
Sorption Dynamics of Uranium onto Anion Exchangers

Abstract
:1. Introduction
2. Materials and Methods
2.1. Exchangers
2.2. Experimental Methods
2.2.1. Column Experiments
2.2.2. Regeneration
2.3. Modelling
3. Theoretical Description of Sorption Dynamics
- No loading on the initial adsorbent:
- Symmetry of the loading at the centre of the particle:
- Film mass flow is equal to the mass flow at the exterior of the particle:
- Increase of the mean solid phase loading is given by the mass flow through the film:
- Initial and boundary conditions for the concentration for the filter mass balance:
4. Results
4.1. Filter Dynamics
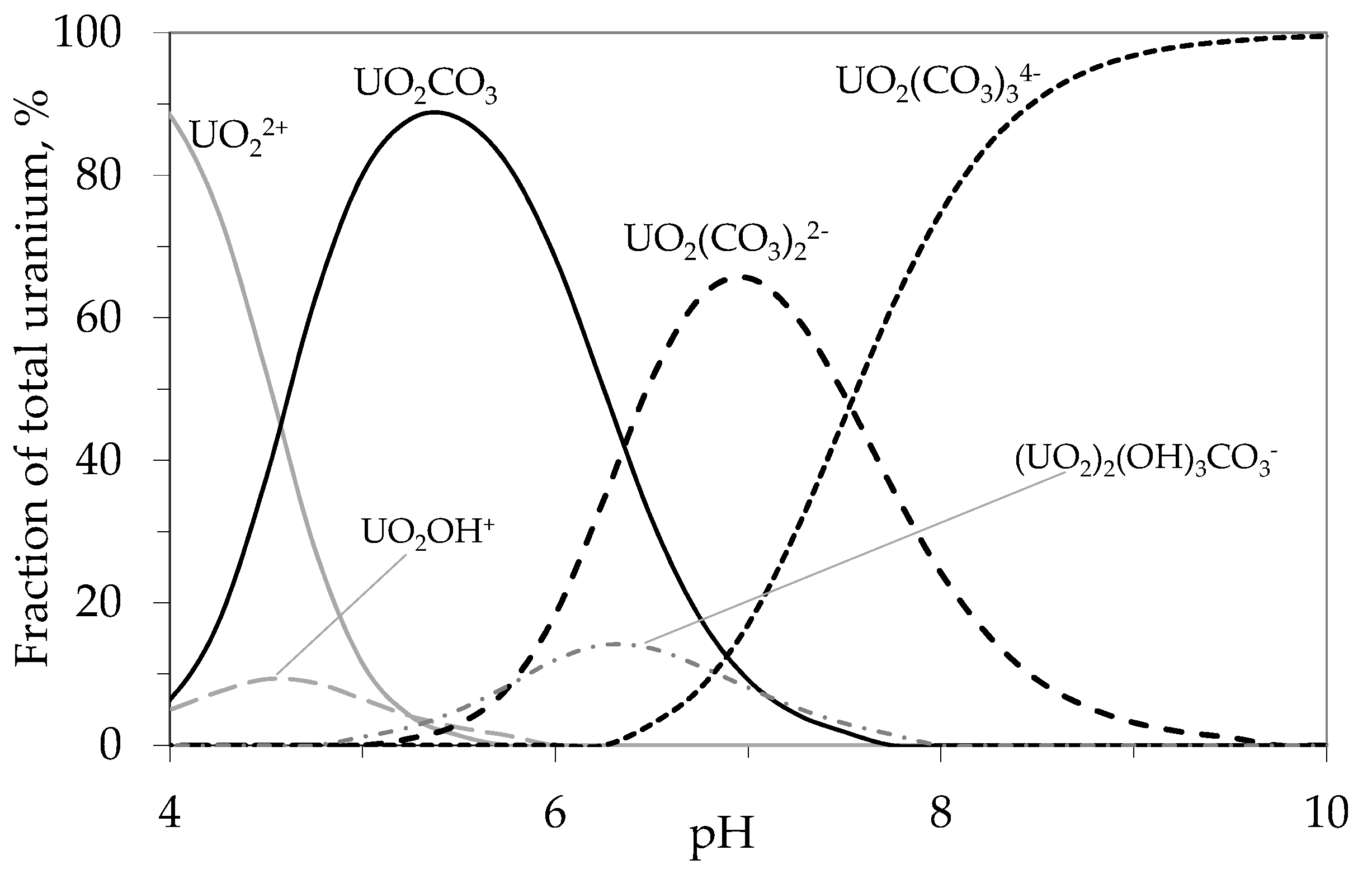
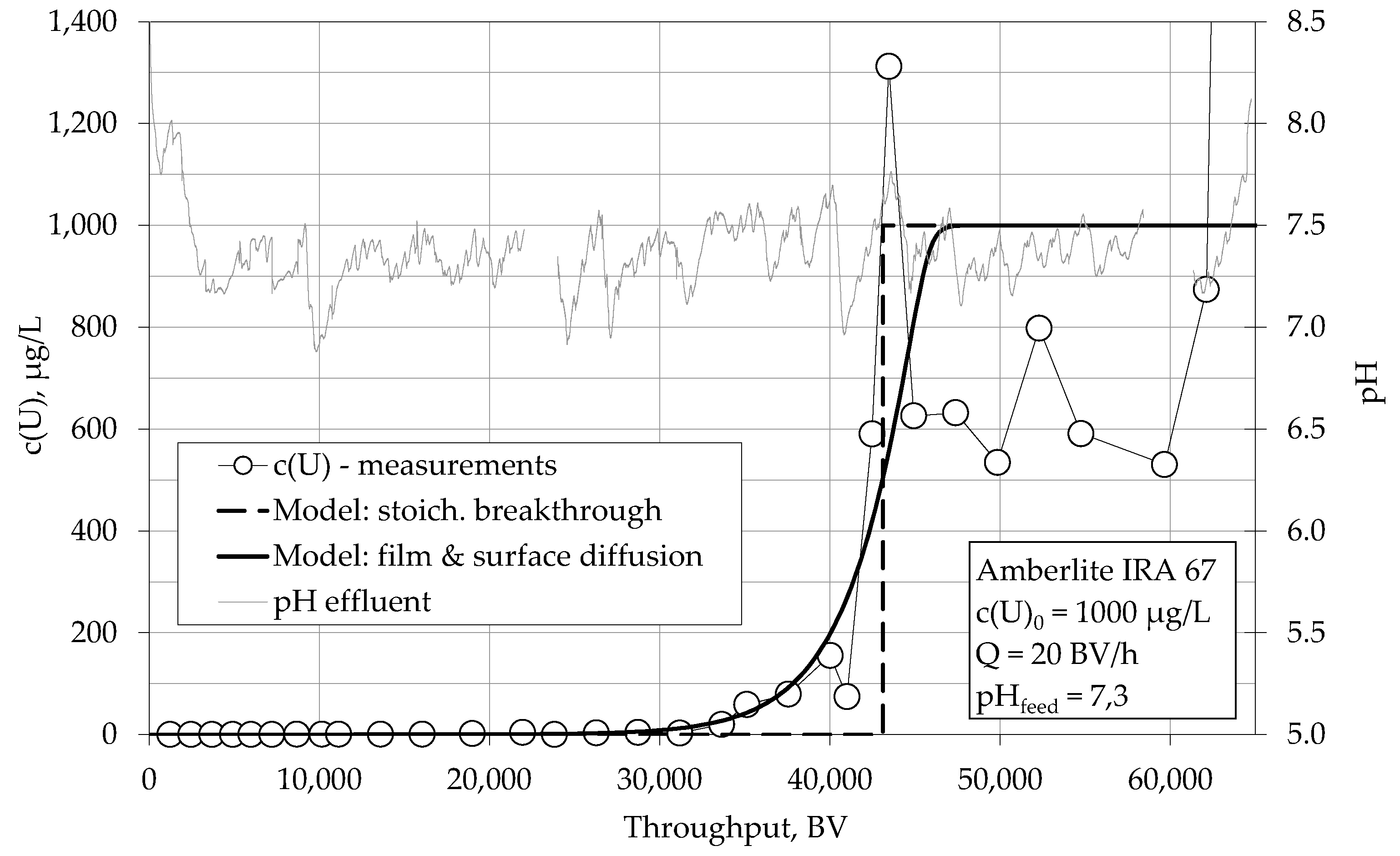
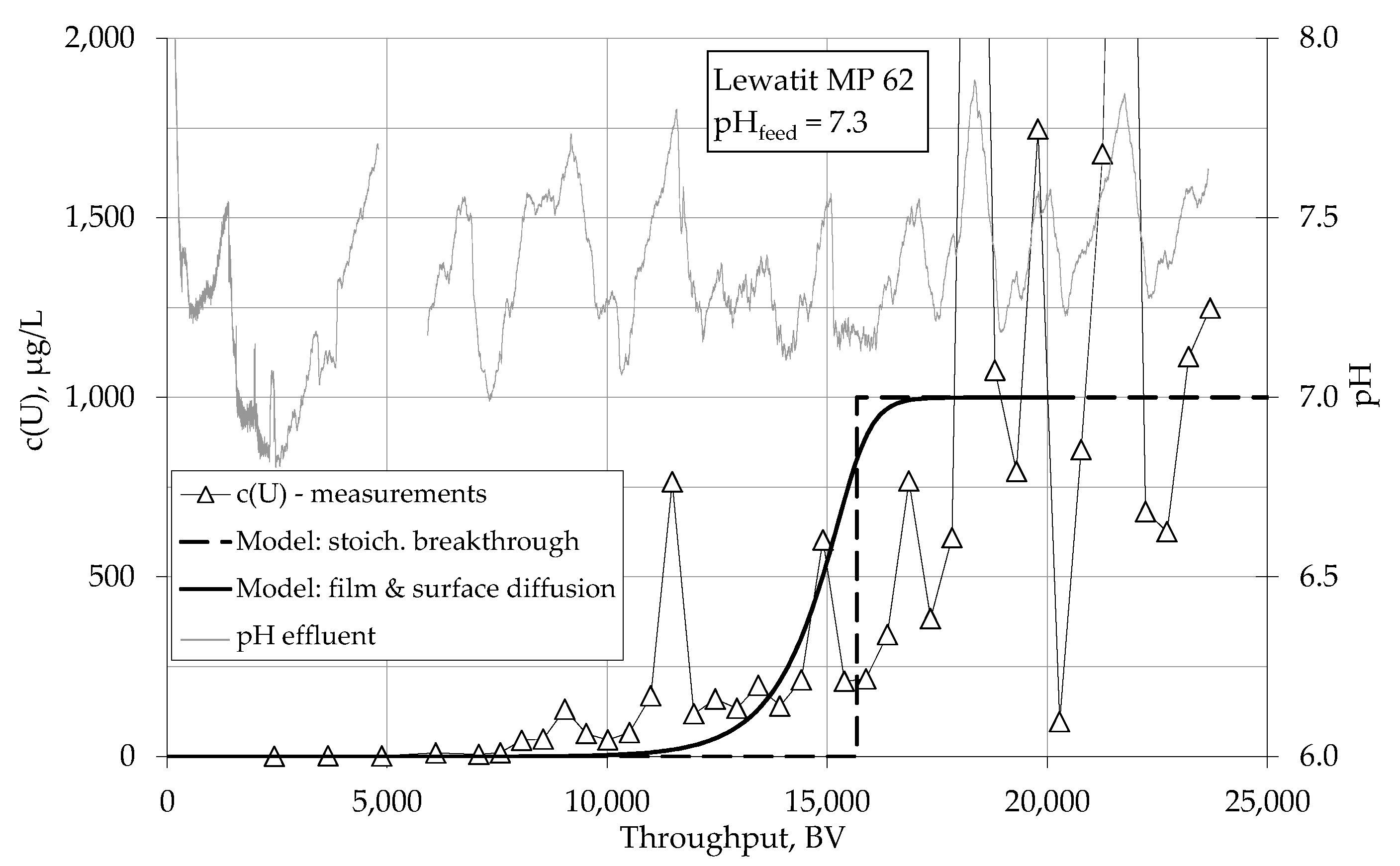
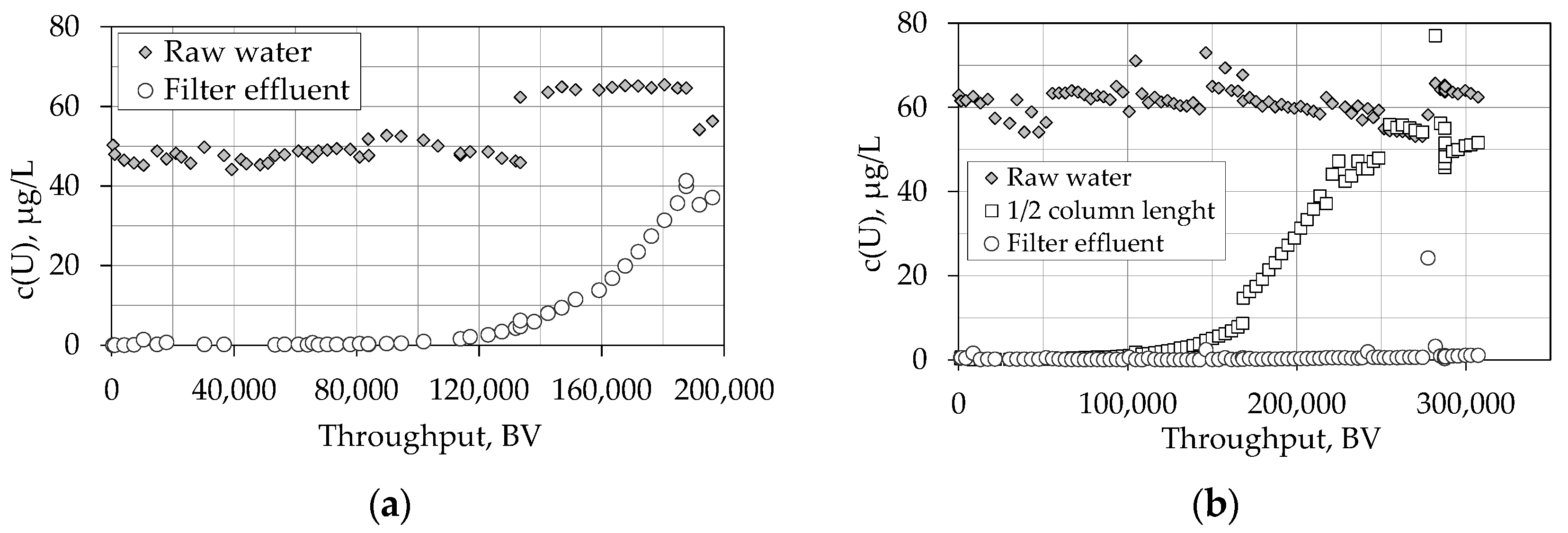
4.1.1. Breakthrough Behaviour of Uranium Species in Bench Scale Experiments
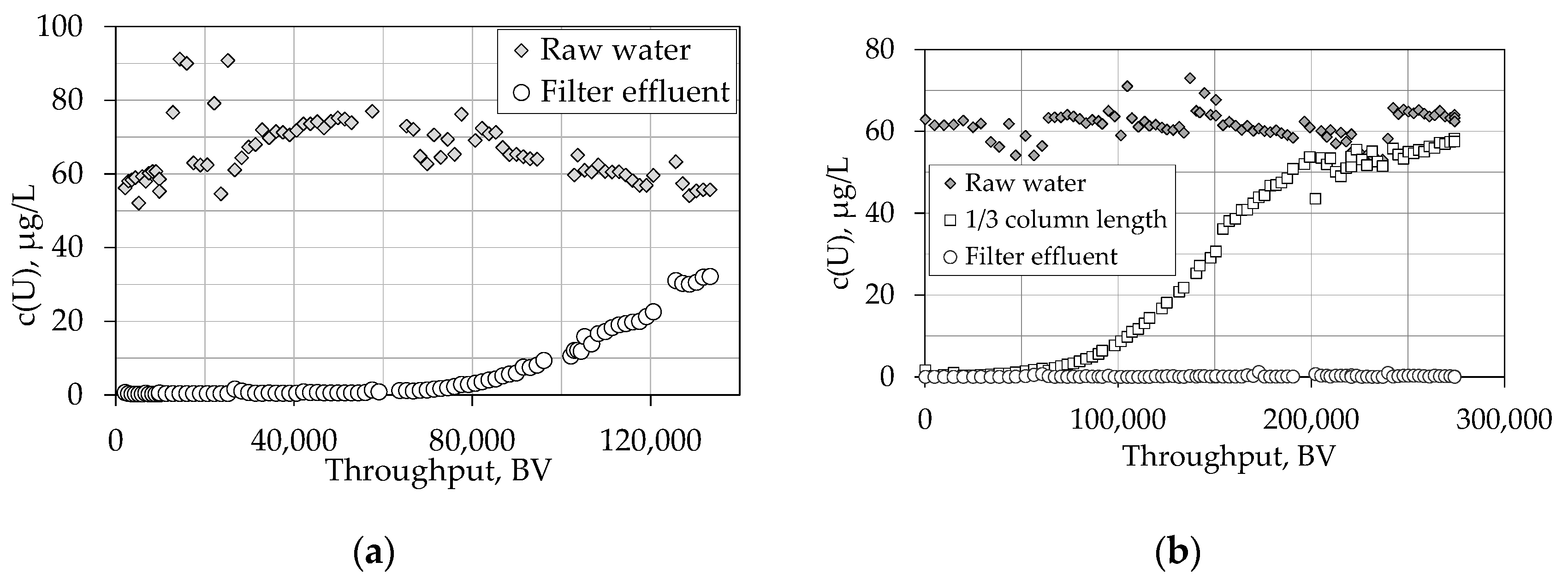
4.1.2. Breakthrough Behaviour of Uranium Species in Pilot Scale Experiments
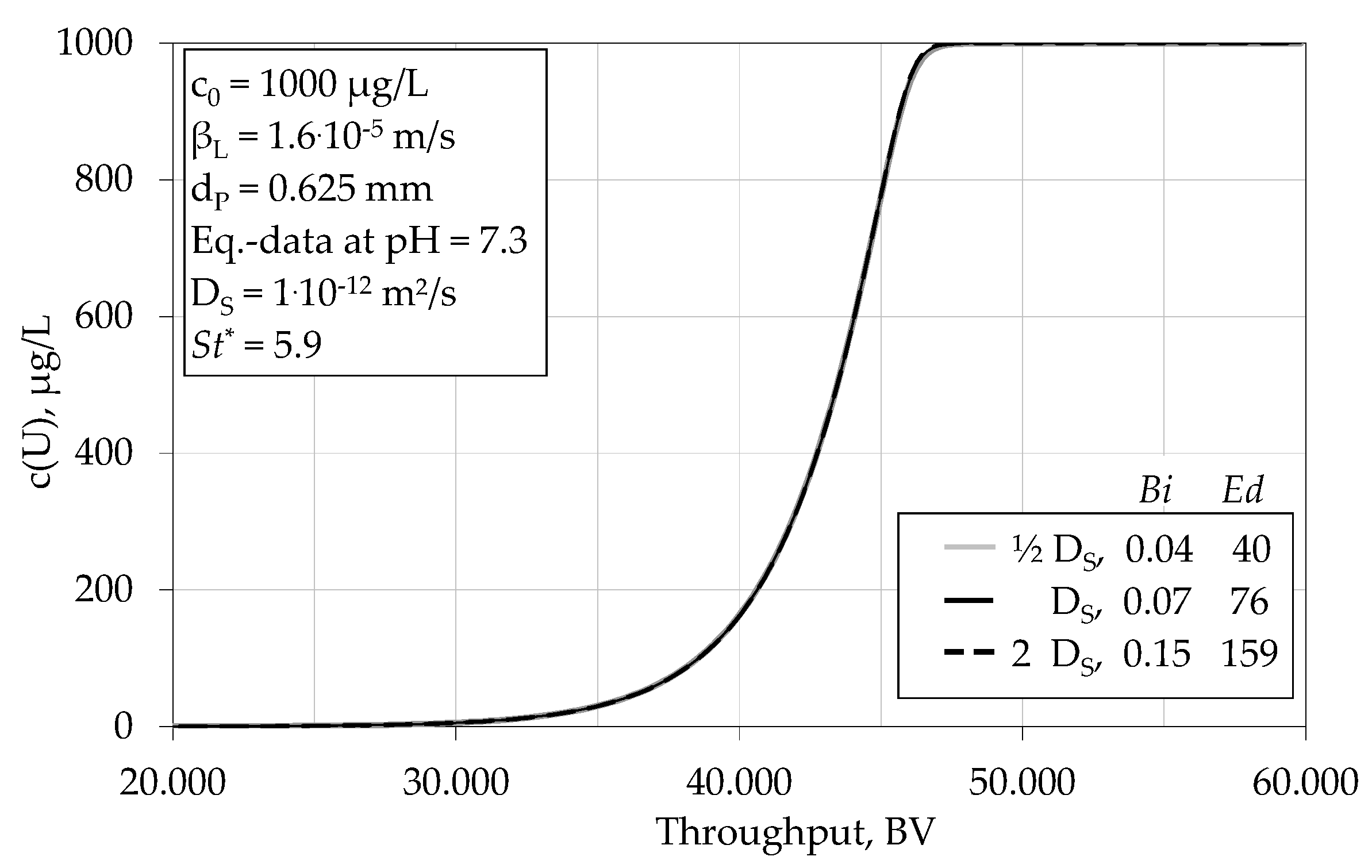
