
Structure and Dynamics of Interfacial Water on Muscovite Surface under Different Temperature Conditions (298 K to 673 K): Molecular Dynamics Investigation


Abstract
:1. Introduction
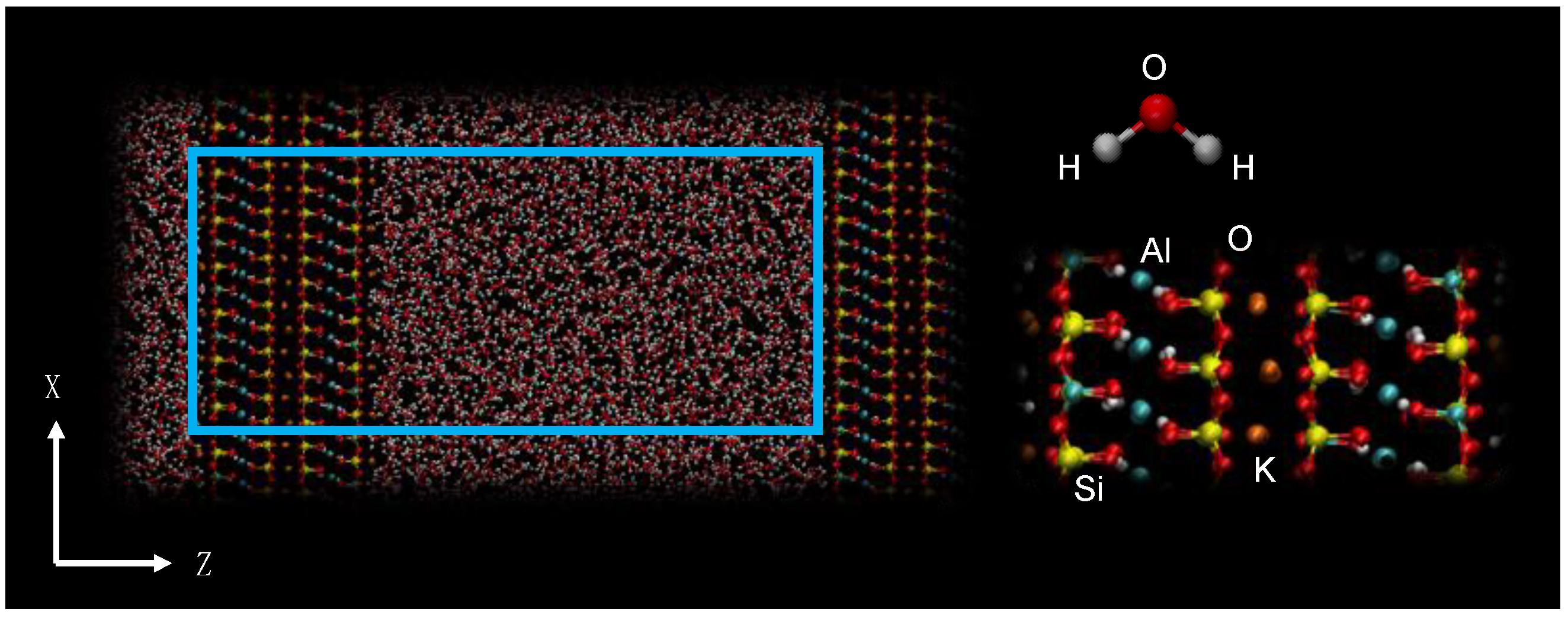
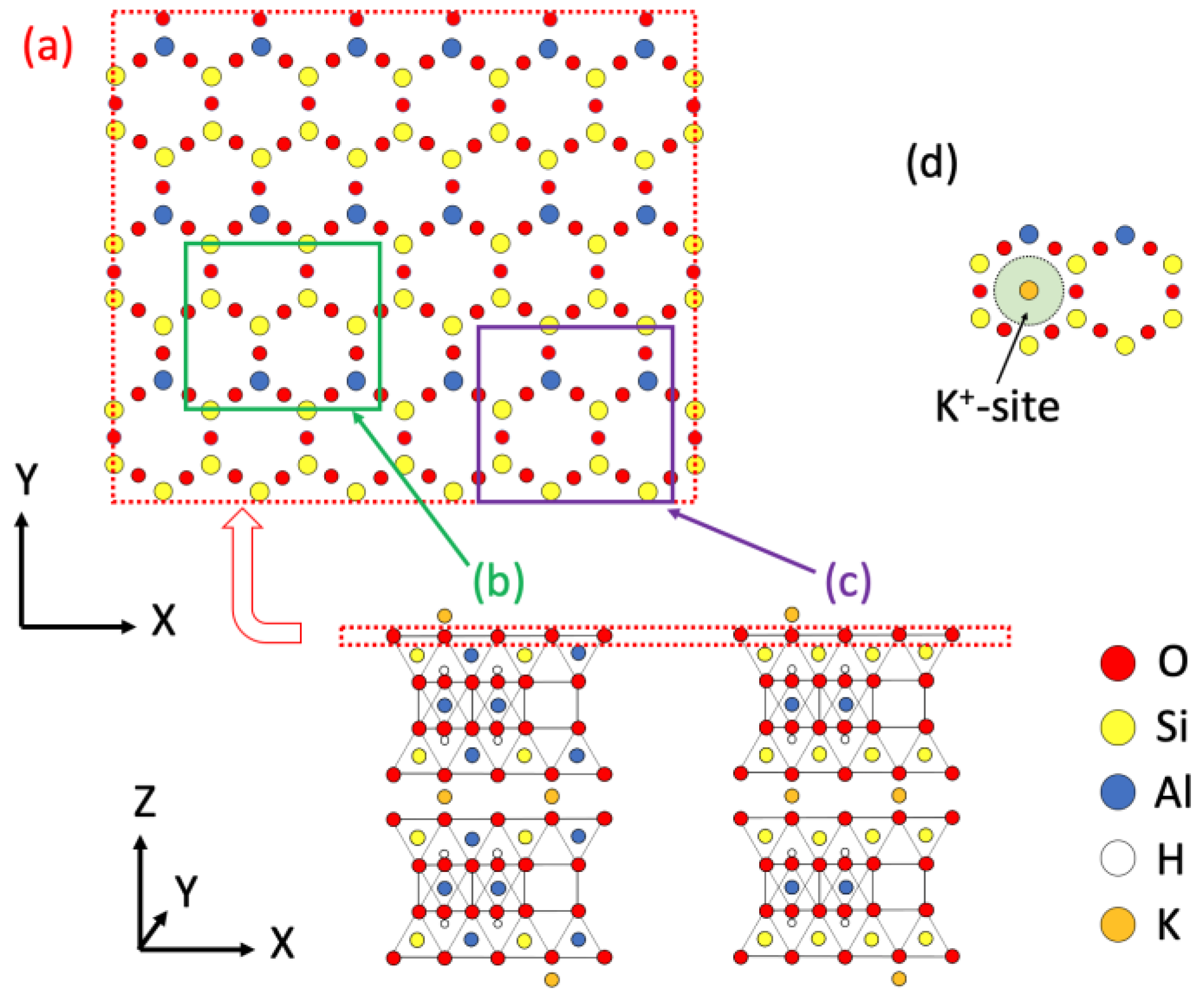
2. Materials and Methods
3. Results and Discussion
3.1. Structural Properties
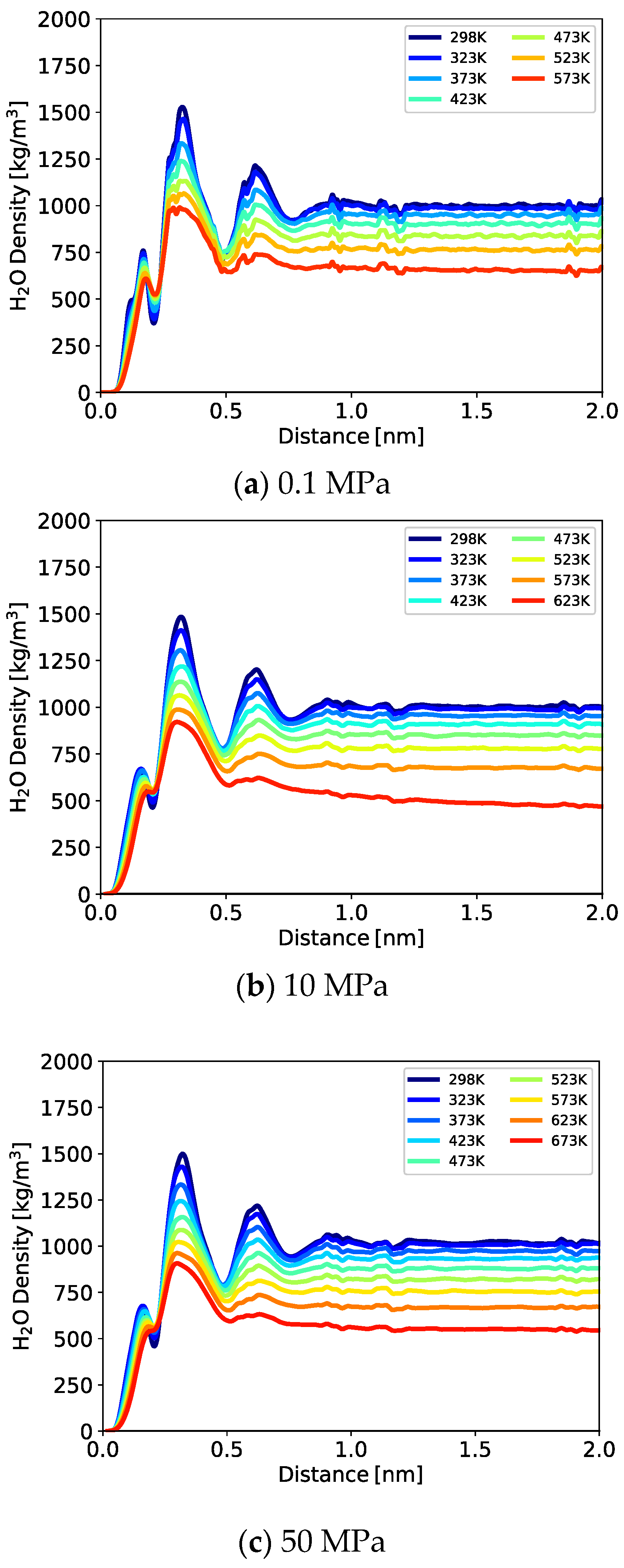
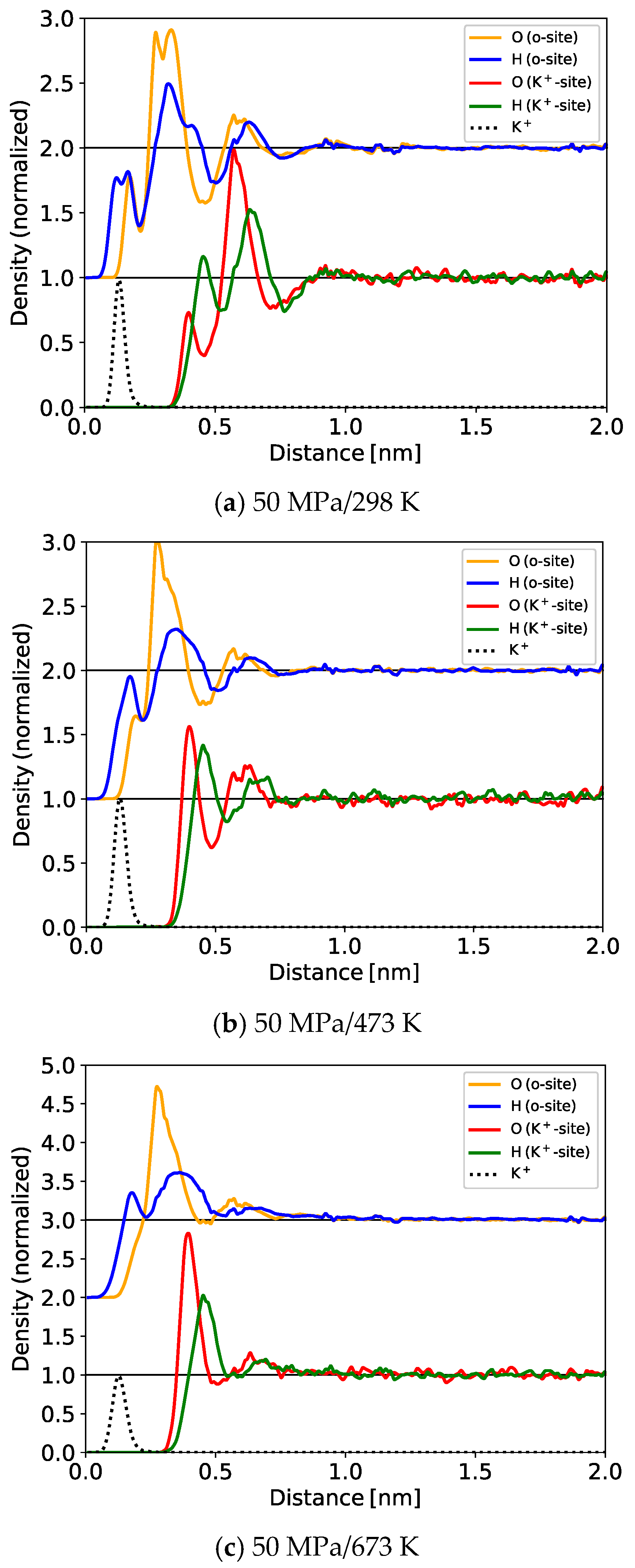
3.1.1. One-Dimensional (1D) Density Profile
3.1.2. Two-Dimensional (2D) Density Map
- The first layer
- The second layer
- The third layer
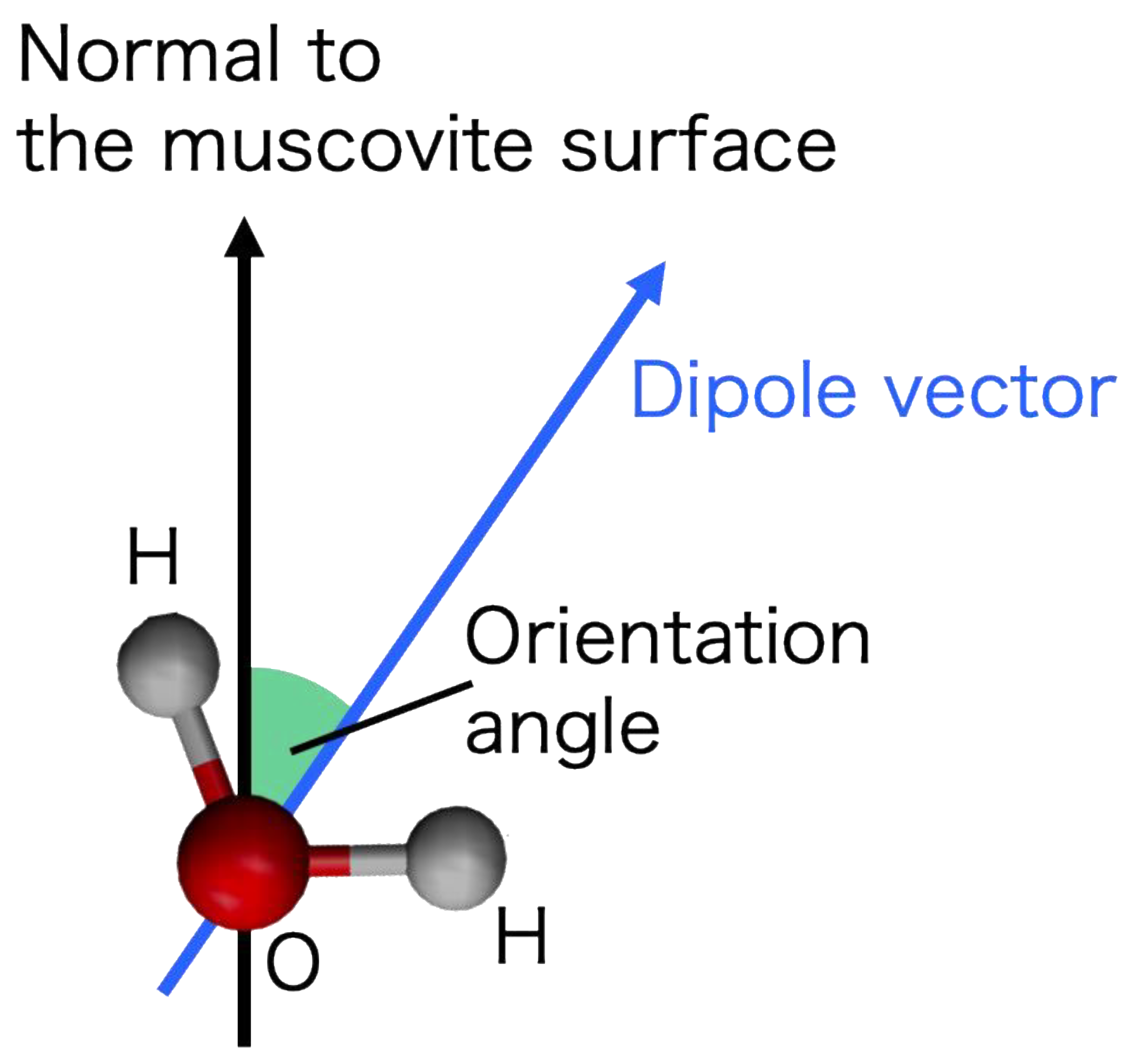
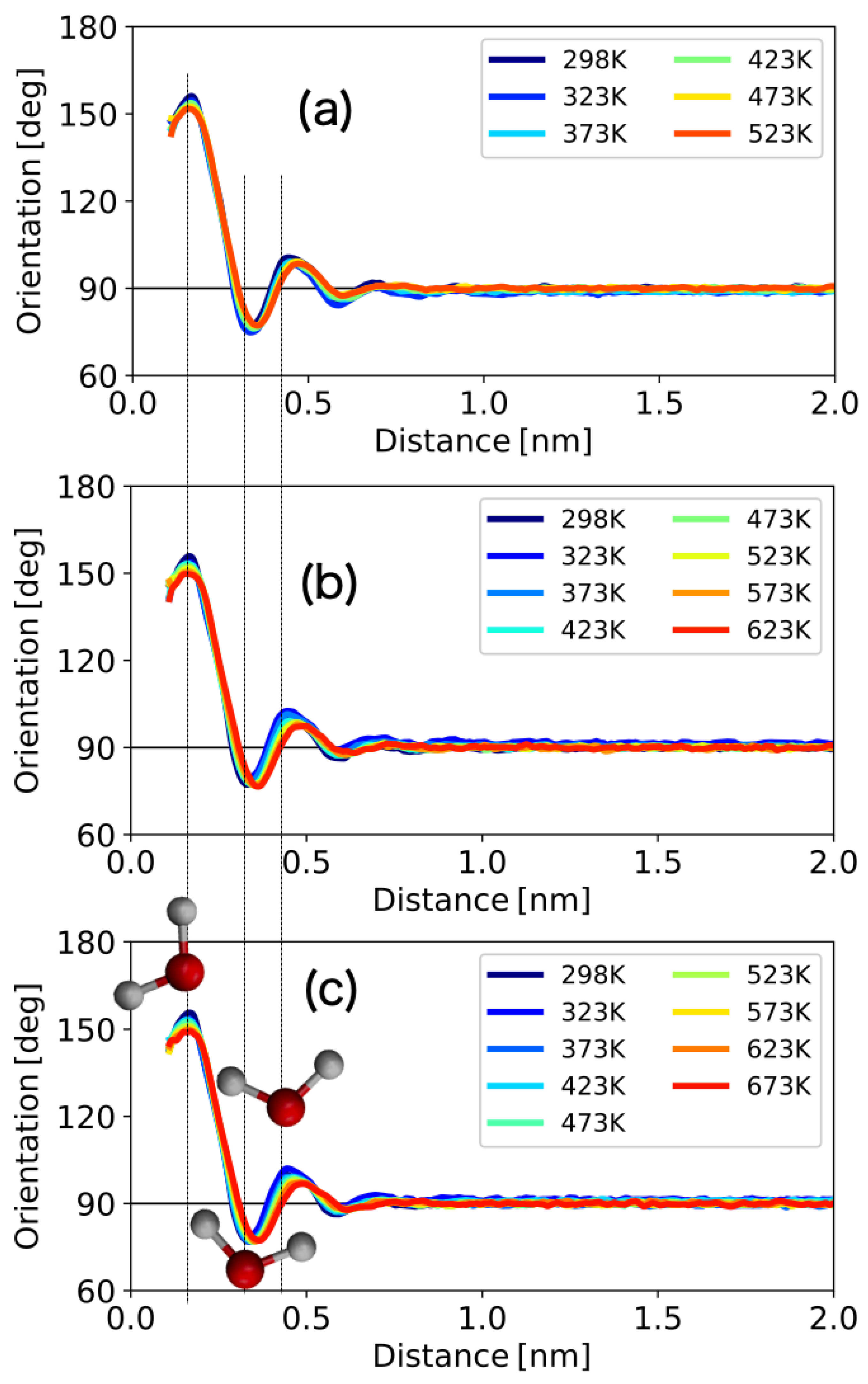
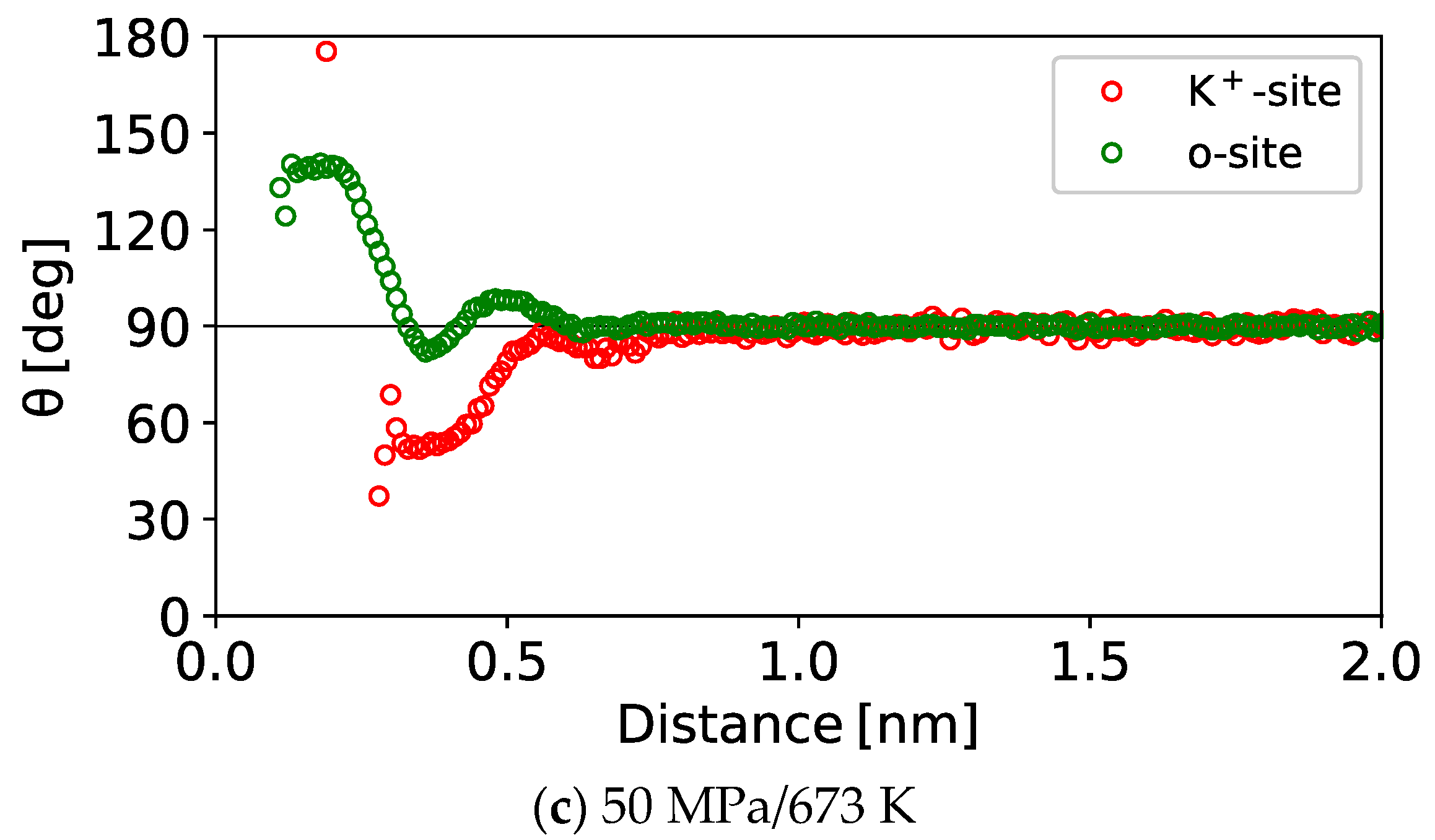
3.1.3. 1D Profile of the Orientation Angle
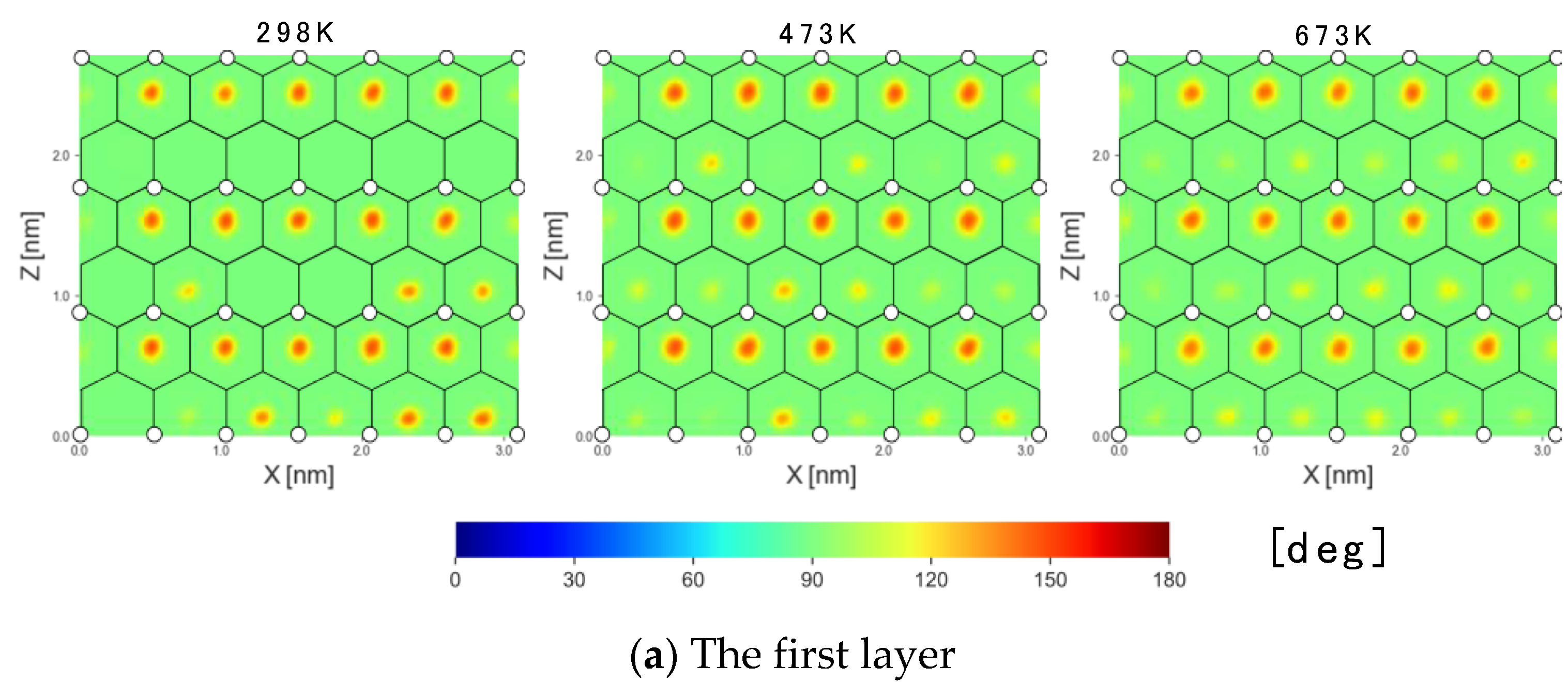
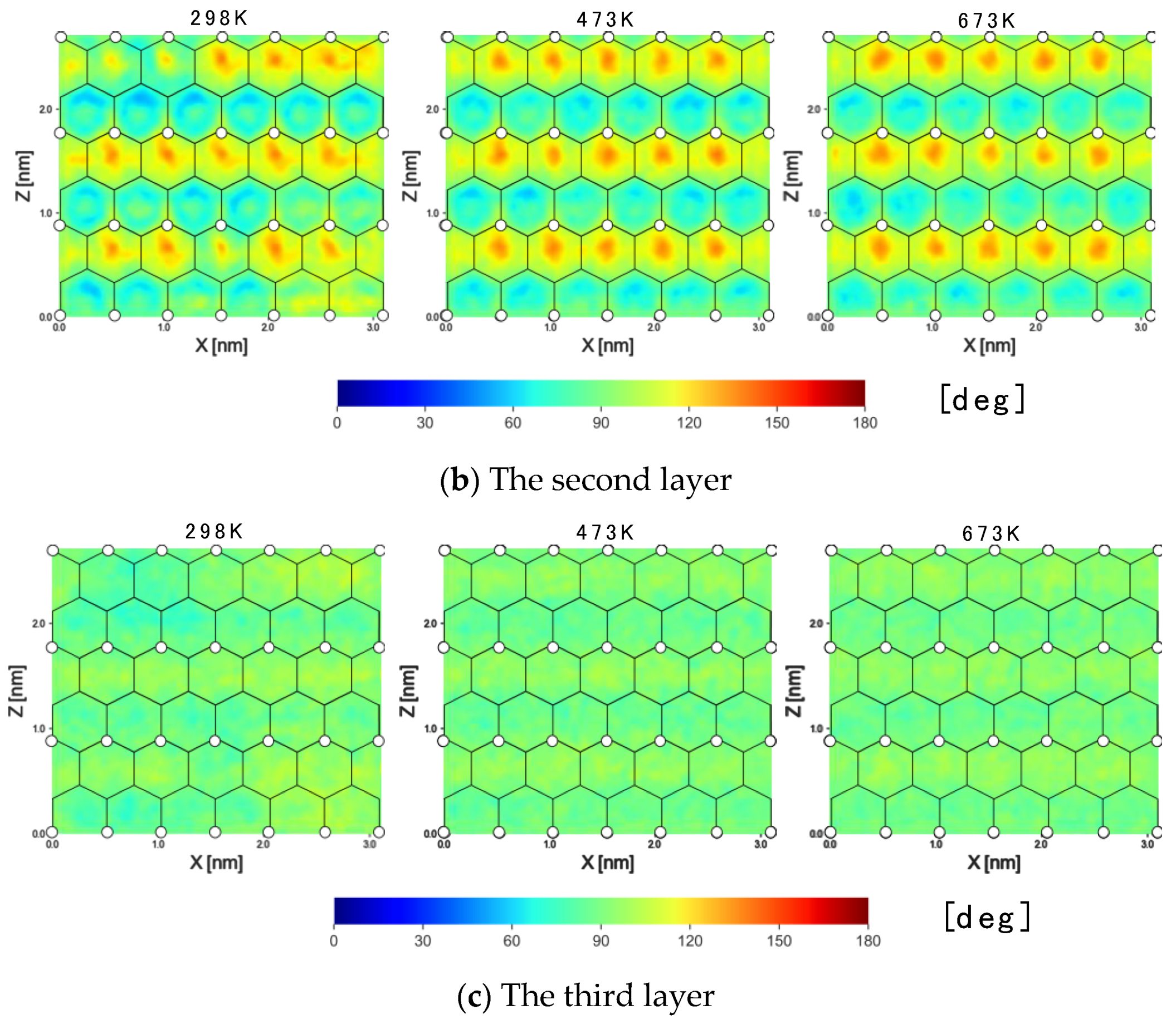
3.1.4. 2D Maps of the Orientation Angle
- The first layer
- The second layer
- The third layer
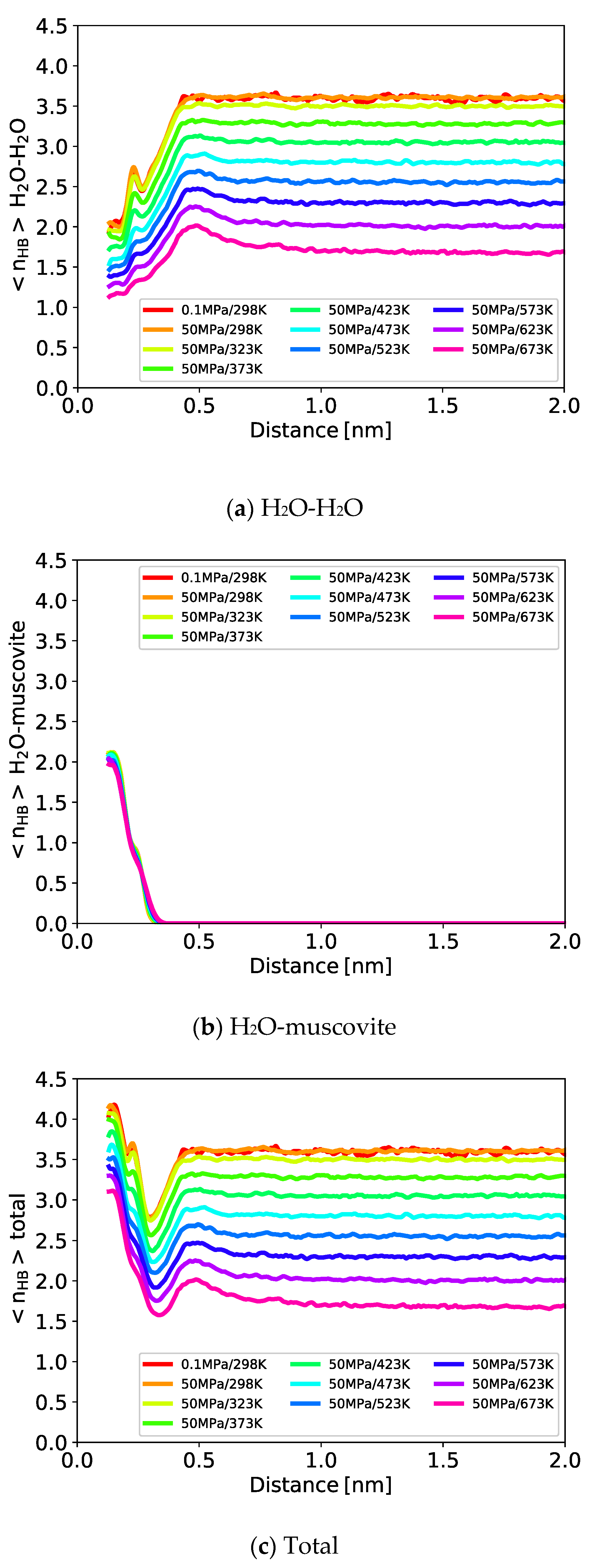
3.1.5. Hydrogen Bonding
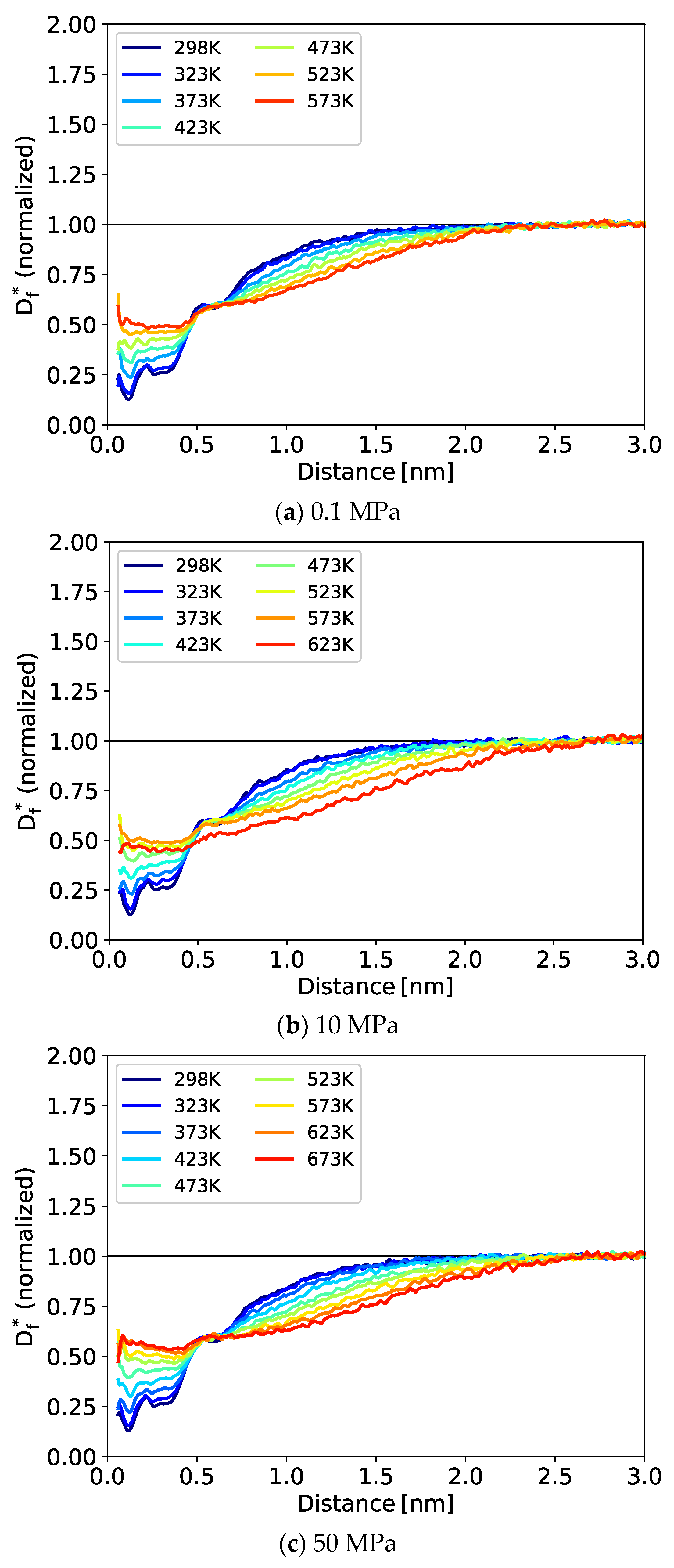
3.2. Dynamic Properties
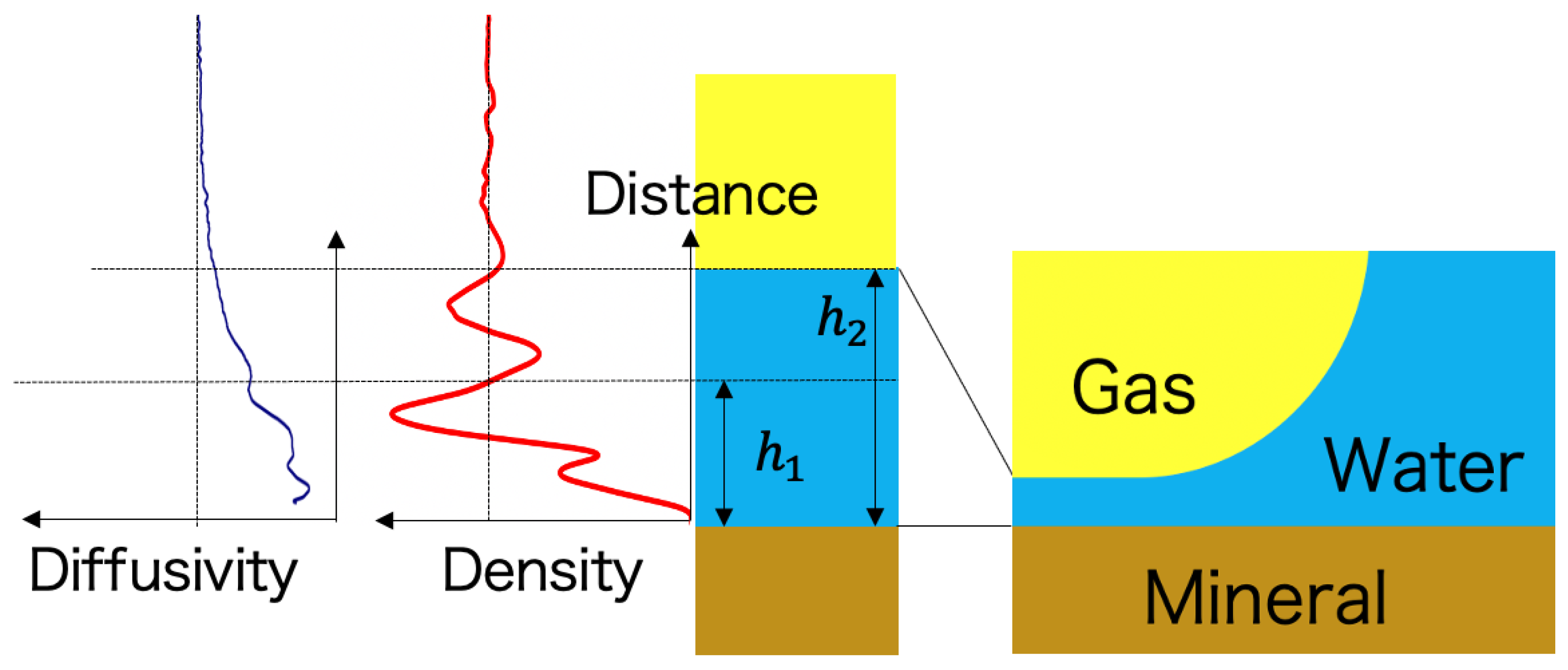
4. Implication to Hydration Forces and Wettability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sposito, G.; Skipper, N.T.; Sutton, R.; Park, S.-H.; Soper, A.K.; Greathouse, J.A. Surface geochemistry of the clay minerals. Proc. Natl. Acad. Sci. USA 1999, 96, 3358–3364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokunaga, T.K. Hydraulic properties of adsorbed water films in unsaturated porous media. Water Resour. Res. 2009, 45. [Google Scholar] [CrossRef] [Green Version]
- Gardés, E.; Wunder, B.; Marquardt, K.; Heinrich, W. The effect of water on intergranular mass transport: New insights from diffusion-controlled reaction rims in the MgO–SiO2 system. Contrib. Mineral. Petrol. 2012, 164, 1–16. [Google Scholar] [CrossRef]
- Broseta, D.; Tonnet, N.; Shah, V. Are rocks still water-wet in the presence of dense CO2or H2S? Geofluids 2012, 12, 280–294. [Google Scholar] [CrossRef]
- DePaolo, D.J.; Cole, D.R. Geochemistry of Geologic Carbon Sequestration: An Overview. Rev. Miner. Geochem. 2013, 77, 1–14. [Google Scholar] [CrossRef]
- Iglauer, S.; Pentland, C.H.; Busch, A. CO2 wettability of seal and reservoir rocks and the implications for carbon geo-sequestration. Water Resour. Res. 2015, 51, 729–774. [Google Scholar] [CrossRef] [Green Version]
