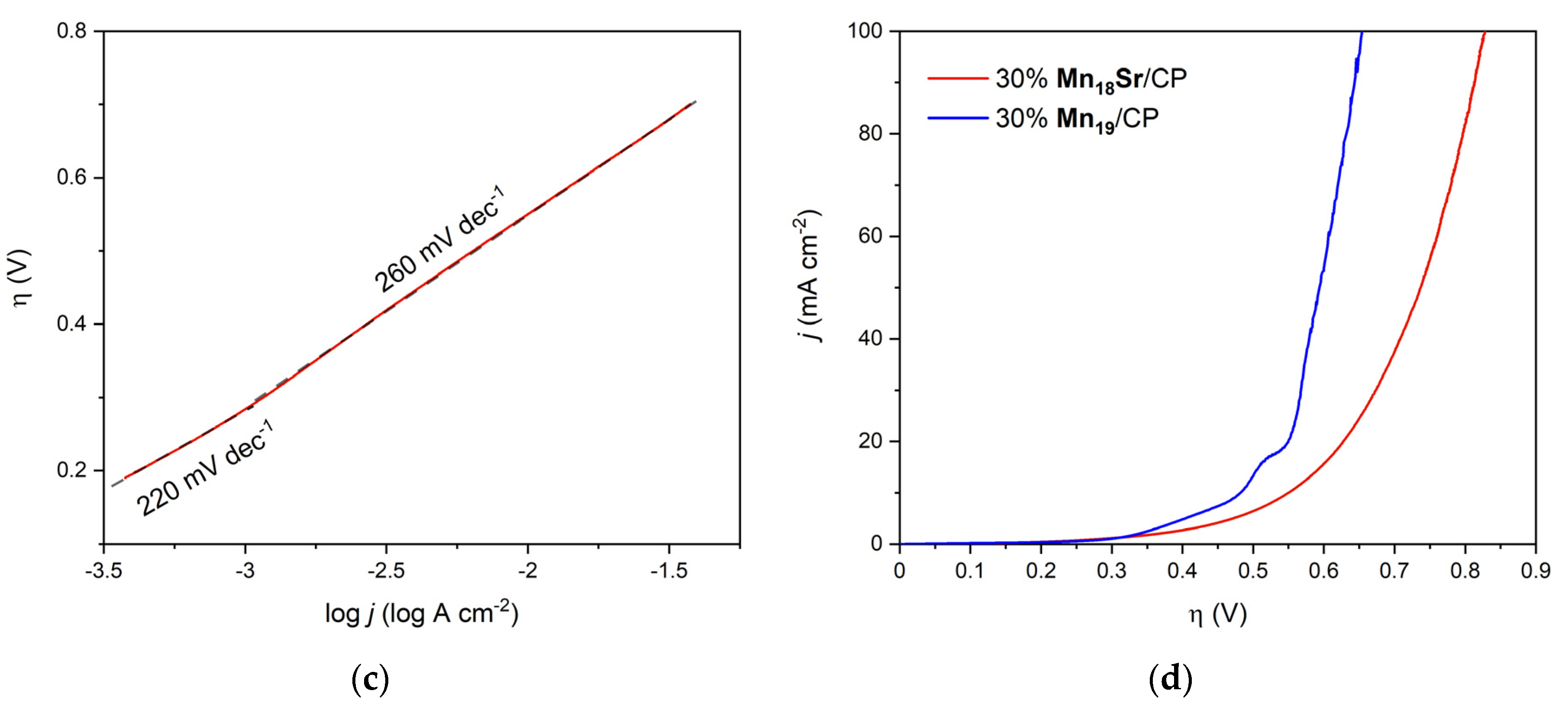
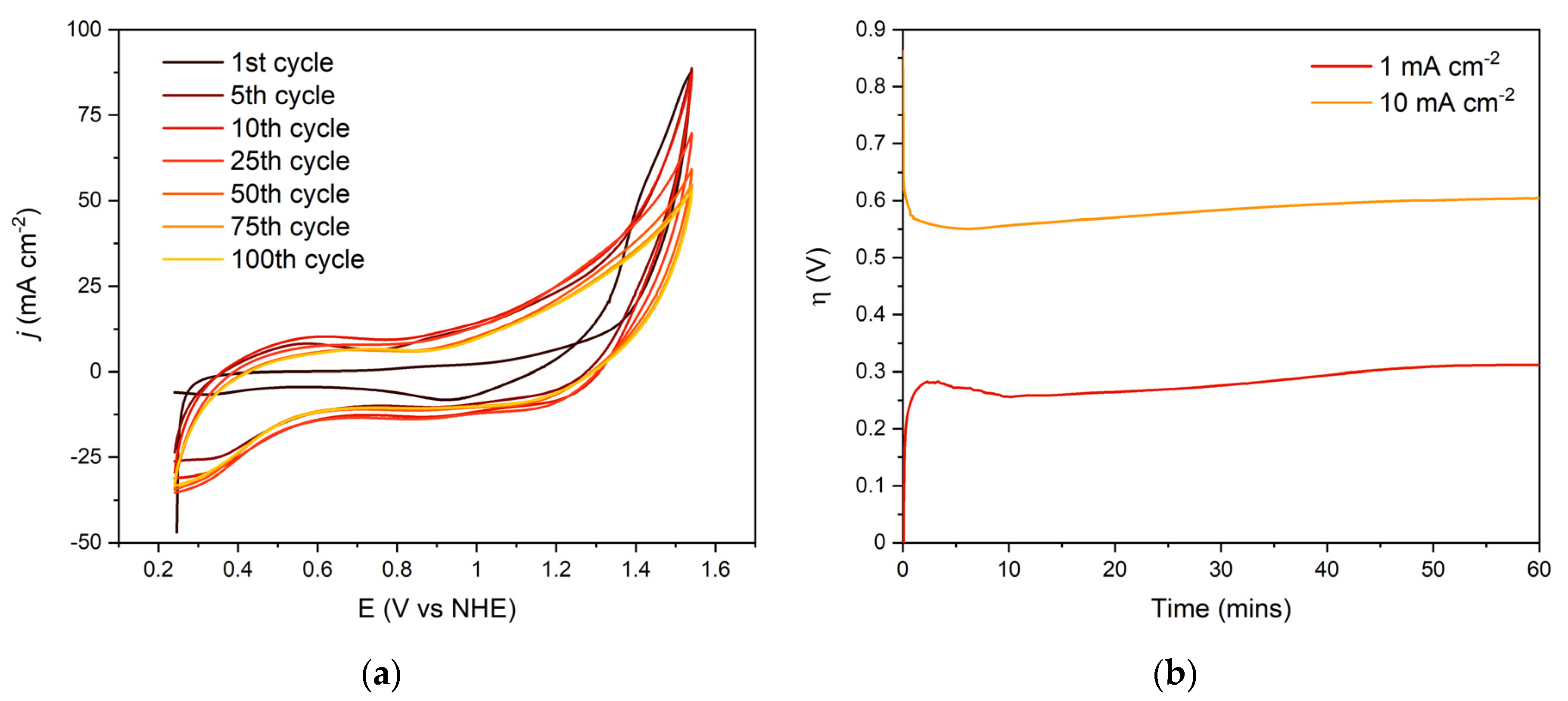
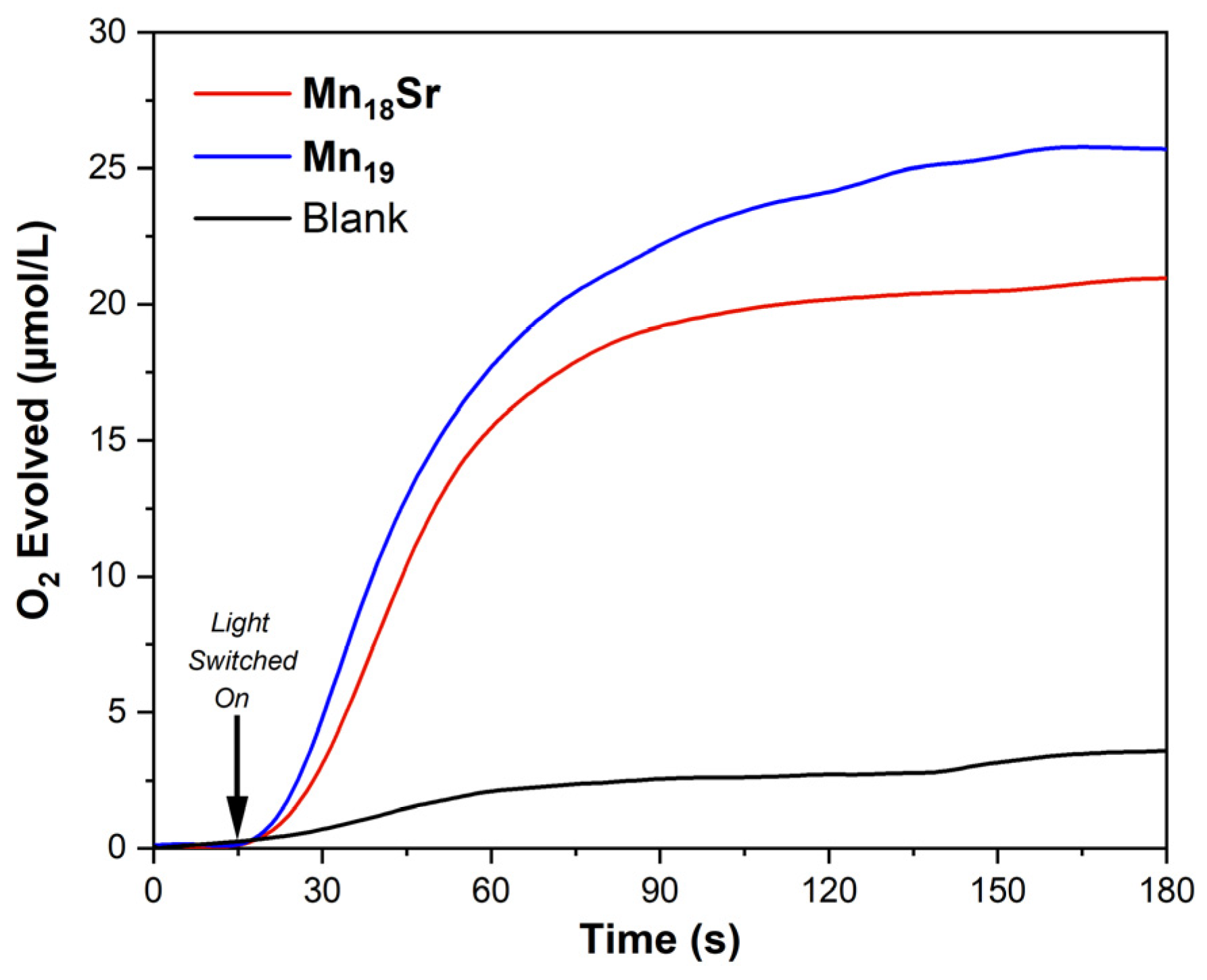
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zahran, Z.N.; Tsubonouchi, Y.; Mohamed, E.A.; Yagi, M. Recent advances in the development of molecular catalyst-based anodes for water oxidation toward artificial photosynthesis. ChemSusChem 2019, 12, 1775–1793. [Google Scholar] [CrossRef]
- Li, J.; Triana, C.A.; Wan, W.; Adiyeri Saseendran, D.P.A.; Zhao, Y.; Balaghi, S.E.; Heidari, S.; Patzke, G.R. Molecular and heterogeneous water oxidation catalysts: Recent progress and joint perspectives. Chem. Soc. Rev. 2021, 50, 2444–2485. [Google Scholar] [CrossRef]
- Sherif, S.A.; Barbir, F.; Veziroglu, T.N. Wind energy and the hydrogen economy—Review of the technology. Sol. Energy 2005, 78, 647–660. [Google Scholar] [CrossRef]
- Mazloomi, K.; Gomes, C. Hydrogen as an energy carrier: Prospects and challenges. Renew. Sustain. Energy Rev. 2012, 16, 3024–3033. [Google Scholar] [CrossRef]
- McKone, J.R.; Lewis, N.S.; Gray, H.B. Will solar-driven water-splitting devices see the light of day? Chem. Mater. 2014, 26, 407–414. [Google Scholar] [CrossRef]
- Soriano-López, J.; Schmitt, W.; García-Melchor, M. Computational modelling of water oxidation catalysts. Curr. Opin. Electrochem. 2018, 7, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Dau, H.; Zaharieva, I. Principles, efficiency, and blueprint character of solar-energy conversion in photosynthetic water oxidation. Acc. Chem. Res. 2009, 42, 1861–1870. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-P.; Wang, H.-Y.; Zheng, H.; Zhang, W.; Cao, R. O–O bond formation mechanisms during the oxygen evolution reaction over synthetic molecular catalysts. Chin. J. Catal. 2021, 42, 1253–1268. [Google Scholar] [CrossRef]
- Lyons, M.E.G.; Doyle, R.L.; Browne, M.P.; Godwin, I.J.; Rovetta, A.A.S. Recent developments in electrochemical water oxidation. Curr. Opin. Electrochem. 2017, 1, 40–45. [Google Scholar] [CrossRef]
- Blakemore, J.D.; Crabtree, R.H.; Brudvig, G.W. Molecular catalysts for water oxidation. Chem. Rev. 2015, 115, 12974–13005. [Google Scholar] [CrossRef]
