
Effect of Sewage and Industrial Effluents on Bacterial and Archaeal Communities of Creek Sediments in the Taihu Basin

Abstract
:1. Introduction
2. Materials and Methods
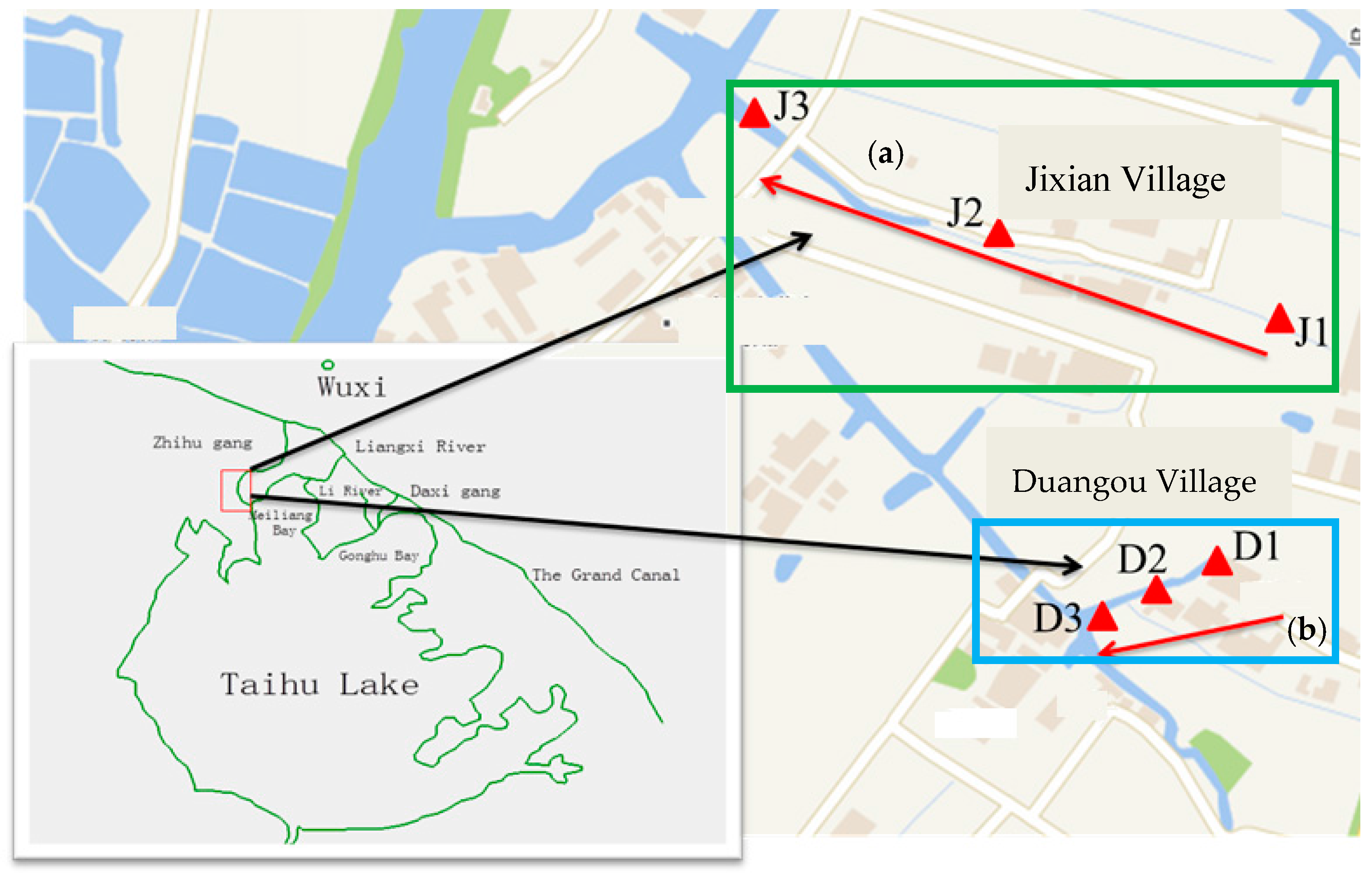
2.1. Study Area
2.2. Sample Collection and Measurement
2.3. DNA Extraction and Illumina MiSeq Sequencing
2.4. Sequencing Data Processing and Statistical Analysis
3. Results
3.1. Water Quality and Sediment Heavy Metal
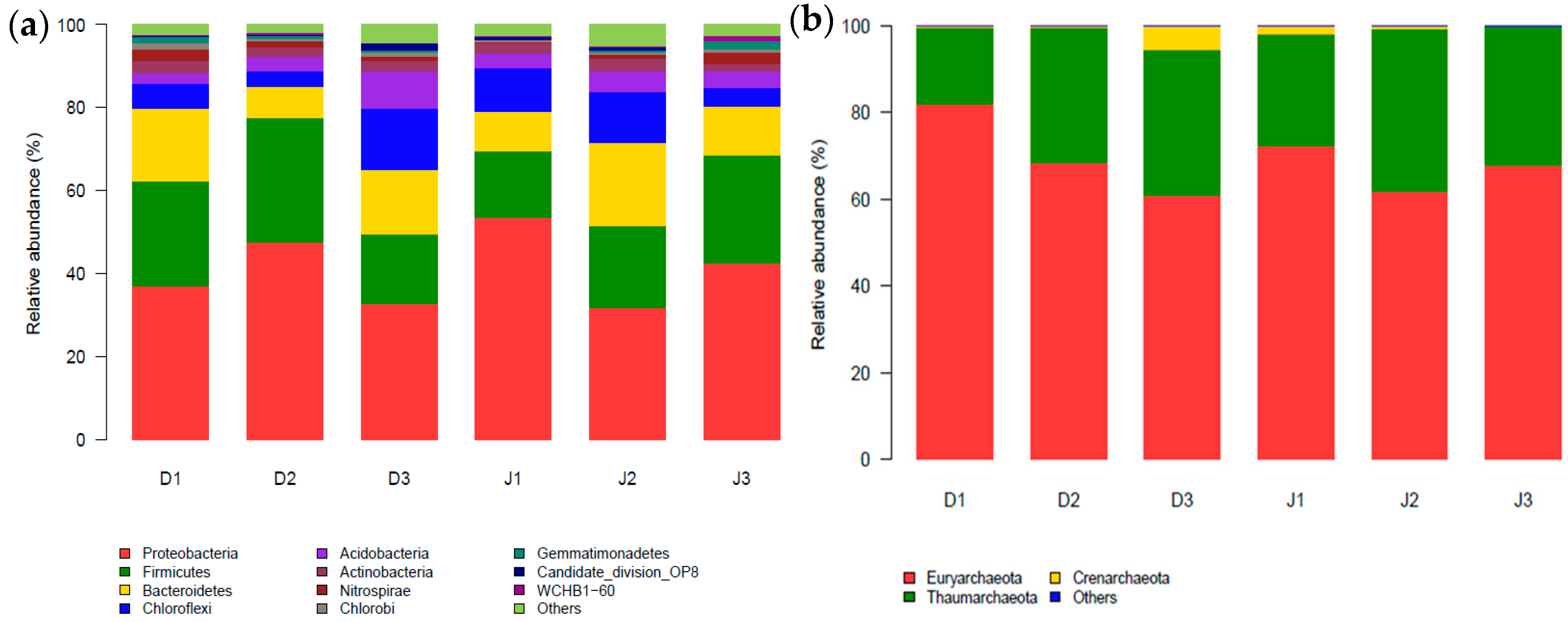
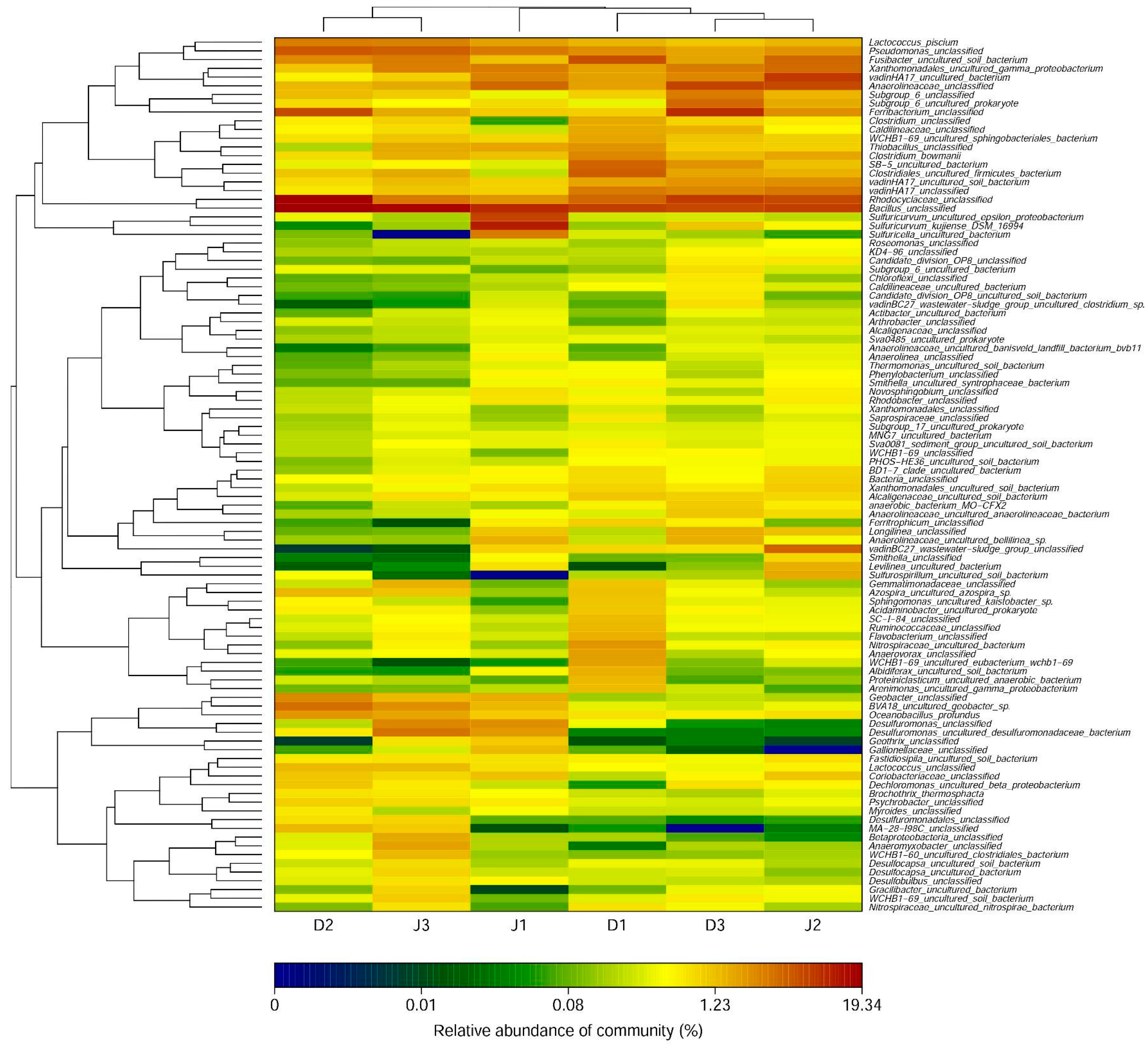
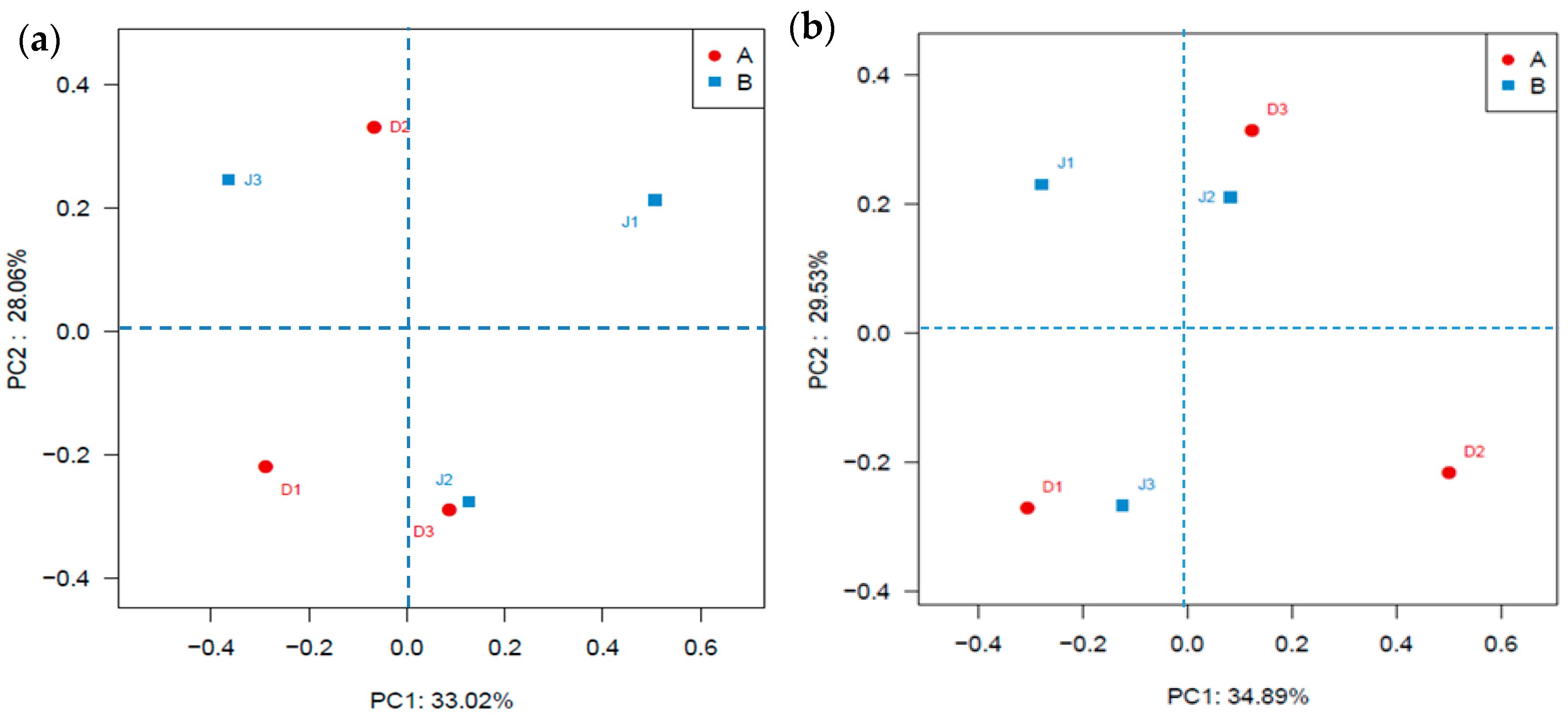
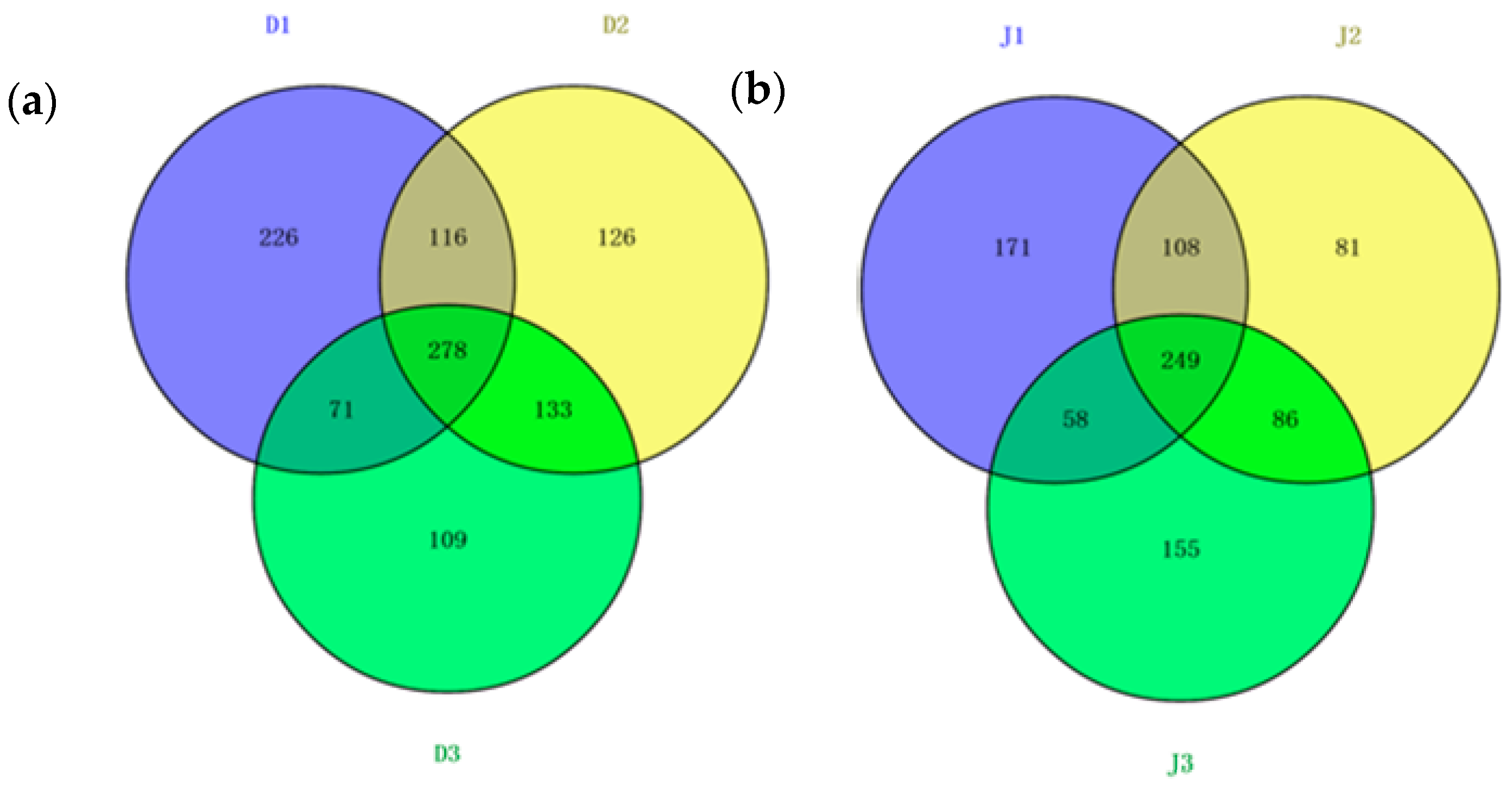
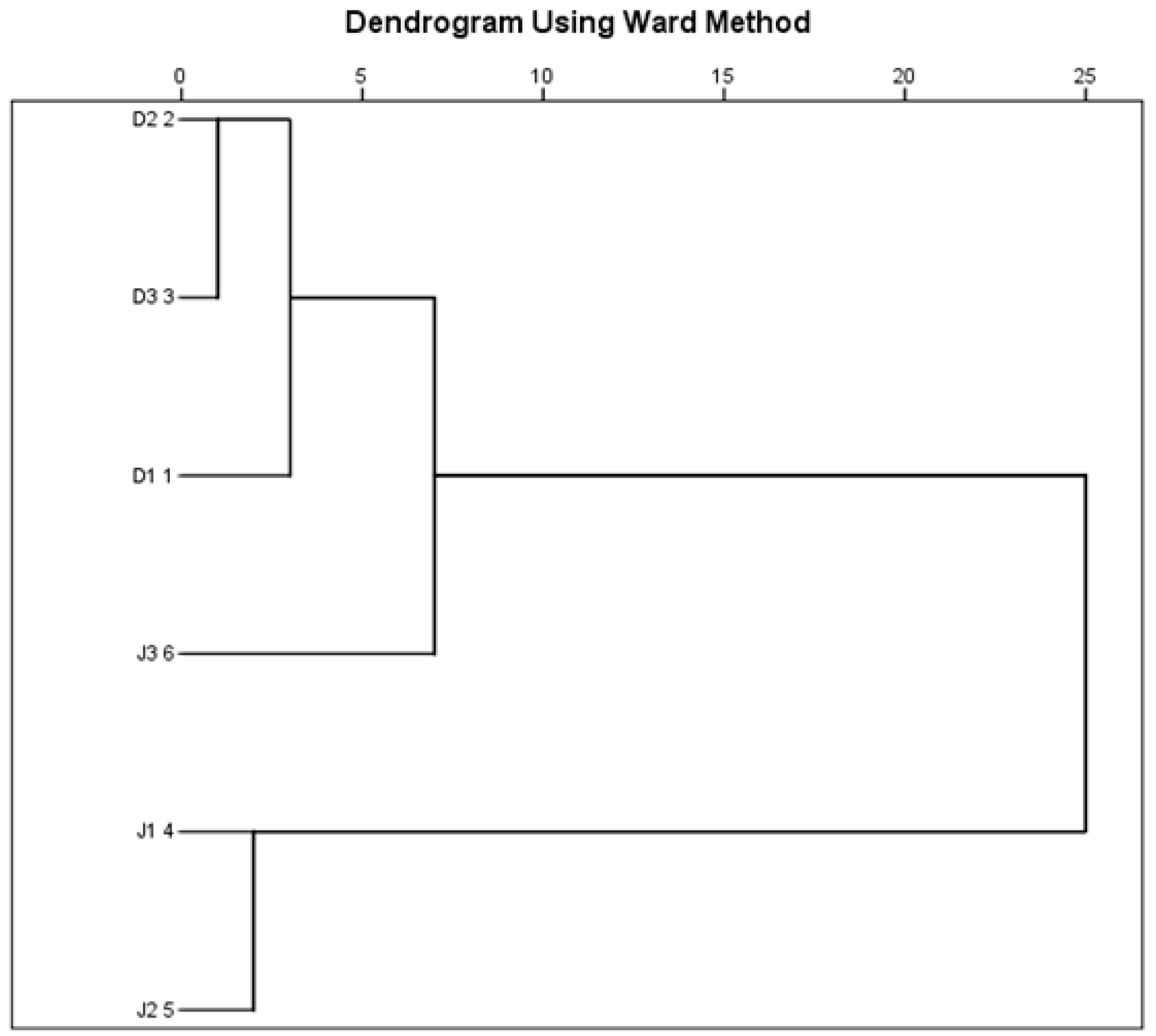
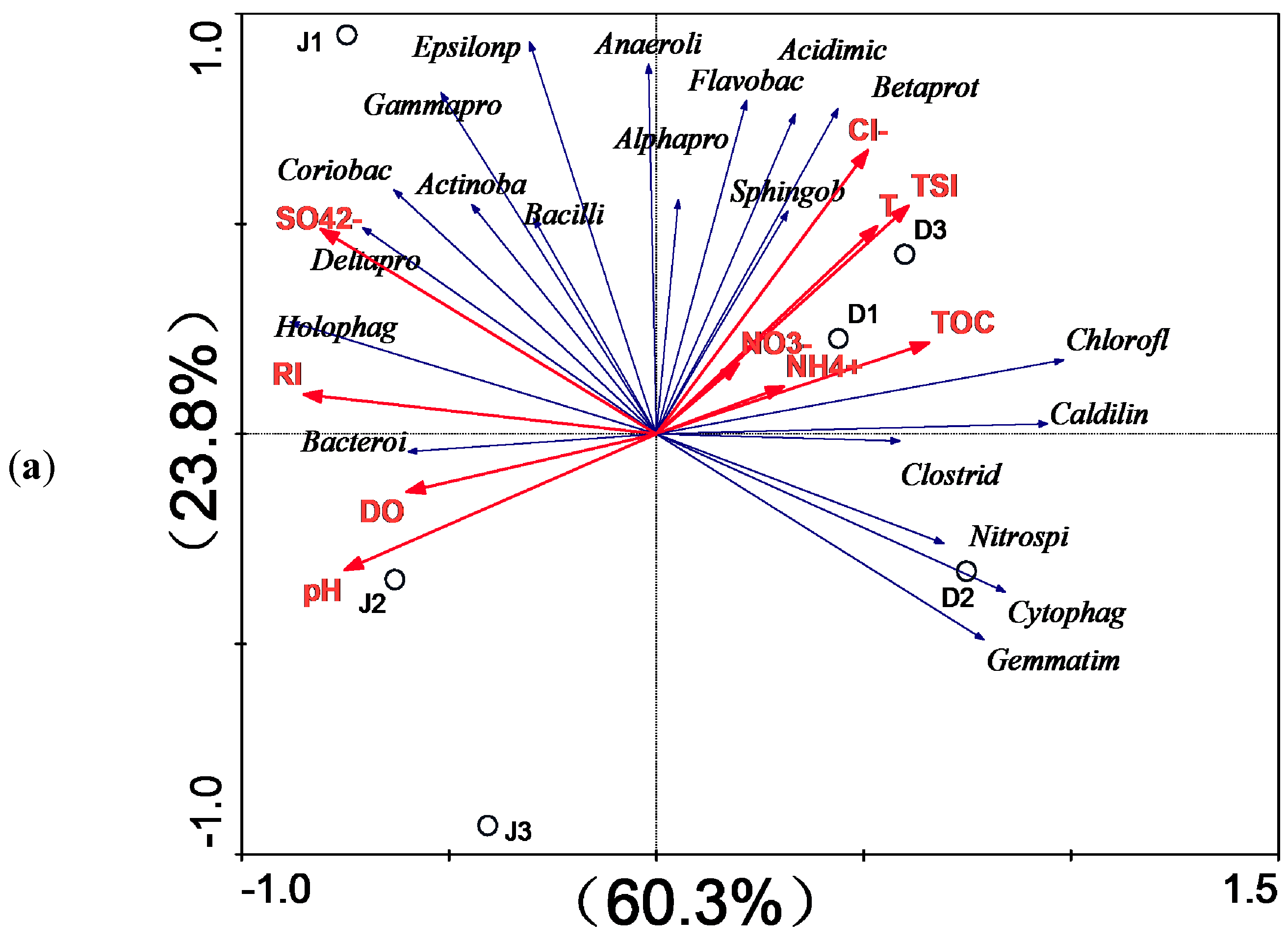
3.2. Bacterial Diversity of the Creeks
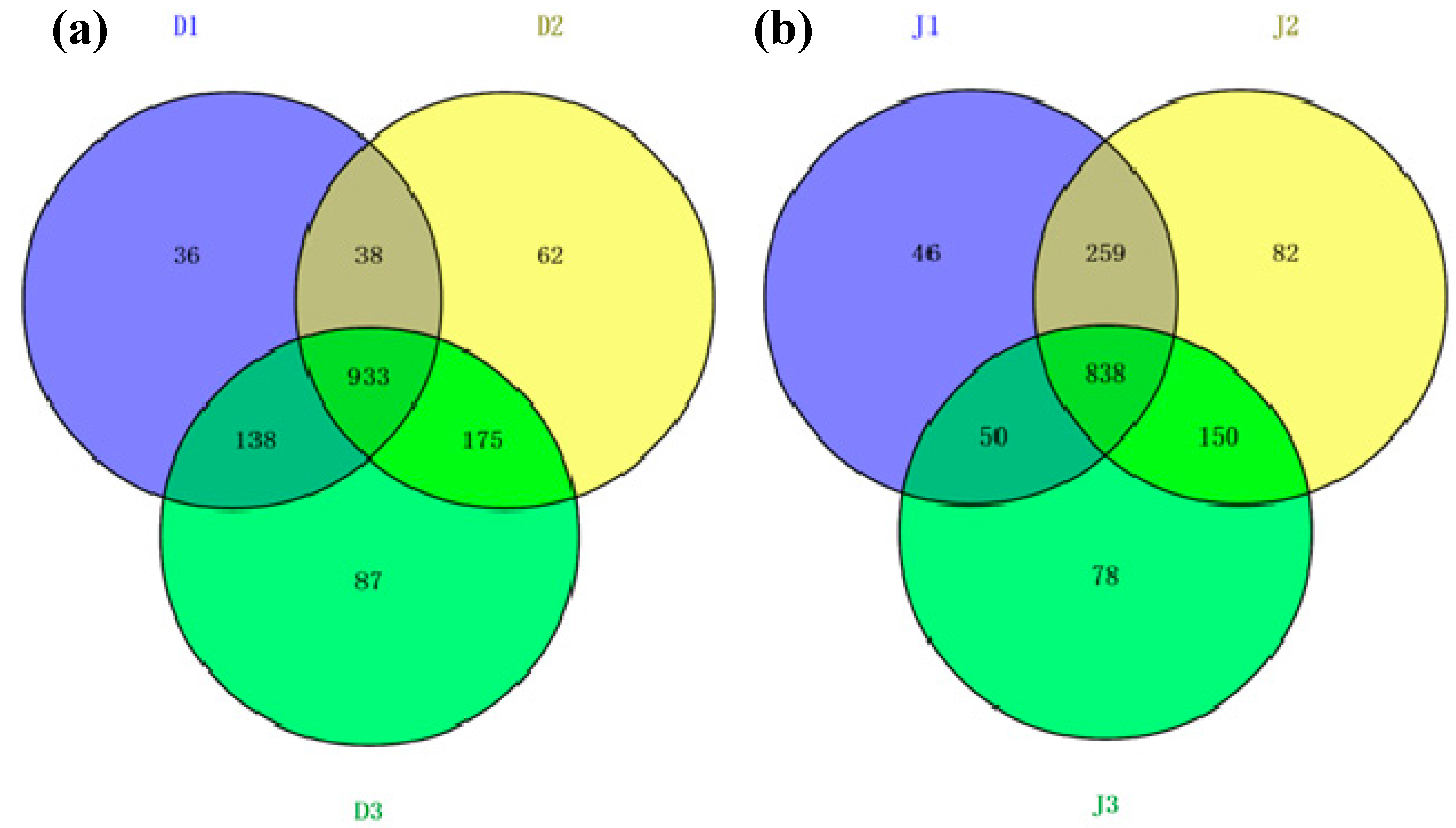