- Anderson, W. Wettability Literature Survey- Part 4: Effects of Wettability on Capillary Pressure. J. Pet. Technol. 1987, 39, 1283–1300. [Google Scholar] [CrossRef]
- Morrow, N.R. Wettability and Its Effect on Oil Recovery. J. Pet. Technol. 1990, 42, 1476–1484. [Google Scholar] [CrossRef]
- Romero, E.; Gens, A.; Lloret, A. Water permeability, water retention and microstructure of unsaturated compacted Boom clay. Eng. Geol. 1999, 54, 117–127. [Google Scholar] [CrossRef]
- Mock, J.E.; Tester, J.W.; Wright, P.M. Geothermal Energy from The Earth: Its Potential Impact as an Environmentally Sustainable Resource. Annu. Rev. Energy Environ. 1997, 22, 305–356. [Google Scholar] [CrossRef]
- Zimmermann, G.; Reinicke, A. Hydraulic stimulation of a deep sandstone reservoir to develop an Enhanced Geo-thermal System: Laboratory and field experiments. Geothermics 2010, 39, 70–77. [Google Scholar] [CrossRef] [Green Version]
- Eggertsson, G.; Lavallée, Y.; Kendrick, J.; Markússon, S. Improving fluid flow in geothermal reservoirs by thermal and mechanical stimulation: The case of Krafla volcano, Iceland. J. Volcanol. Geotherm. Res. 2020, 391, 106351. [Google Scholar] [CrossRef]
- Fan, K.; Li, Y.; Elsworth, D.; Dong, M.; Yin, C.; Li, Y.; Chen, Z. Three stages of methane adsorption capacity affected by moisture content. Fuel 2018, 231, 352–360. [Google Scholar] [CrossRef]
- Klewiah, I.; Berawala, D.S.; Walker, H.C.A.; Andersen, P.Ø.; Nadeau, P.H. Review of experimental sorption studies of CO2 and CH4 in shales. J. Nat. Gas Sci. Eng. 2020, 73, 103045. [Google Scholar] [CrossRef]
- Tokunaga, T.K. Physicochemical controls on adsorbed water film thickness in unsaturated geological media. Water Resour. Res. 2011, 47. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, T.K. DLVO-Based Estimates of Adsorbed Water Film Thicknesses in Geologic CO2 Reservoirs. Langmuir 2012, 28, 8001–8009. [Google Scholar] [CrossRef]
- Israelachvili, J.N. Intermolecular and Surface Forces; Academic Press: Cambridge, MA, USA, 2011. [Google Scholar]
- Butt, H.-J.; Kappl, M. Surface and Interfacial Forces; Wiley: Weinheim, Germany, 2010. [Google Scholar]