- McCrory, C.C.L.; Jung, S.; Ferrer, I.M.; Chatman, S.M.; Peters, J.C.; Jaramillo, T.F. Benchmarking hydrogen evolving reaction and oxygen evolving reaction electrocatalysts for solar water splitting devices. J. Am. Chem. Soc. 2015, 137, 4347–4357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galán-Mascarós, J.R. Water oxidation at electrodes modified with earth-abundant transition-metal catalysts. ChemElectroChem 2015, 2, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Roger, I.; Shipman, M.A.; Symes, M.D. Earth-abundant catalysts for electrochemical and photoelectrochemical water splitting. Nat. Rev. Chem. 2017, 1, 0003. [Google Scholar] [CrossRef]
- Du, H.-Y.; Chen, S.-C.; Su, X.-J.; Jiao, L.; Zhang, M.-T. Redox-active ligand assisted multielectron catalysis: A case of CoIII complex as water oxidation catalyst. J. Am. Chem. Soc. 2018, 140, 1557–1565. [Google Scholar] [CrossRef]
- Nguyen, A.I.; Darago, L.E.; Balcells, D.; Tilley, T.D. Influence of a “dangling” Co(II) ion bound to a [MnCo3O4] oxo cubane. J. Am. Chem. Soc. 2018, 140, 9030–9033. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Tan, J.M.; Besson, C.; Geletii, Y.V.; Musaev, D.G.; Kuznetsov, A.E.; Luo, Z.; Hardcastle, K.I.; Hill, C.L. A fast soluble carbon-free molecular water oxidation catalyst based on abundant metals. Science 2010, 328, 342–345. [Google Scholar] [CrossRef] [Green Version]
- Martin-Sabi, M.; Soriano-López, J.; Winter, R.S.; Chen, J.-J.; Vilà-Nadal, L.; Long, D.-L.; Galán-Mascarós, J.R.; Cronin, L. Redox tuning the Weakley-type polyoxometalate archetype for the oxygen evolution reaction. Nat. Catal. 2018, 1, 208–213. [Google Scholar] [CrossRef]
- Han, X.-B.; Wang, D.-X.; Gracia-Espino, E.; Luo, Y.-H.; Tan, Y.-Z.; Lu, D.-F.; Li, Y.-G.; Wågberg, T.; Wang, E.-B.; Zheng, L.-S. Fe-substituted cobalt-phosphate polyoxometalates as enhanced oxygen evolution catalysts in acidic media. Chin. J. Catal. 2020, 41, 853–857. [Google Scholar] [CrossRef]