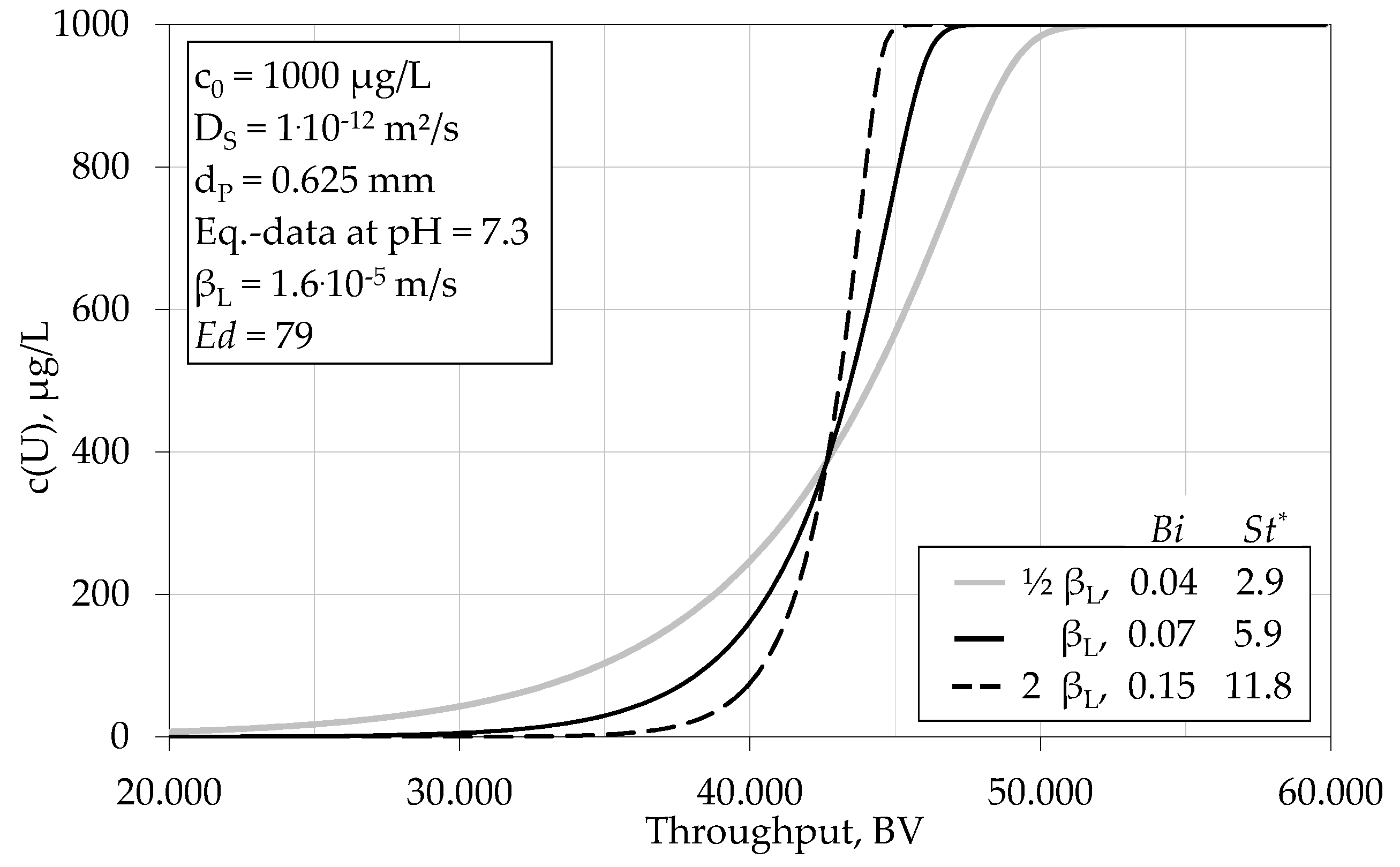
4.1.3. Impact of Various Parameters on the Breakthrough Behaviour
4.1.4. Theoretical Scale-Up and Consequences for Waterworks
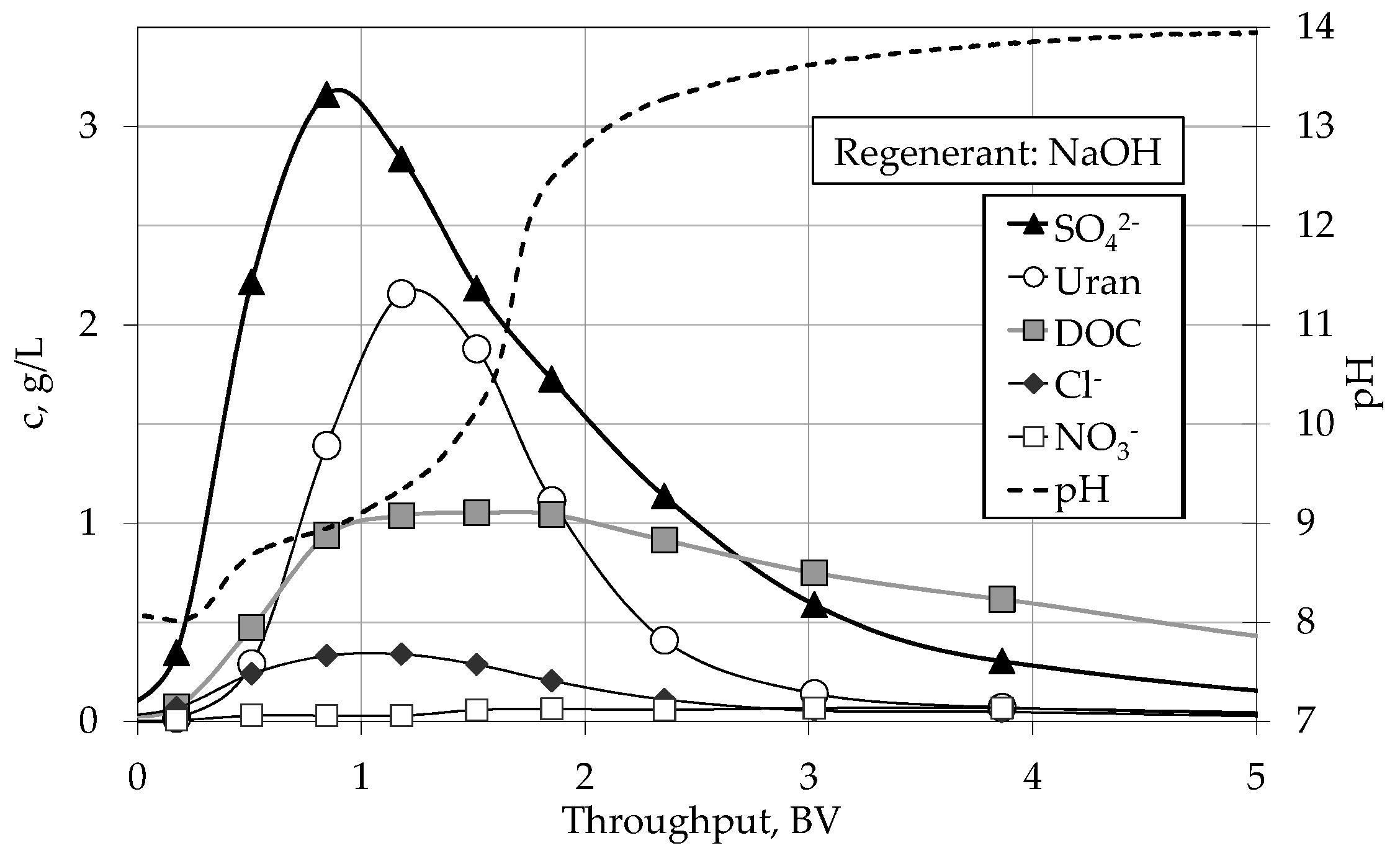
4.2. Regeneration
4.2.1. Batch Experiments
4.2.2. Column Experiments
5. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nolan, J.; Weber, K.A. Natural uranium contamination in major U.S. aquifers linked to nitrate. Environ. Sci. Technol. Lett. 2015, 2, 215–220. [Google Scholar] [CrossRef]
