
Evaluation of Factors Influencing Lab-Scale Studies to Determine Heavy Metal Removal by Six Sorbents for Stormwater Treatment


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Sorbents and Chemicals
2.2. Experimental Setup
2.2.1. Batch Experiments with Heavy Metals
2.2.2. Column Experiments
2.2.3. Batch Experiments According to Standard Methods
2.3. Analytical Procedures
2.4. Data Evaluation
3. Results and Discussion
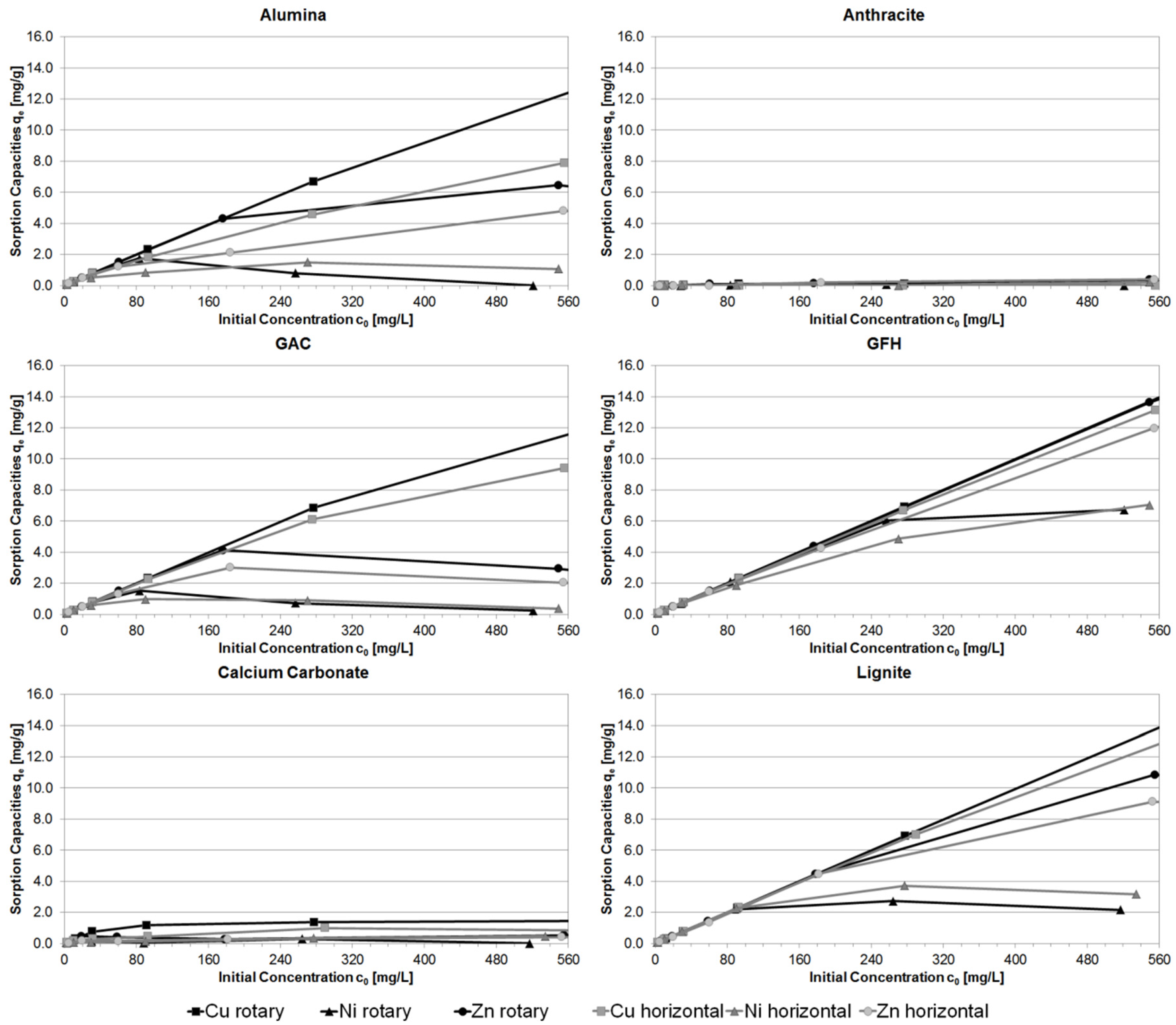
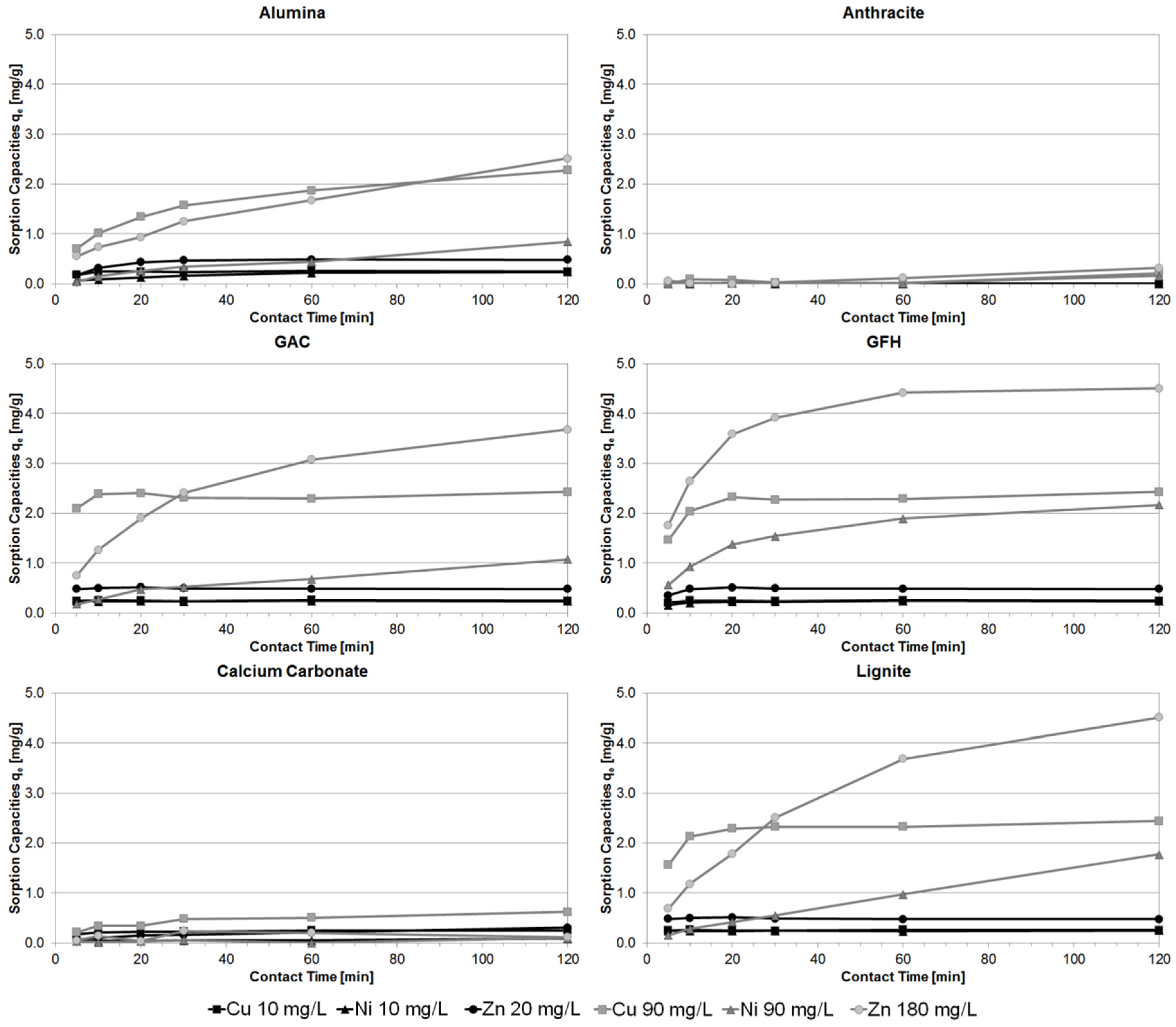
3.1. Batch Experiments
3.2. Column Experiments