- Azmani, K.; Besora, M.; Soriano-López, J.; Landolsi, M.; Teillout, A.-L.; de Oliveira, P.; Mbomekallé, I.-M.; Poblet, J.M.; Galán-Mascarós, J.-R. Understanding polyoxometalates as water oxidation catalysts through iron vs. cobalt reactivity. Chem. Sci. 2021, 12, 8755–8766. [Google Scholar] [CrossRef]
- Schwarz, B.; Forster, J.; Anjass, M.H.; Daboss, S.; Kranz, C.; Streb, C. From molecular to colloidal manganese vanadium oxides for water oxidation catalysis. Chem. Commun. 2017, 53, 11576–11579. [Google Scholar] [CrossRef]
- Arsalan, M.; Babar, N.-U.-A.; Sadiqa, A.; Mansha, S.; Baig, N.; Nisar, L.; Ashiq, M.N.; Saleh, T.A.; Joya, K.S. Surface-assembled Fe-oxide colloidal nanoparticles for high performance electrocatalytic water oxidation. Int. J. Hydrogen Energy 2021, 46, 5207–5222. [Google Scholar] [CrossRef]
- Trotochaud, L.; Ranney, J.K.; Williams, K.N.; Boettcher, S.W. Solution-cast metal oxide thin film electrocatalysts for oxygen evolution. J. Am. Chem. Soc. 2012, 134, 17253–17261. [Google Scholar] [CrossRef]
- Kanan, M.W.; Nocera, D.G. In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and Co2+. Science 2008, 321, 1072–1075. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wei, X.; Wang, X.; Song, W.; Zhong, W.; Wang, M.; Ju, J.; Tang, Y. Plasmonic Au nanoparticle@Ti3C2Tx heterostructures for improved oxygen evolution performance. Inorg. Chem. 2021, 60, 5890–5897. [Google Scholar] [CrossRef]
- Gutiérrez-Tarriño, S.; Olloqui-Sariego, J.L.; Calvente, J.J.; Palomino, M.; Mínguez Espallargas, G.; Jordá, J.L.; Rey, F.; Oña-Burgos, P. Cobalt metal–organic framework based on two dinuclear secondary building units for electrocatalytic oxygen evolution. ACS Appl. Mater. Interfaces 2019, 11, 46658–46665. [Google Scholar] [CrossRef]
- Zhao, S.; Wang, Y.; Dong, J.; He, C.-T.; Yin, H.; An, P.; Zhao, K.; Zhang, X.; Gao, C.; Zhang, L.; et al. Ultrathin metal–organic framework nanosheets for electrocatalytic oxygen evolution. Nat. Energy 2016, 1, 16184. [Google Scholar] [CrossRef]