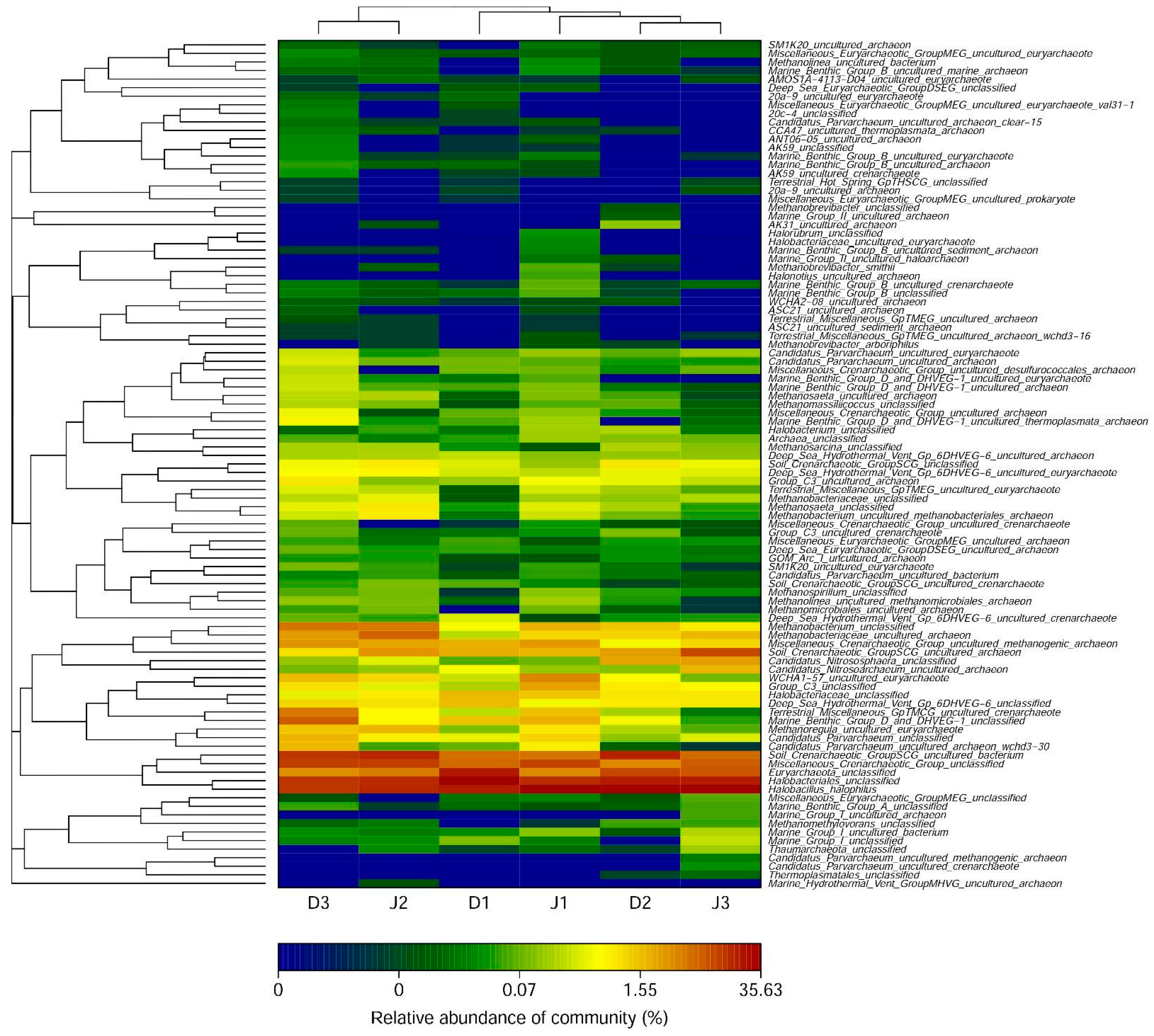
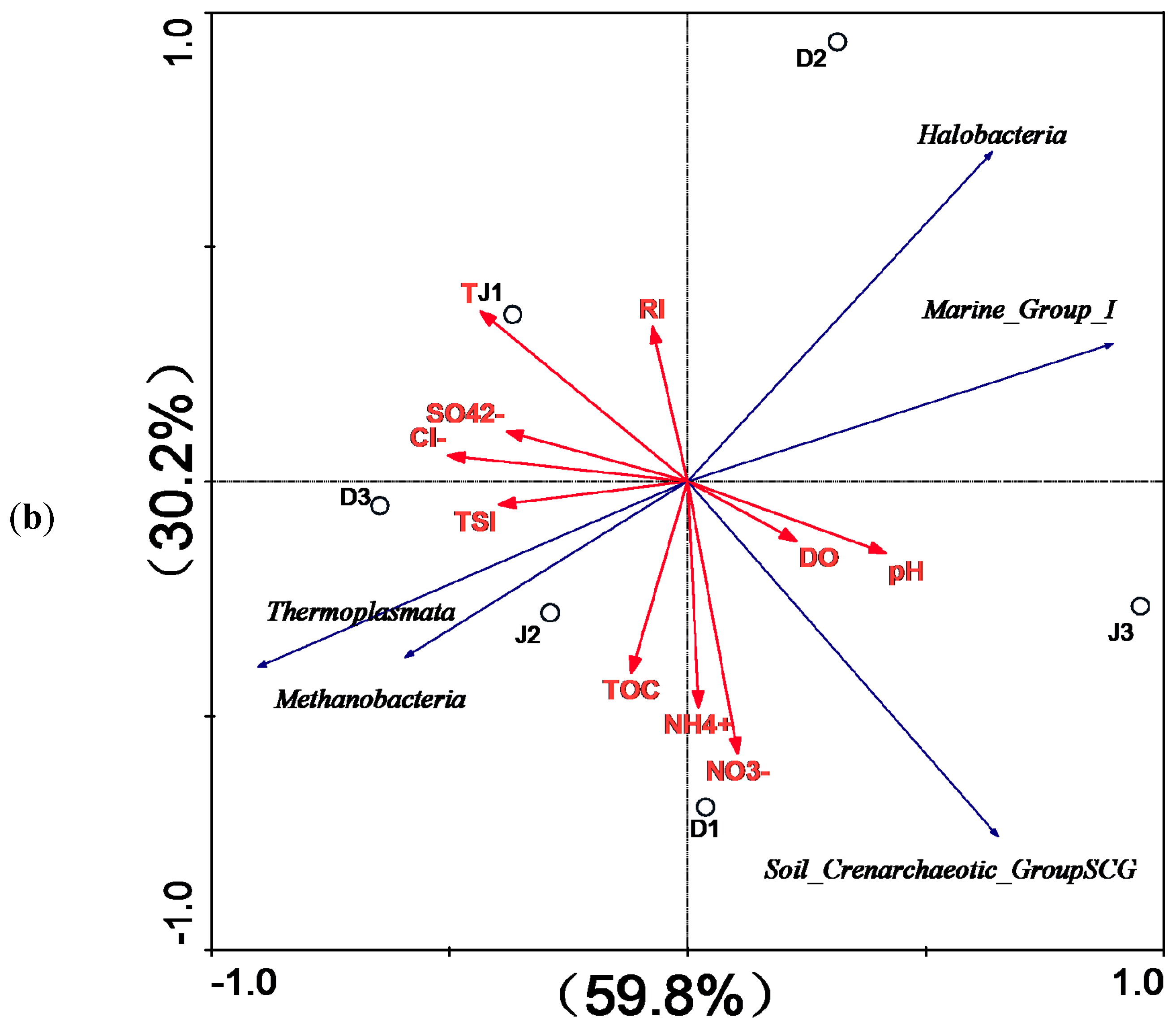
3.3. Archaeal Diversity of the Creeks
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Ethical Statement
References
- Baken, L.R. Separation and purification of bacteria from soil. Appl. Environ. Microbiol. 1983, 49, 1482–1487. [Google Scholar]
- Medeirospm, M.; Fernandes, M.F.; Dick, R.P.; Simoneit, B.R.T. Seasonal variations in sugar contents and microbial community in a ryegrass soil. Chemosphere 2006, 65, 832–839. [Google Scholar] [CrossRef] [PubMed]
- Syakti, A.D.; Mazzella, N.; Nerini, D.; Guiliano, M.; Bertrand, J.C.; Doumenq, P. Phospholipid fatty acids of a marine sedimentary microbial community in a laboratory microcosm: Responses to petroleum hydrocarbon contamination. Org. Geochem. 2006, 37, 1617–1628. [Google Scholar] [CrossRef]