3.3. Comparison of Batch and Column Experiment Capacities
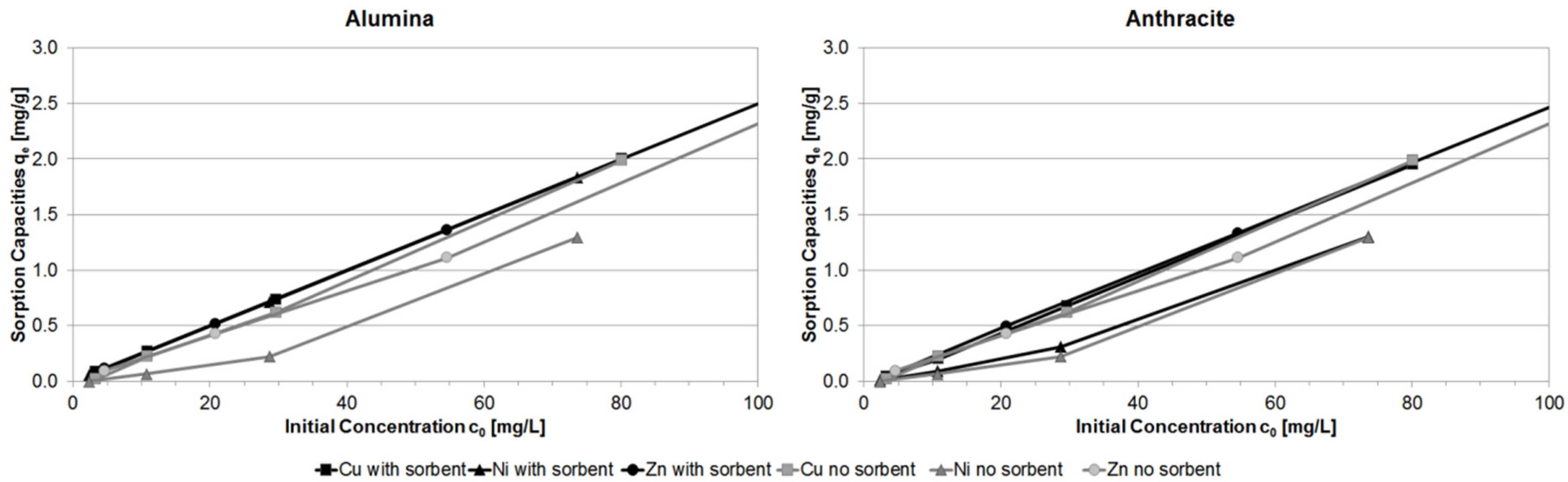
3.4. Comparison of Different Batch Experiment Capacities
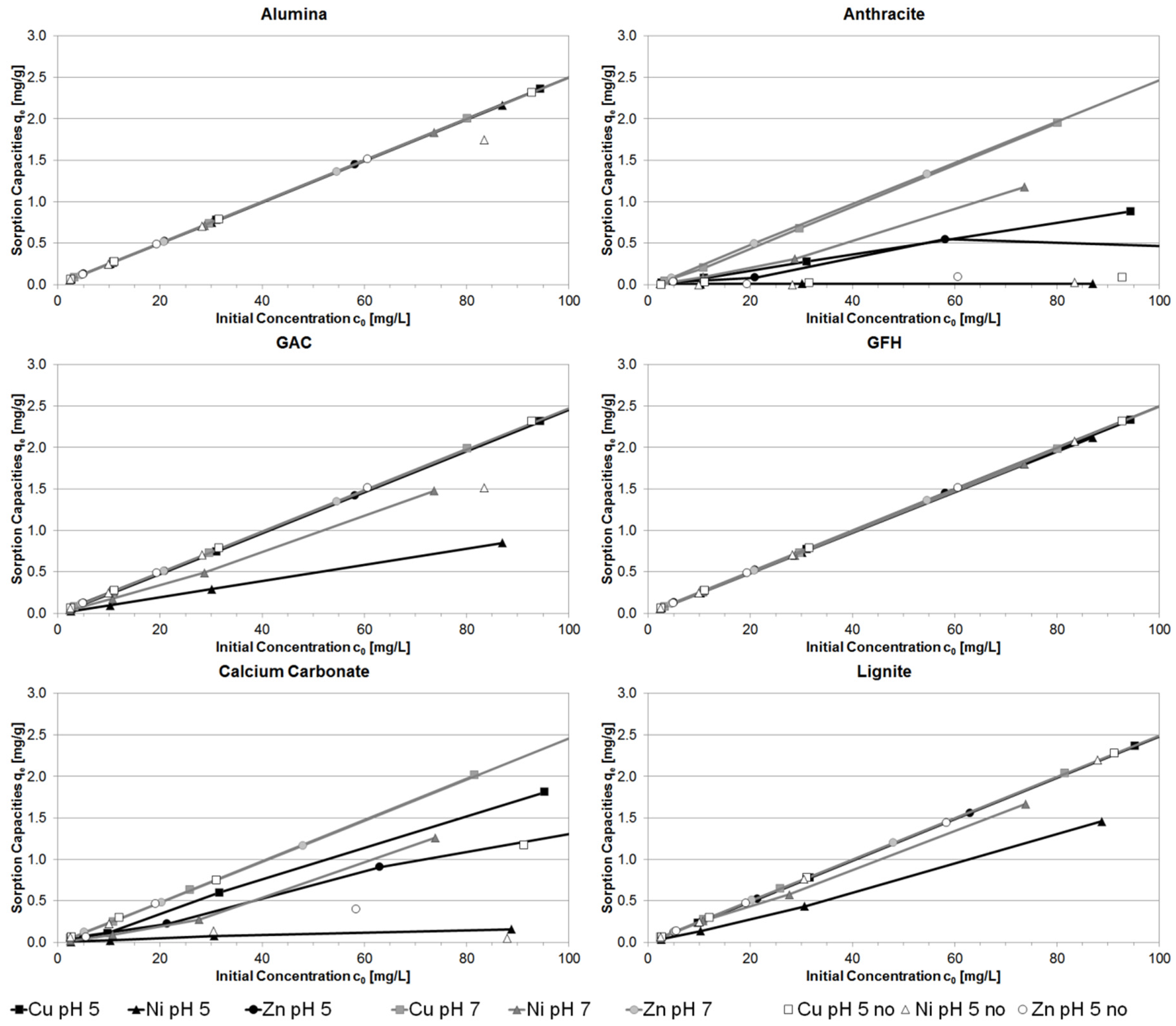
3.5. Implementation of Ionic Strength and the pH value of Stormwater Runoff