- Li, X.; Fang, Y.; Zhao, S.; Wu, J.; Li, F.; Tian, M.; Long, X.; Jin, J.; Ma, J. Nitrogen-doped mesoporous carbon nanosheet/carbon nanotube hybrids as metal-free bi-functional electrocatalysts for water oxidation and oxygen reduction. J. Mater. Chem. A 2016, 4, 13133–13141. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, K.-H.; Lu, Q.; Gu, Q.; Zhang, L.; Zhang, B.; Su, D.; Plodinec, M.; Schlögl, R.; Heumann, S. Electrocatalytic water oxidation at quinone-on-carbon: A model system study. J. Am. Chem. Soc. 2018, 140, 14717–14724. [Google Scholar] [CrossRef]
- Arens, J.T.; Blasco-Ahicart, M.; Azmani, K.; Soriano-López, J.; García-Eguizábal, A.; Poblet, J.M.; Galan-Mascaros, J.R. Water oxidation electrocatalysis in acidic media with Co-containing polyoxometalates. J. Catal. 2020, 389, 345–351. [Google Scholar] [CrossRef]
- Garrido-Barros, P.; Gimbert-Suriñach, C.; Moonshiram, D.; Picón, A.; Monge, P.; Batista, V.S.; Llobet, A. electronic π-delocalization boosts catalytic water oxidation by Cu(II) molecular catalysts heterogenized on graphene sheets. J. Am. Chem. Soc. 2017, 139, 12907–12910. [Google Scholar] [CrossRef] [PubMed]
- Haumann, M. Photosynthetic O2 formation tracked by time-resolved X-ray experiments. Science 2005, 310, 1019–1021. [Google Scholar] [CrossRef] [PubMed]
- Barber, J. Biological solar energy. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2007, 365, 1007–1023. [Google Scholar] [CrossRef]
- Barber, J. Crystal structure of the oxygen-evolving complex of Photosystem II. Inorg. Chem. 2008, 47, 1700–1710. [Google Scholar] [CrossRef]
- Vogt, L.; Vinyard, D.J.; Khan, S.; Brudvig, G.W. Oxygen-evolving complex of Photosystem II: An analysis of second-shell residues and hydrogen-bonding networks. Curr. Opin. Chem. Biol. 2015, 25, 152–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.N.; Eriksson, L.A. B3LYP studies of the formation of neutral tyrosyl radical Yz and regeneration of neutral tyrosine Yz in PSII. Int. J. Quantum Chem. 2001, 83, 220–229. [Google Scholar] [CrossRef]
- Baranov, S.V.; Ananyev, G.M.; Klimov, V.V.; Dismukes, G.C. Bicarbonate accelerates assembly of the inorganic core of the water-oxidizing complex in manganese-depleted Photosystem II: A proposed biogeochemical role for atmospheric carbon dioxide in oxygenic photosynthesis. Biochemistry 2000, 39, 6060–6065. [Google Scholar] [CrossRef] [PubMed]