- Gómez, P.; Garralón, A.; Buil, B.; Turrero, M.J.; Sánchez, L.; de la Cruz, B. Modeling of geochemical processes related to uranium mobilization in the groundwater of a uranium mine. Sci. Total Environ. 2006, 366, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Birke, M.; Rauch, U.; Lorenz, H. Uranium in stream and mineral water of the Federal Republic of Germany. Environ. Geochem. Health 2009, 31, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Liesch, T.; Hinrichsen, S.; Goldscheider, N. Uranium in groundwater—Fertilizers versus geogenic sources. Sci. Total Environ. 2015, 536, 981–995. [Google Scholar] [CrossRef] [PubMed]
- United States Environmental Protection Agency (US-EPA). National Primary Drinking Water Regulations; Radionuclides; Environmental Protection Agency: Washington, DC, USA, 2000.
- World Health Organization (WHO). Guidelines for Drinking-Water Quality, 1st Addendum to 3rd ed.; World Health Organization: Geneva, Switzerland, 2006. [Google Scholar]
- Verordnung über die Qualität von Wasser für den Menschlichen Gebrauch (Trinkwasserverordnung—TrinkwV) (Ordinance on the Quality of Water Intended for Human Consumption) vom 21. Mai 2001. Neufassung vom 10. März 2016. Available online: http://www.gesetze-im-internet.de/bundesrecht/trinkwv_2001/gesamt.pdf (accessed on 24 November 2016).
- Hanson, S.W.; Wilson, D.B.; Gunaji, N.N. Removal of Uranium from Drinking Water by Ion Exchange and Chemical Clarification; U.S. Environmental Protection Agency: Washington, DC, USA, 1987.