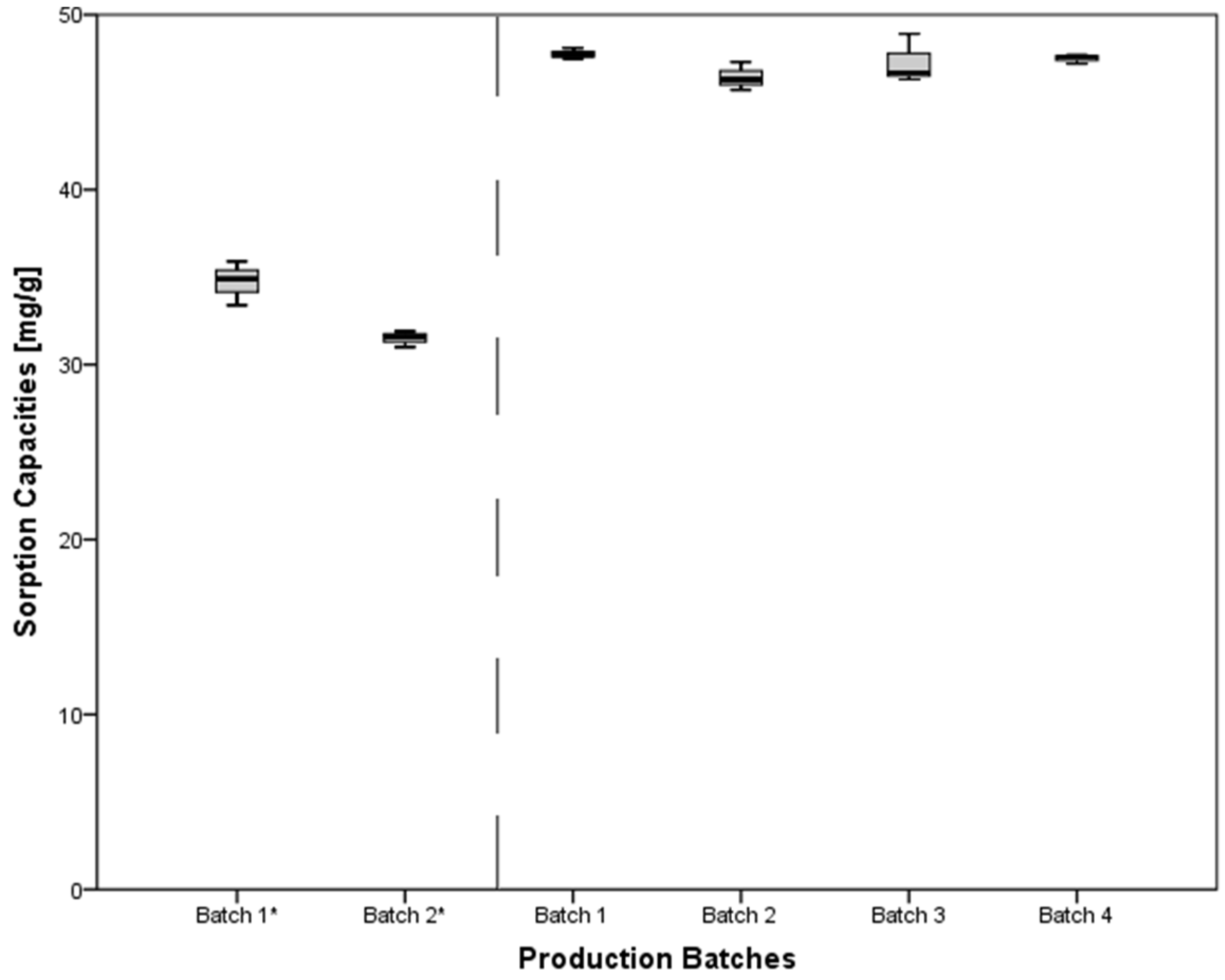
3.6. Evaluation of Different Production Batches
4. Conclusions
- Experiments with the dissolved heavy metals of interest using synthetic water at pH 5 is suggested and the influence of the type and speed of the shaker must be considered.