- Cheng, L.; Fenter, P.; Nagy, K.L.; Schlegel, M.L.; Sturchio, N.C. Molecular-Scale Density Oscillations in Water Adjacent to a Mica Surface. Phys. Rev. Lett. 2001, 87, 156103. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, H.; Kondo, T.; Nakao, H.; Shiraki, K.; Kawamura, K. Structure of Hydrated Sodium Ions and Water Molecules Adsorbed on the Mica/Water Interface. J. Phys. Chem. C 2011, 115, 15959–15964. [Google Scholar] [CrossRef]
- Lee, S.S.; Fenter, P.; Nagy, K.L.; Sturchio, N.C. Monovalent Ion Adsorption at the Muscovite (001)–Solution Interface: Relationships among Ion Coverage and Speciation, Interfacial Water Structure, and Substrate Relaxation. Langmuir 2012, 28, 8637–8650. [Google Scholar] [CrossRef] [PubMed]
- Van Lin, S.R.; Grotz, K.K.; Siretanu, I.; Schwierz, N.; Mugele, F. Ion-Specific and pH-Dependent Hydration of Mica–Electrolyte Interfaces. Langmuir 2019, 35, 5737–5745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlig, M.R.; Benaglia, S.; Thakkar, R.; Comer, J.; Garcia, R. Atomically resolved interfacial water structures on crystalline hydrophilic and hydrophobic surfaces. Nanoscale 2021, 13, 5275–5283. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kalinichev, A.G.; Kirkpatrick, R.J.; Cygan, R.T. Structure, Energetics, and Dynamics of Water Adsorbed on the Muscovite (001) Surface: A Molecular Dynamics Simulation. J. Phys. Chem. B 2005, 109, 15893–15905. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, H.; Kawamura, K. Structure and dynamics of water on muscovite mica surfaces. Geochim. Cosmochim. Acta 2009, 73, 4100–4110. [Google Scholar] [CrossRef]
- Sakuma, H.; Kawamura, K. Structure and dynamics of water on Li+-, Na+-, K+-, Cs+-, H3O+-exchanged muscovite surfaces: A molecular dynamics study. Geochim. Cosmochim. Acta 2011, 75, 63–81. [Google Scholar] [CrossRef]
- Kobayashi, K.; Liang, Y.; Amano, K.-I.; Murata, S.; Matsuoka, T.; Takahashi, S.; Nishi, N.; Sakka, T. Molecular Dynamics Simulation of Atomic Force Microscopy at the Water–Muscovite Interface: Hydration Layer Structure and Force Analysis. Langmuir 2016, 32, 3608–3616. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Liang, Y.; Murata, S.; Matsuoka, T.; Takahashi, S.; Nishi, N.; Sakka, T. Ion Distribution and Hydration Structure in the Stern Layer on Muscovite Surface. Langmuir 2017, 33, 3892–3899. [Google Scholar] [CrossRef] [PubMed]
- Bourg, I.C.; Lee, S.S.; Fenter, P.; Tournassat, C. Stern Layer Structure and Energetics at Mica–Water Interfaces. J. Phys. Chem. C 2017, 121, 9402–9412. [Google Scholar] [CrossRef]