- Wazne, M.; Korfiatis, G.P.; Meng, X. Carbonate effects on hexavalent uranium adsorption by iron oxyhydroxide. Environ. Sci. Technol. 2003, 37, 3619–3624. [Google Scholar] [CrossRef] [PubMed]
- Katsoyiannis, I.A. Carbonate effects and pH-dependence of uranium sorption onto bacteriogenic iron oxides: Kinetic and equilibrium studies. J. Hazard. Mater. 2007, 139, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Kalmykov, S.N.; Choppin, G.R. Mixed Ca2+/UO22+/CO32− complex formation at different ionic strengths. Radiochim. Acta 2000, 88, 603–606. [Google Scholar] [CrossRef]
- Dong, W.; Brooks, S.C. Determination of the formation constants of ternary complexes of uranyl and carbonate with alkaline earth metals (Mg2+, Ca2+, Sr2+, and Ba2+) using anion exchange method. Environ. Sci. Technol. 2006, 40, 4689–4695. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Brooks, S.C. Formation of aqueous MgUO2(CO3)32− complex and uranium anion exchange mechanism onto an exchange resin. Environ. Sci. Technol. 2008, 42, 1979–1983. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.D.; Kemner, K.M.; Brooks, S.C.; Fredrickson, J.K.; Carroll, S.L.; Kennedy, D.W.; Zachara, J.M.; Plymale, A.E.; Fendorf, S. Ca-UO2-CO3 complexation—Implications for bioremediation of U(VI). Phys. Scr. 2005, T115, 915–917. [Google Scholar] [CrossRef]
- Kelly, S.D.; Kemner, K.M.; Brooks, S.C. X-ray absorption spectroscopy identifies calcium-uranyl-carbonate complexes at environmental concentrations. Geochim. Cosmochim. Acta 2007, 71, 821–834. [Google Scholar] [CrossRef]
- Riegel, M.; Höll, W.H. Removal of natural uranium from groundwater by means of weakly basic anion exchanges. In Proceedings of the IEX 2008, Cambridge, UK, 9–11 July 2008; Cox, M., Ed.; Society of Chemical Industry: London, UK, 2008; pp. 331–338. [Google Scholar]
- Riegel, M. Untersuchungen zur Elimination von Natürlichen Uranspezies aus Wässern mit Hilfe Schwach Basischer Anionenaustauscher (Investigation of Elimination of Natural Occurring Uranium Species from Water by Means of Weak Basic Anion Exchangers). Ph.D. Thesis, Technical University of Karlsruhe, Karlsruhe, Germany, 2009. [Google Scholar]
- Langmuir, D. Uranium solution-mineral equilibria at low temperatures with applications to sedimentary ore deposits, Geochim. Cosmochim. Acta 1978, 42, 547–569. [Google Scholar] [CrossRef]
- Fox, P.M.; Davis, J.A.; Zachara, J.M. The effect of calcium on aqueous uranium(VI) speciation and adsorption to ferrihydrite and quartz. Geochim. Cosmochim. Acta 2006, 70, 1379–1387. [Google Scholar] [CrossRef]
- Jekel, M.; Bahr, C.; Riegel, M.; Schlitt, V.; Baldauf, G.; Höll, W.H. Uranentfernung in der Trinkwasseraufbereitung (Elimination of uranium during drinking water treatment). Energ. Wasser Prax. 2010, 6, 54–59. [Google Scholar]
- Umweltbundesamt (Federal Environmental Agency). Liste der Aufbereitungsstoffe und Desinfektionsverfahren gemäß § 11 Trinkwasserverordnung 2001 (List of Preparation Substances and Disinfection Processes in Compliance with § 11 German Drinking Water Ordinance). 12. Änderung, Stand Dezember 2009. Available online: https://www.umweltbundesamt.de/sites/default/files/medien/481/dokumente/17_aenderung_aufbereitungsstoffe_desinfektionsverfahren_11_trinkwv_11_2012.pdf (accessed on 24 November 2016).
- Water Quality–Application of Inductively Coupled Plasma Mass Spectrometry (ICP-MS)—Part 2: Determination of 62 Elements (ISO 17294-2:2003); Beuth: Berlin, Germany, 2003.