- Adjustments of ionic strength and pH > 5 are more sensitive and should be avoided because they mostly lead to an overestimation of the removal capacities of sorbents.
- The filtration step at the end of the batch experiments has an effect on the results and does not simulate real situations.
- Batch experiments with other cations (e.g., magnesium and ammonium) should not be used as surrogates.
- Batch experiments with heavy metals can only be used under identical and well-defined conditions to prove the comparability of different production batches and to select sorbents for subsequent column experiments.
- Column experiments can be used as an indicator to determine the efficiencies and service lives of treatment systems.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Roeva, N.N.; Rovinskii, F.Y.; Kononov, E.Y. Special features of the behavior of heavy metals in various natural environments. J. Anal. Chem. 1996, 51, 352–364. [Google Scholar]
- von Uexküll, O.; Skerfving, S.; Doyle, R.; Braungart, M. Antimony in brake pads-a carcinogenic component? J. Clean. Prod. 2005, 13, 19–31. [Google Scholar] [CrossRef]
- Thorpe, A.; Harrison, R.M. Sources and properties of non-exhaust particulate matter from road traffic: A review. Sci. Total Environ. 2008, 400, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Kayhanian, M.; Fruchtman, B.D.; Gulliver, J.S.; Montanaro, C.; Ranieri, E.; Wuertz, S. Review of highway runoff characteristics: Comparative analysis and universal implications. Water Res. 2012, 46, 6609–6624. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.; Welker, A.; Helmreich, B. Critical review of heavy metal pollution of traffic area runoff: Occurrence, influencing factors, and partitioning. Sci. Total Environ. 2016, 541, 895–919. [Google Scholar] [CrossRef] [PubMed]
- Sansalone, J.; Glenn, D.W. Temporal variations in heavy metal partitioning and loading in urban highway pavement sheet flow: Implications for in situ treatment design. Transp. Res. Rec. 2000, 1720, 100–111. [Google Scholar] [CrossRef]
- Helmreich, B.; Hilliges, R.; Schriewer, A.; Horn, H. Runoff pollutants of a highly trafficked urban road—Correlation analysis and seasonal influences. Chemosphere 2010, 80, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Roubicek, V.; Raclavska, H.; Juchelkova, D.; Filip, P. Wear and environmental aspects of composite materials for automotive braking industry. Wear 2008, 265, 167–175. [Google Scholar] [CrossRef]
- Ball, J.E.; Jenks, R.; Aubourg, D. An assessment of the availability of pollutant constituents on road surfaces. Sci. Total Environ. 1998, 209, 243–254. [Google Scholar] [CrossRef]
- Mangani, G.; Berloni, A.; Bellucci, F.; Tatàno, F.; Maione, M. Evaluation of the pollutant content in road runoff first flush waters. Water Air Soil Pollut. 2005, 160, 213–228. [Google Scholar] [CrossRef]
- Denkhaus, E.; Salnikow, K. Nickel essentiality, toxicity, and carcinogenicity. Critical Rev. Oncol. Hematol. 2002, 42, 35–56. [Google Scholar] [CrossRef]
- Davis, A.P.; Shokouhian, M.; Ni, S. Loading estimates of lead, copper, cadmium, and zinc in urban runoff from specific sources. Chemosphere 2001, 44, 997–1009. [Google Scholar] [CrossRef]
- Legret, M.; Pagotto, C. Evaluation of pollutant loadings in the runoff waters from a major rural highway. Sci. Total Environ. 1999, 235, 143–150. [Google Scholar] [CrossRef]
- Sansalone, J.J.; Buchberger, S.G. Partitioning and first flush of metals in urban roadway storm water. J. Environ. Eng. 1997, 123, 134–143. [Google Scholar] [CrossRef]
- Dierkes, C.; Geiger, W.F. Decontaminating effect of greened highway embankments. In Proceedings of the NOVATECH-Conference, Lyon, France, 4–6 May 1998; pp. 497–504.