- Loganathan, N.; Kalinichev, A.G. Quantifying the Mechanisms of Site-Specific Ion Exchange at an Inhomogeneously Charged Surface: Case of Cs+/K+ on Hydrated Muscovite Mica. J. Phys. Chem. C 2017, 121, 7829–7836. [Google Scholar] [CrossRef] [Green Version]
- Adapa, S.; Malani, A. Role of hydration energy and co-ions association on monovalent and divalent cations adsorption at mica-aqueous interface. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Churaev, N. Wetting films and wetting. Rev. Phys. Appl. 1988, 23, 975–987. [Google Scholar] [CrossRef]
- Derjaguin, B.; Churaev, N. The current state of the theory of long-range surface forces. Colloids Surf. 1989, 41, 223–237. [Google Scholar] [CrossRef]
- IPCC. Underground Geological Storage. In IPCC Special Report on Carbon Dioxide Capture and Storage; Cambridge University Press: Cambridge, UK, 2005; pp. 195–276. [Google Scholar]
- Watanabe, N.; Numakura, T.; Sakaguchi, K.; Saishu, H.; Okamoto, A.; Ingebritsen, S.E.; Tsuchiya, N. Potentially exploitable supercritical geothermal resources in the ductile crust. Nat. Geosci. 2017, 10, 140–144. [Google Scholar] [CrossRef]
- Chase, M.W., Jr.; Tables, N.J.T. Data reported in NIST standard reference database 69, June 2005 release: NIST Chemistry WebBook. J. Phys. Chem. Ref. Data Monogr. 1998, 9, 1–1951. [Google Scholar]
- Lammers, K.; Smith, M.M.; Carroll, S.A. Muscovite dissolution kinetics as a function of pH at elevated temperature. Chem. Geol. 2017, 466, 149–158. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Cygan, R.T.; Liang, J.-J.; Kalinichev, A.G. Molecular Models of Hydroxide, Oxyhydroxide, and Clay Phases and the Development of a General Force Field. J. Phys. Chem. B 2004, 108, 1255–1266. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Richardson, S.M.; Richardson, J.W. Crystal structure of a pink muscovite from Archer’s Post, Kenya: Implications for reverse pleochroism in dioctahedral micas. Am. Mineral. 1982, 67, 69–75. [Google Scholar]
- Calero, C.; Franzese, G. Water under extreme confinement in graphene: Oscillatory dynamics, structure, and hydration pressure explained as a function of the confinement width. J. Mol. Liq. 2020, 317, 114027. [Google Scholar] [CrossRef]