- Mondini, C.; Insam, H. Community level physiological profiling as a tool to evaluate compost maturity: A kinetic approach. Eur. J. Soil Biol. 2003, 39, 141–148. [Google Scholar] [CrossRef]
- Princic, A.; Mahne, I.; Megusar, F.; Paul, E.A.; Tiedje, J.M. Effects of pH and oxygen and ammonium concentrations on the community structure of nitrifying bacteria from wastewater. Appl. Environ. Microbiol. 1998, 64, 3584–3590. [Google Scholar] [PubMed]
- McCaig, A.E.; Phillips, C.J.; Stephen, J.R.; Kowalchuk, G.A.; Harvey, S.M.; Herbert, R.A.; Embley, T.M.; Prosser, J.I. Nitrogen cycling and community structure of proteobacterial β-subgroup ammonia-oxidizing bacteria within polluted marine fish farm sediments. Appl. Environ. Microbiol. 1999, 65, 213–220. [Google Scholar] [PubMed]
- Luo, C.W.; Tsementzi, D.; Kyrpides, N.; Read, T.; Konstantinidis, K.T. Direct comparisons of Illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PLoS ONE 2012, 7, e30087. [Google Scholar]
- Schuster, S.C. Next-generation sequencing transforms today’s biology. Nat. Methods 2008, 5, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Guan, D.W.; Zhou, B.K.; Zhao, B.S.; Ma, M.C.; Qin, J.; Jiang, X.; Chen, S.F.; Cao, F.M.; Shen, D.L.; et al. Influence of 34-years of fertilization on bacterial communities in an intensively cultivated black soil in northeast China Soil. Soil Biol. Biochem. 2015, 90, 42–51. [Google Scholar] [CrossRef]