- Goberna-Ferrón, S.; Soriano-López, J.; Galán-Mascarós, J. Activity and stability of the tetramanganese polyanion [Mn4(H2O)2(PW9O34)2]10− during electrocatalytic water oxidation. Inorganics 2015, 3, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Tandon, S.; Soriano-López, J.; Kathalikkattil, A.C.; Jin, G.; Wix, P.; Venkatesan, M.; Lundy, R.; Morris, M.A.; Watson, G.W.; Schmitt, W. A cubane-type manganese complex with H2O oxidation capabilities. Sustain. Energy Fuels 2020, 4, 4464–4468. [Google Scholar] [CrossRef]
- Al-Oweini, R.; Sartorel, A.; Bassil, B.S.; Natali, M.; Berardi, S.; Scandola, F.; Kortz, U.; Bonchio, M. Photocatalytic water oxidation by a mixed-valent MnIII3MnIVO3 manganese oxo core that mimics the natural oxygen-evolving center. Angew. Chem. Int. Ed. 2014, 53, 11182–11185. [Google Scholar] [CrossRef]
- Brimblecombe, R.; Swiegers, G.F.; Dismukes, G.C.; Spiccia, L. Sustained water oxidation photocatalysis by a bioinspired manganese cluster. Angew. Chem. Int. Ed. 2008, 47, 7335–7338. [Google Scholar] [CrossRef]
- Schwarz, B.; Forster, J.; Goetz, M.K.; Yücel, D.; Berger, C.; Jacob, T.; Streb, C. Visible-light-driven water oxidation by a molecular manganese vanadium oxide cluster. Angew. Chemie Int. Ed. 2016, 55, 6329–6333. [Google Scholar] [CrossRef] [PubMed]
- Haxel, G.B.; Hedrick, J.B.; Orris, G.J.; Stauffer, P.H.; Hendley, J.W., II. Rare Earth Elements: Critical Resources for High Technology; U.S. Geological Survey (USGS): Renton, VA, USA, 2002. [CrossRef] [Green Version]
- Hocking, R.K.; Brimblecombe, R.; Chang, L.Y.; Singh, A.; Cheah, M.H.; Glover, C.; Casey, W.H.; Spiccia, L. Water-oxidation catalysis by manganese in a geochemical-like cycle. Nat. Chem. 2011, 3, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Limburg, J.; Vrettos, J.S.; Liable-Sands, L.M.; Rheingold, A.L.; Crabtree, R.H.; Brudvig, G.W. A functional model for O-O bond formation by the O2-evolving complex in Photosystem II. Science 1999, 283, 1524–1527. [Google Scholar] [CrossRef] [PubMed]