- Schlegel, M.L.; Nagy, K.L.; Fenter, P.; Cheng, L.; Sturchio, N.C.; Jacobsen, S.D. Cation sorption on the muscovite (001) surface in chloride solutions using high-resolution X-ray reflectivity. Geochim. Cosmochim. Acta 2006, 70, 3549–3565. [Google Scholar] [CrossRef]
- Ricci, M.; Spijker, P.; Voïtchovsky, K. Water-induced correlation between single ions imaged at the solid–liquid in-terface. Nat. Commun. 2014, 5, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morishita, T. Liquid-Liquid Phase Transitions of Phosphorus via Constant-Pressure First-Principles Molecular Dynamics Simulations. Phys. Rev. Lett. 2001, 87, 105701. [Google Scholar] [CrossRef]
- Morishita, T. Polymeric liquid of phosphorus at high pressure: First-principles molecular-dynamics simulations. Phys. Rev. B 2002, 66, 054204. [Google Scholar] [CrossRef]
- Morishita, T. High Density Amorphous Form and Polyamorphic Transformations of Silicon. Phys. Rev. Lett. 2004, 93, 055503. [Google Scholar] [CrossRef]
- Eisenberg, D.; Kauzmann, W. The Structure and Properties of Water; Oxford University Press: Oxford, UK, 2005. [Google Scholar]
- Ohmine, I.; Tanaka, H. Fluctuation, relaxations, and hydration in liquid water. Hydrogen-bond rearrangement dynamics. Chem. Rev. 1993, 93, 2545–2566. [Google Scholar] [CrossRef]
- Matsumoto, M.; Saito, S.; Ohmine, I. Molecular dynamics simulation of the ice nucleation and growth process leading to water freezing. Nat. Cell Biol. 2002, 416, 409–413. [Google Scholar] [CrossRef]
- Hassanpouryouzband, A.; Joonaki, E.; Farahani, M.V.; Takeya, S.; Ruppel, C.; Yang, J.; English, N.J.; Schicks, J.M.; Edlmann, K.; Mehrabian, H.; et al. Gas hydrates in sustainable chemistry. Chem. Soc. Rev. 2020, 49, 5225–5309. [Google Scholar] [CrossRef] [PubMed]
- Luzar, A.; Chandler, D. Effect of Environment on Hydrogen Bond Dynamics in Liquid Water. Phys. Rev. Lett. 1996, 76, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Gorbaty, Y.; Demianets, Y. The pair-correl.Ation functions of water at a pressure of 1000 bar in the temperature range 25–500 °C. Chem. Phys. Lett. 1983, 100, 450–454. [Google Scholar] [CrossRef]
- Kalinichev, A.G.; Bass, J.D. Hydrogen Bonding in Supercritical Water. 2. Computer Simulations. J. Phys. Chem. A 1997, 101, 9720–9727. [Google Scholar] [CrossRef]