- Van Bohemen, H.D.; Van De Laak, W.H.J. The influence of road infrastructure and traffic on soil, water, and air quality. Environ. Manage. 2003, 31, 50–68. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, E.R.; Money, J.E.; Green, P.G.; Young, T.M. Metals associated with stormwater-relevant brake and tire samples. Sci. Total Environ. 2009, 407, 5855–5860. [Google Scholar] [CrossRef] [PubMed]
- Dierkes, C.; Lucke, T.; Helmreich, B. General technical approvals for decentralised sustainable urban drainage systems (SUDS)—The current situation in Germany. Sustainability 2015, 7, 3031–3051. [Google Scholar] [CrossRef]
- Liu, D.; Sansalone, J.J.; Cartledge, F.K. Comparison of sorptive filter media for treatment of metals in runoff. J. Environ. Eng. 2005, 131, 1178–1186. [Google Scholar] [CrossRef]
- Hilliges, R.; Schriewer, A.; Helmreich, B. A three-stage treatment system for highly polluted urban road runoff. J. Environ. Manage. 2013, 128, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Maniquiz-Redillas, M.; Kim, L.-H. Fractionation of heavy metals in runoff and discharge of a stormwater management system and its implications for treatment. J. Environ. Sci. 2014, 26, 1214–1222. [Google Scholar] [CrossRef]
- Hendershot, W.H.; Duquette, M. A simple barium chloride method for determining cation exchange capacity and exchangeable cations. Soil Sci. Soc. Am. J. 1986, 50, 605–608. [Google Scholar] [CrossRef]
- Genç-Fuhrman, H.; Mikkelsen, P.S.; Ledin, A. Simultaneous removal of As, Cd, Cr, Cu, Ni and Zn from stormwater: Experimental comparison of 11 different sorbents. Water Res. 2007, 41, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Zhou, Y.S. Simultaneous removal of coexistent heavy metals from simulated urban stormwater using four sorbents: A porous iron sorbent and its mixtures with zeolite and crystal gravel. J.Hazard. Mater. 2009, 168, 674–680. [Google Scholar] [CrossRef]
- Liu, D.; Sansalone, J.J.; Cartledge, F.K. Adsorption characteristics of oxide coated buoyant media (ρs < 1.0) for storm water treatment. I: Batch equilibria and kinetics. J. Environ. Eng. 2004, 130, 374–382. [Google Scholar]
- Summers, S.R. Standardized Protocol for the Evaluation of GAC; AWWA Research Foundation and American Water Works Association: Denver, CO, USA, 1992. [Google Scholar]
- Crittenden, J.C.; Berrigan, J.K.; Hand, D.W. Design of rapid small-scale adsorption tests for a constant diffusivity. Water Pollut. Control Fed. 1986, 58, 312–319. [Google Scholar]
- Li, Y.; Helmreich, B. Simultaneous removal of organic and inorganic pollutants from synthetic road runoff using a combination of activated carbon and activated lignite. Sep. Purif. Technol. 2014, 122, 6–11. [Google Scholar] [CrossRef]
- Doula, M.; Ioannou, A.; Dimirkou, A. Copper adsorption and Si, Al, Ca, Mg, and Na release from clinoptilolite. J. Colloid Interface Sci. 2002, 245, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Athanasiadis, K.; Helmreich, B. Influence of chemical conditioning on the ion exchange capacity and on kinetic of zinc uptake by clinoptilolite. Water Res. 2005, 39, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.H. Modeling batch kinetics of cadmium removal: By a recycled iron adsorbent. Sep. Sci. Technol. 1998, 33, 149–168. [Google Scholar] [CrossRef]
- Soil Quality—Determination of Effective Cation Exchange Capacity and Base Saturation Level Using Barium Chloride Solution (ISO 11260:1994+Cor. 1:1996), Normenausschuss Wasserwesen (NAW) im DIN; DIN EN ISO 11260:2011-09; DIN Deutsches Institut für Normung e. V.: Berlin, Germany, 2011; pp. 1–16.
- Products Used for Treatment of Water Intended for Human Consumption—Natural Zeolite, Normenausschuss Wasserwesen (NAW) im DIN; DIN EN 16070:2014-06; DIN Deutsches Institut für Normung e. V.: Berlin, Germany, 2014; pp. 1–20.