- Agarwal, R.P. Difference Equation and Inequalities; Marcel Dekker: New York, NY, USA, 2000. [Google Scholar]
- Bjoerck, A.; Dahlquist, G. Numerical Mathematics and Scientific Computation; Siam: Philadelphia, PA, USA, 1999. [Google Scholar]
- Sontheimer, H.; Crittenden, J.C.; Summers, R.S. Activated Carbon for Water Treatment; DVGW-Forschungsstelle: Karlsruhe, Germany, 1988. [Google Scholar]
- Crittenden, J.C.; Weber, W.J. Predictive model for design of fixed-bed adsorbers: Single-component model verification. J. Environ. Eng. Div. 1978, 104, 433–443. [Google Scholar]
- Crittenden, J.C.; Wong, B.W.C.; Thacker, W.E.; Snoeyink, V.L.; Hinrichs, R.L. Mathematical model of sequential loading in fixed-bed adsorbers. J. Water Pollut. Control Fed. 1980, 52, 2780–2795. [Google Scholar]
- Sperlich, A.; Schimmelpfennig, S.; Baumgarten, B.; Genz, A.; Amy, G.; Worch, E.; Jekel, M. Predicting anion breakthrough in granular ferric hydroxide (GFH) adsorption filters. Water Res. 2008, 42, 2073–2082. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Crittenden, J.; Hristovski, K.; Hand, D.; Westerhoff, P. User-oriented batch reactor solutions to the homogeneous surface diffusion model for different activated carbon dosages. Water Res. 2009, 43, 1859–1866. [Google Scholar] [CrossRef] [PubMed]
- Riegel, M.; Tokmachev, M.; Höll, W.H. Kinetics of uranium sorption onto weakly basic anion exchangers. React. Funct. Polym. 2008, 68, 1072–1080. [Google Scholar] [CrossRef]
- Grossmann, J.J.; Adamson, A.W. The diffusion process for organolite exchangers. J. Phys. Chem. 1952, 56, 97–100. [Google Scholar] [CrossRef]
- Helfferich, F. Kinetik des Ionenaustauschs. Angew. Chem. 1956, 68, 693–698. [Google Scholar] [CrossRef]
- Schlögl, R.; Helfferich, F. Comment on the significance of diffusion potentials in ion exchange kinetics. J. Chem. Phys. 1957, 26, 5–7. [Google Scholar] [CrossRef]
- Helfferich, F.; Plesset, M.S. Ion exchange kinetics. A nonlinear diffusion problem. J. Chem. Phys. 1958, 28, 418–424. [Google Scholar] [CrossRef]
- Gnielinski, V. Gleichungen zur Berechnung des Wärme- und Stoffaustausches in durchströmten Kugelschüttungen bei mittleren und großen Peclet-Zahlen. Verfahrenstechnik 1978, 12, 363–366. [Google Scholar]
- Worch, E. Eine neue Gleichung zur Berechnung von Diffusionskoeffizienten gelöster Stoffe. Vom Wasser 1993, 81, 289–297. [Google Scholar]
- Wilson, E.J.; Geankoplis, C.J. Liquid mass transfer at very low Reynolds numbers in packed beds. Ind. Eng. Chem. Fundam. 1966, 5, 9–14. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amberlite IRA 67 | Lewatit MP 62 | Lewatit S 6368 | ||
|---|---|---|---|---|
| WBA | WBA | SBA | ||
| Matrix 1 | Acrylic acid divinylbenzene copolymer | Styrene divinylbenzene copolymer | Styrene divinylbenzene copolymer | |
| Functional groups 1 | Tertiary and secondary amine | Tertiary amine | Quaternary amine | |
| Effective size dP 1 | mm | 0.47 | 0.5–0.75 | 0.62 |
| Particle density ρP 2 | g/mL | 1.06 | 1.03 | - |
| Bulk density ρF 2 | g/mL | 0.68 | 0.65 | - |
| Bulk porosity ε 2 | - | 0.36 | 0.37 | - |
| Waterworks I | Waterworks II | ||
|---|---|---|---|
| pH | - | 6.9 | 7.4 |
| Ca2+ | mg/L | 79 | 48 |
| SO42− | mg/L | 92 | 38 |
| HCO3− | mg/L | 350 | 221 |
| Cl− | mg/L | 65 | 19 |
| DOC | mg/L | 1.8 | 0.6 |
| Bench Scale | Full Scale | ||
|---|---|---|---|
| Filter geometry | |||
| Filter diameter | dF | 2 cm | 1 m |
| Filter height | hF | 8.5 cm | 1.5 m |
| Filter volume | VF | 27 mL | 1.2 m3 |
| Ion exchanger | |||
| Resin amount | m | 18 g | 800 kg |
| Kinetics | |||
| Solid diffusion coefficient | DS | 10–12 m2/s | 10–12 m2/s |
| Liquid mass transfer coefficient | βL | 1.5 10–5 m/s | 5 10–5 m/s |
| Raw water | |||
| Initial concentration | c0 | 1000 µg/L | 60 µg/L |
| Loading in equilibrium with c0 | q0 | 267 µmol/L | 42 µmol/L |
| Filter conditions | |||
| Flux | Q | 20 BV/h | 20 BV/h |
| Q | 0.5 L/h | 24 m3/h | |
| Filter velocity | VF | 1.7 m/h | 30 m/h |
| Stoichiometric throughput | Vstoich | 43,000 BV | 113,000 BV |
| Time of stoichiometric breakthrough | tstoich | 90 d | 235 d |
| Surface diffusion modulus | Ed | 86 | 226 |
| Modified Stanton number | St* | 6 | 19 |
| Biot number | Bi | 0.07 | 0.08 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riegel, M.; Schlitt, V. Sorption Dynamics of Uranium onto Anion Exchangers. Water 2017, 9, 268. https://doi.org/10.3390/w9040268
Riegel M, Schlitt V. Sorption Dynamics of Uranium onto Anion Exchangers. Water. 2017; 9(4):268. https://doi.org/10.3390/w9040268
Chicago/Turabian StyleRiegel, Marcel, and Volker Schlitt. 2017. "Sorption Dynamics of Uranium onto Anion Exchangers" Water 9, no. 4: 268. https://doi.org/10.3390/w9040268
APA StyleRiegel, M., & Schlitt, V. (2017). Sorption Dynamics of Uranium onto Anion Exchangers. Water, 9(4), 268. https://doi.org/10.3390/w9040268