- Dubey, V.; Erimban, S.; Indra, S.; Daschakraborty, S. Understanding the Origin of the Breakdown of the Stokes–Einstein Relation in Supercooled Water at Different Temperature–Pressure Conditions. J. Phys. Chem. B 2019, 123, 10089–10099. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chan, K.-Y.; Quirke, N. Molecular dynamics simulation of water confined in a nanopore of amorphous silica. Mol. Simul. 2009, 35, 1215–1223. [Google Scholar] [CrossRef]
- Milischuk, A.A.; Ladanyi, B.M. Structure and dynamics of water confined in silica nanopores. J. Chem. Phys. 2011, 135, 174709. [Google Scholar] [CrossRef]
- Lee, S.H.; Rossky, P.J. A comparison of the structure and dynamics of liquid water at hydrophobic and hydrophilic surfaces—a molecular dynamics simulation study. J. Chem. Phys. 1994, 100, 3334–3345. [Google Scholar] [CrossRef]
- Hanasaki, I.; Nakatani, A. Hydrogen bond dynamics and microscopic structure of confined water inside carbon nanotubes. J. Chem. Phys. 2006, 124, 174714. [Google Scholar] [CrossRef]
- Zielkiewicz, J. Structural properties of water: Comparison of the SPC, SPCE, TIP4P, and TIP5P models of water. J. Chem. Phys. 2005, 123, 104501. [Google Scholar] [CrossRef]
- Ishikawa, S.; Sakuma, H.; Tsuchiya, N. Self–diffusion of water molecules confined between quartz surfaces at elevated temperatures by molecular dynamics simulations. J. Mineral. Petrol. Sci. 2016, 111, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Guissani, Y.; Guillot, B. A computer simulation study of the liquid–vapor coexistence curve of water. J. Chem. Phys. 1993, 98, 8221–8235. [Google Scholar] [CrossRef]
- Guevara-Carrion, G.; Vrabec, J.; Hasse, H. Prediction of self-diffusion coefficient and shear viscosity of water and its binary mixtures with methanol and ethanol by molecular simulation. J. Chem. Phys. 2011, 134, 074508. [Google Scholar] [CrossRef] [Green Version]
- Moultos, O.A.; Tsimpanogiannis, I.N.; Panagiotopoulos, A.Z.; Economou, I.G. Atomistic molecular dynamics simulations of CO2 diffusivity in H2O for a wide range of temperatures and pressures. J. Phys. Chem. B 2014, 118, 5532–5541. [Google Scholar] [CrossRef] [PubMed]
- Tsimpanogiannis, I.N.; Moultos, O.A.; Franco, L.F.; Spera, M.B.D.M.; Erdős, M.; Economou, I.G. Self-diffusion coefficient of bulk and confined water: A critical review of classical molecular simulation studies. Mol. Simul. 2019, 45, 425–453. [Google Scholar] [CrossRef] [Green Version]