- American Public Health Association (APHA). Standard Methods for the Examination of Water and Wastewater, 21st ed.; APHA: Washington, DC, USA, 2005; pp. 1–184. [Google Scholar]
- Determination of the Specific Surface Area of Solids by Gas Adsorption—BET Method; Normenausschuss Bauwesen (NABau) im DIN; ISO 9277:2010-09; DIN Deutsches Institut für Normung e. V.: Berlin, Germany, 2014; pp. 1–30.
- Bürgisser, C.S.; Cernik, M.; Borkovec, M.; Sticher, H. Determination of nonlinear adsorption isotherms from column experiments: An alternative to batch studies. Environ.Sci. Technol. 1993, 27, 943–948. [Google Scholar] [CrossRef]
- Quevauviller, P.; Rauret, G.; Muntau, H.; Ure, A.M.; Rubio, R.; López-Sánchez, J.F.; Fiedler, H.D.; Griepink, B. Evaluation of a sequential extraction procedure for the determination of extractable trace metal contents in sediments. Fresenius J. Anal. Chem. 1994, 349, 808–814. [Google Scholar] [CrossRef]
- Reed, B.E.; Jamil, M.; Thomas, B. Effect of pH, empty bed contact time and hydraulic loading rate on lead removal by granular activated carbon columns. Water Environ. Res. 1996, 68, 877–882. [Google Scholar] [CrossRef]
- Pehlivan, E.; Arslan, G. Removal of metal ions using lignite in aqueous solution—low cost biosorbents. Fuel Process. Technol. 2007, 88, 99–106. [Google Scholar] [CrossRef]
- Mohan, D.; Chander, S. Single, binary, and multicomponent sorption of iron and manganese on lignite. J. Colloid Interface Sci. 2006, 299, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Mohan, D.; Chander, S. Removal and recovery of metal ions from acid mine drainage using lignite—a low cost sorbent. J.Hazard. Mater. 2006, 137, 1545–1553. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Hoadley, A.F.A.; Chaffee, A.L.; Garnier, G. Characterisation of lignite as an industrial adsorbent. Fuel 2011, 90, 1567–1574. [Google Scholar] [CrossRef]
- Inglezakis, V.J.; Loizidou, M.D.; Grigoropoulou, H.P. Equilibrium and kinetic ion exchange studies of Pb2+, Cr3+, Fe3+ and Cu2+ on natural clinoptilolite. Water Res. 2002, 36, 2784–2792. [Google Scholar] [CrossRef]
- Jochová, M.; Punčochář, M.; Horáček, J.; Štamberg, K.; Vopálka, D. Removal of heavy metals from water by lignite-based sorbents. Fuel 2004, 83, 1197–1203. [Google Scholar] [CrossRef]
- Yan, G.; Viraraghavan, T.; Chen, M. A new model for heavy metal removal in a biosorption column. Adsorption Sci. Technol. 2001, 19, 25–43. [Google Scholar] [CrossRef]
- Plassard, F.; Winiarski, T.; Petit-Ramel, M. Retention and distribution of three heavy metals in a carbonated soil: Comparison between batch and unsaturated column studies. J. Contam. Hydrol. 2000, 42, 99–111. [Google Scholar] [CrossRef]
- Poddar, M.; Nair, A.N.B.; Mahindrakar, A.B. A review on the use of rapid small scale column test (RSSCT) on predicting adsorption of various contaminants. J. Environ. Sci., Toxicol. Food Technol. 2013, 3, 77–85. [Google Scholar] [CrossRef]
- Boekhold, A.E.; Temminghoff, E.J.M.; Van der Zee, S.E.A.T.M. Influence of electrolyte composition and pH on cadmium sorption by an acid sandy soil. J. Soil Sci. 1993, 44, 85–96. [Google Scholar] [CrossRef]