- Bai, Y.; Shi, Q.; Wen, D.; Li, Z.; Jefferson, W.A.; Feng, C.; Tang, X. Bacterial communities in the sediments of Dianchi Lake, a partitioned eutrophic waterbody in China. PLoS ONE 2012, 7, e37796. [Google Scholar] [CrossRef] [PubMed]
- Nogales, B.; Lanfranconi, M.P.; Pina-Villalonga, J.M.; Bosch, R. Anthropogenic perturbations in marine microbial communities. FEMS Microbiol. Rev. 2011, 35, 275–298. [Google Scholar] [CrossRef] [PubMed]
- Azarbad, H.; van Straalen, N.M.; Laskowski, R.; Nikiel, K.; Röling, W.F.M.; Niklińska, M. Susceptibility to additional stressors in metal-tolerant soil microbial communities from two pollution gradients. Appl. Soil Ecol. 2016, 98, 233–242. [Google Scholar] [CrossRef]
- Hamilton, S.K.; Kellndorfer, J.; Lehner, B.; Tobler, M. Remote sensing of floodplain geomorphology as a surrogate for biodiversity in a tropical river system (Madre de Dios, Peru). Geomorphology 2007, 89, 23–38. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, M.M. The analysis of govern contaminate measure about mainly pollution enter Tailake. North Environ. 2011, 23, 100–101. [Google Scholar]
- Ma, L.S.; Wang, Z.Q.; Zhang, S.M.; Ma, X.F.; Zhang, G.Y. Pollution from agricultural non point sources and its control in river system of Taihu Lake, JiangSu. Acta Sci. Circumstantiae 1997, 17, 39–47. [Google Scholar]
- Ahn, C.; Gillevet, P.M.; Sikaroodi, M. Molecular characterization of microbial communities in treatment microcosm wetlands as influenced by macrophytes and phosphorus loading. Ecol. Indic. 2007, 7, 852–863. [Google Scholar] [CrossRef]