- Israelachvili, J.N.; Pashley, R.M. Molecular layering of water at surfaces and origin of repulsive hydration forces. Nat. Cell Biol. 1983, 306, 249–250. [Google Scholar] [CrossRef]
- Hirasaki, G. Wettability: Fundamentals and Surface Forces. SPE Form. Eval. 1991, 6, 217–226. [Google Scholar] [CrossRef]
- Hirasaki, G.J. Thermodynamics of Thin Films and Three-Phase Contact Regions. In Interfacial Phenomena in Oil Recovery; Morrow, N.R., Ed.; Marcel Dekker: New York, NY, USA, 1991; pp. 23–75. [Google Scholar]
- Mugele, F.G.; Bera, B.; Cavalli, A.; Siretanu, I.; Maestro, A.; Duits, M.H.; Stuart, M.A.C.; Ende, D.V.D.; Stocker, I.N.; Collins, I.R. Ion adsorption-induced wetting transition in oil-water-mineral systems. Sci. Rep. 2015, 5, 10519. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Ángeles, F.; Firoozabadi, A. Contact Angle, Liquid Film, and Liquid–Liquid and Liquid–Solid Interfaces in Model Oil–Brine–Substrate Systems. J. Phys. Chem. C 2016, 120, 11910–11917. [Google Scholar] [CrossRef]
- Jiménez-Ángeles, F.; Firoozabadi, A. Tunable substrate wettability by thin water layer. J. Phys. Chem. C 2016, 120, 24688–24696. [Google Scholar] [CrossRef]
- Alshakhs, M.J.; Kovscek, A.R. Understanding the role of brine ionic composition on oil recovery by assessment of wettability from colloidal forces. Adv. Colloid Interface Sci. 2016, 233, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Sanaei, A.; Sepehrnoori, K. Implication of Oil/Brine/Rock Surface Interactions in Modeling Modified Salinity Waterflooding in Carbonate and Sandstone Reservoirs. In Proceedings of the SPE Annual Technical Conference and Exhibition, Dallas, TX, USA, 26 September 2018. [Google Scholar]
- Sanaei, A.; Tavassoli, S.; Sepehrnoori, K. Investigation of modified Water chemistry for improved oil recovery: Application of DLVO theory and surface complexation model. Colloids Surf. A Physicochem. Eng. Asp. 2019, 574, 131–145. [Google Scholar] [CrossRef]
- Shiga, M.; Aichi, M.; Sorai, M. Quantitative Investigation on the Contributing Factors to the Contact Angle of the CO2/H2O/Muscovite Systems Using the Frumkin-Derjaguin Equation. Geofluids 2020, 2020, 6656460. [Google Scholar] [CrossRef]
- Bordeaux-Rego, F.; Mehrabi, M.; Sanaei, A.; Sepehrnoori, K. Improvements on modelling wettability alteration by Engineered water injection: Surface complexation at the oil/brine/rock contact. Fuel 2021, 284, 118991. [Google Scholar] [CrossRef]