- Bäckström, M.; Karlsson, S.; Bäckman, L.; Folkeson, L.; Lind, B. Mobilisation of heavy metals by deicing salts in a roadside environment. Water Res. 2004, 38, 720–732. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.; Yonge, D.; Barber, M. Effects of road salts on heavy metal mobility in two eastern Washington soils. J. Environ. Eng. 2009, 135, 505–510. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Filter Material | Surface Area m²/g | TC % | TS % | Al mg/kg | Ca mg/kg | Fe mg/kg | Mn mg/kg | Cu mg/kg | Ni mg/kg | Zn mg/kg |
|---|---|---|---|---|---|---|---|---|---|---|
| Alumina | 263 | - | - | 462000 | 87.6 | 1270 | 7.43 | 18.6 | 1.98 | 9.11 |
| Anthracite | 1.4 | 88.2 | 0.91 | 931 | 1010 | 2260 | 20.5 | 16.0 | 2.57 | 1.56 |
| Calcium carbonate | 3.1 | - | - | 3520 | 318500 | 4370 | 78.7 | 3.50 | 2.91 | 5.04 |
| GAC | 896 | 91.6 | 0.83 | 6300 | 3050 | 3260 | 61.8 | 458 | 28.4 | 7.92 |
| GFH | 224 | - | - | 753 | 56300 | 431000 | 9090 | 2.00 | 41.3 | 87.6 |
| Lignite | 262 | 89.2 | 0.46 | 1090 | 18800 | 2330 | 83.1 | 0.17 | 1.69 | 0.37 |
| Filter Material | Cu | Ni | Zn | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| qe,2.5 | qe,540 | qm | q10% | q50% | qe,2.5 | qe,540 | qm | q10% | q50% | qe,2.5 | qe,540 | qm | q10% | q50% | |
| Alumina | 0.06 | 12.7 | 13.5 | 1.46 | 3.07 | 0.06 | 1.75 | 1.77 | 0.25 | 0.71 | 0.12 | 6.45 | 6.47 | 1.35 | 2.01 |
| Anthracite | 0 | 0.09 | - | - | - | 0 | 0.06 | - | - | - | 0 | 0.36 | - | - | - |
| Calcium carbonate | 0.06 | 1.45 | 1.45 | 1.60 | >2.27 | 0.06 | 0.30 | 0.31 | 0.13 | 0.42 | 0.12 | 0.53 | 0.53 | 1.06 | 2.25 |
| GAC | 0.06 | 11.9 | 12.0 | 1.40 | 1.81 | 0.06 | 1.51 | 1.52 | 0.28 | 0.41 | 0.12 | 4.11 | 4.44 | 0.71 | 1.22 |
| GFH | 0.06 | 14.4 | 21.8 | >8.92 | >8.92 | 0.06 | 6.73 | 6.82 | 4.90 | >7.57 | 0.12 | 24.0 | 23.5 | 11.1 | >11.3 |
| Lignite | 0.06 | 14.6 | 16.6 | 1.27 | 2.00 | 0.06 | 2.73 | 2.73 | 0.44 | 0.60 | 0.12 | 10.9 | 11.2 | 1.26 | 1.62 |
| Filter Material | Four Sorption Capacities | CEC 1 | CEC 2 | |||
|---|---|---|---|---|---|---|
| 120 mg/L Cu | 120 mg/L Ni | 240 mg/L Zn | 480 mg/L ∑(Cu+Ni+Zn) | 480 mg/L Mg | 180 mg/L NH4+ | |
| Alumina | 8.42 | 5.93 | 15.0 | 29.4 | 28.5 | 7.33 |
| Anthracite | 0 | 0 | 0 | 0 | 0 | 5.37 |
| Calcium carbonate | 3.00 | 1.34 | 0 | 4.34 | 7.94 | 3.45 |
| GAC | 8.38 | 3.39 | 12.4 | 24.2 | 3.99 | 3.63 |
| GFH | 8.44 | 8.96 | 17.0 | 34.4 | 40.2 | 10.7 |
| Lignite | 8.44 | 8.93 | 17.0 | 34.4 | 9.39 | 2.98 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huber, M.; Badenberg, S.C.; Wulff, M.; Drewes, J.E.; Helmreich, B. Evaluation of Factors Influencing Lab-Scale Studies to Determine Heavy Metal Removal by Six Sorbents for Stormwater Treatment. Water 2016, 8, 62. https://doi.org/10.3390/w8020062
Huber M, Badenberg SC, Wulff M, Drewes JE, Helmreich B. Evaluation of Factors Influencing Lab-Scale Studies to Determine Heavy Metal Removal by Six Sorbents for Stormwater Treatment. Water. 2016; 8(2):62. https://doi.org/10.3390/w8020062
Chicago/Turabian StyleHuber, Maximilian, Sophia C. Badenberg, Moritz Wulff, Jörg E. Drewes, and Brigitte Helmreich. 2016. "Evaluation of Factors Influencing Lab-Scale Studies to Determine Heavy Metal Removal by Six Sorbents for Stormwater Treatment" Water 8, no. 2: 62. https://doi.org/10.3390/w8020062
APA StyleHuber, M., Badenberg, S. C., Wulff, M., Drewes, J. E., & Helmreich, B. (2016). Evaluation of Factors Influencing Lab-Scale Studies to Determine Heavy Metal Removal by Six Sorbents for Stormwater Treatment. Water, 8(2), 62. https://doi.org/10.3390/w8020062