- Maayan, G.; Gluz, N.; Christou, G. A bioinspired soluble manganese cluster as a water oxidation electrocatalyst with low overpotential. Nat. Catal. 2018, 1, 48–54. [Google Scholar] [CrossRef]
- Ghosh, T.; Maayan, G. Efficient homogeneous electrocatalytic water oxidation by a manganese cluster with an overpotential of only 74 mV. Angew. Chem. Int. Ed. 2019, 58, 2785–2790. [Google Scholar] [CrossRef]
- Soriano-López, J.; Elliott, R.; Kathalikkattil, A.C.; Ako, A.M.; Mulahmetović, M.; Venkatesan, M.; Schmitt, W. Bioinspired water oxidation using a Mn-oxo cluster stabilized by non-innocent organic tyrosine Y161 and plastoquinone mimics. ACS Sustain. Chem. Eng. 2020, 8, 13648–13659. [Google Scholar] [CrossRef]
- Ako, A.M.; Hewitt, I.J.; Mereacre, V.; Clérac, R.; Wernsdorfer, W.; Anson, C.E.; Powell, A.K. A ferromagnetically coupled Mn19 aggregate with a record S = 83/2 ground spin state. Angew. Chem. Int. Ed. 2006, 45, 4926–4929. [Google Scholar] [CrossRef]
- Boussac, A.; Rappaport, F.; Carrier, P.; Verbavatz, J.-M.; Gobin, R.; Kirilovsky, D.; Rutherford, A.W.; Sugiura, M. Biosynthetic Ca2+/Sr2+ exchange in the Photosystem II oxygen-evolving enzyme of Thermosynechococcus elongatus. J. Biol. Chem. 2004, 279, 22809–22819. [Google Scholar] [CrossRef] [Green Version]
- Chevallot-Beroux, E.; Ako, A.M.; Schmitt, W.; Twamley, B.; Moran, J.; Corinne, B.; Ruhlmann, L.; Mameri, S. Synthesis of new Mn19 analogues and their structural, electrochemical and catalytic properties. Dalton Trans. 2019, 48, 4830–4836. [Google Scholar] [CrossRef]
- Xia, H.; Zhu, Y.; Lu, D.; Li, M.; Zhang, C.; Yang, B.; Ma, Y. Ruthenium(II) complexes with the mixed ligands 2,2′-Bipyridine and 4,4′-Dialkyl Ester-2,2′-bipyridine as pure red dopants for a single-layer electrophosphorescent device. J. Phys. Chem. B 2006, 110, 18718–18723. [Google Scholar] [CrossRef]
- Sullivan, B.P.; Salmon, D.J.; Meyer, T.J. Mixed phosphine 2,2′-bipyridine complexes of ruthenium. Inorg. Chem. 1978, 17, 3334–3341. [Google Scholar] [CrossRef]