- Whalen, J.W.; Lai, K.-Y. Adhesional wetting on modified soda-lime glass surfaces. J. Colloid Interface Sci. 1977, 59, 483–489. [Google Scholar] [CrossRef]
- Morishita, T.; Itoh, S.G.; Okumura, H.; Mikami, M. Free-energy calculation via mean-force dynamics using a logarithmic energy landscape. Phys. Rev. E 2012, 85, 066702. [Google Scholar] [CrossRef] [PubMed]
- Morishita, T.; Yonezawa, Y.; Ito, A.M. Free Energy Reconstruction from Logarithmic Mean-Force Dynamics Using Multiple Nonequilibrium Trajectories. J. Chem. Theory Comput. 2017, 13, 3106–3119. [Google Scholar] [CrossRef] [PubMed]
- Leoni, F.; Franzese, G. Structural behavior and dynamics of an anomalous fluid between attractive and repulsive walls: Templating, molding, and superdiffusion. J. Chem. Phys. 2014, 141, 174501. [Google Scholar] [CrossRef] [Green Version]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pressure (MPa) | Temperature (K) |
|---|---|
| 0.1 | 298, 323, 373, 423, 473, 523, 573 |
| 10 | 298, 323, 373, 423, 473, 523, 573, 623 |
| 50 | 298, 323, 373, 423, 473, 523, 573, 623, 673 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shiga, M.; Aichi, M.; Sorai, M.; Morishita, T. Structure and Dynamics of Interfacial Water on Muscovite Surface under Different Temperature Conditions (298 K to 673 K): Molecular Dynamics Investigation. Water 2021, 13, 1320. https://doi.org/10.3390/w13091320
Shiga M, Aichi M, Sorai M, Morishita T. Structure and Dynamics of Interfacial Water on Muscovite Surface under Different Temperature Conditions (298 K to 673 K): Molecular Dynamics Investigation. Water. 2021; 13(9):1320. https://doi.org/10.3390/w13091320
Chicago/Turabian StyleShiga, Masashige, Masaatsu Aichi, Masao Sorai, and Tetsuya Morishita. 2021. "Structure and Dynamics of Interfacial Water on Muscovite Surface under Different Temperature Conditions (298 K to 673 K): Molecular Dynamics Investigation" Water 13, no. 9: 1320. https://doi.org/10.3390/w13091320
APA StyleShiga, M., Aichi, M., Sorai, M., & Morishita, T. (2021). Structure and Dynamics of Interfacial Water on Muscovite Surface under Different Temperature Conditions (298 K to 673 K): Molecular Dynamics Investigation. Water, 13(9), 1320. https://doi.org/10.3390/w13091320