- Blasco-Ahicart, M.; Soriano-López, J.; Carbó, J.J.; Poblet, J.M.; Galan-Mascaros, J.R. Polyoxometalate electrocatalysts based on earth-abundant metals for efficient water oxidation in acidic media. Nat. Chem. 2018, 10, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, D.R.; Gagliardi, C.J.; Hull, J.F.; Murphy, C.F.; Kent, C.A.; Westlake, B.C.; Paul, A.; Ess, D.H.; McCafferty, D.G.; Meyer, T.J. Proton-coupled electron transfer. Chem. Rev. 2012, 112, 4016–4093. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Magied, A.F.; Shatskiy, A.; Liao, R.-Z.; Laine, T.M.; Arafa, W.A.A.; Siegbahn, P.E.M.; Kärkäs, M.D.; Åkermark, B.; Johnston, E.V. Chemical and photochemical water oxidation mediated by an efficient single-site ruthenium catalyst. ChemSusChem 2016, 9, 3448–3456. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 30% Mn18Sr/CP | 30% Mn19/CP 2 | |
|---|---|---|
| ηonset 3 (mV) | 192 | 255 |
| η 4 (mV) @ j = 1 mA cm−2 | 284 | 296 |
| η (mV) @ j = 10 mA cm−2 | 550 | 482 |
| η (mV) @ j = 100 mA cm−2 | 827 | 654 |
| Tafel slope (mV dec−1) | 220/260 | 205 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soriano-López, J.; Elliott, R.; Kathalikkattil, A.C.; Ako, A.M.; Schmitt, W. Tuning the Catalytic Water Oxidation Activity through Structural Modifications of High-Nuclearity Mn-oxo Clusters [Mn18M] (M = Sr2+, Mn2+). Water 2021, 13, 2042. https://doi.org/10.3390/w13152042
Soriano-López J, Elliott R, Kathalikkattil AC, Ako AM, Schmitt W. Tuning the Catalytic Water Oxidation Activity through Structural Modifications of High-Nuclearity Mn-oxo Clusters [Mn18M] (M = Sr2+, Mn2+). Water. 2021; 13(15):2042. https://doi.org/10.3390/w13152042
Chicago/Turabian StyleSoriano-López, Joaquín, Rory Elliott, Amal C. Kathalikkattil, Ayuk M. Ako, and Wolfgang Schmitt. 2021. "Tuning the Catalytic Water Oxidation Activity through Structural Modifications of High-Nuclearity Mn-oxo Clusters [Mn18M] (M = Sr2+, Mn2+)" Water 13, no. 15: 2042. https://doi.org/10.3390/w13152042
APA StyleSoriano-López, J., Elliott, R., Kathalikkattil, A. C., Ako, A. M., & Schmitt, W. (2021). Tuning the Catalytic Water Oxidation Activity through Structural Modifications of High-Nuclearity Mn-oxo Clusters [Mn18M] (M = Sr2+, Mn2+). Water, 13(15), 2042. https://doi.org/10.3390/w13152042