- Shipin, O.; Koottatep, T.; Khanh, N.T.T.; Polprasert, C. Integrated natural treatment systems for developing communities: Low-tech N-removal through the fluctuating microbial pathways. Water Sci. Technol. 2005, 51, 299–306. [Google Scholar] [PubMed]
- Ligi, T.; Oopkaup, K.; Truu, M.; Preem, J.-K.; Nõlvak, H.; Mitsch, W.J.; Mander, U.; Truu, J. Characterization of bacterial communities in soil and sediment of a created riverine wetland complex using high-throughput 16S rRNA amplicon sequencing. Ecol. Eng. 2014, 72, 56–66. [Google Scholar] [CrossRef]
- Jiang, Y.; Sun, S.Y. Water environment improvement of Zhihugang River. China Water Res. 2009, 23, 35–38. [Google Scholar]
- Zhen, S.C.; Zhu, W. Analysis of isotope tracing of domestic sewage sources in Taihu Lake—A case study of Meiliang Bay and Gonghu Bay. Ecol. Indic. 2016, 66, 113–120. [Google Scholar] [CrossRef]
- Lin, L.; Wu, J.L.; Zeng, H.A.; Liu, W. Stable nitrogen isotope tracing anthropogenic influence on Lake Taihu. J. Lake Sci. 2012, 24, 546–552. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, Y.Y.; He, T.; Xie, S.G. Change of microbial community structure and functional gene abundance in nonylphenol-degrading sediment. Appl. Microbiol. Biotechnol. 2015, 99, 3259–3268. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Hu, X.; Tao, X.C.; Yu, H.X.; Zhang, X.W. Risk and toxicity assessments of heavy metals in sediments and fishes from the Yangtze River and Taihu Lake, China. Chemosphere 2013, 93, 1887–1895. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.L.; Wu, X.D.; Liu, B.; Yan, D.Z.; Kong, F.X. Dynamics and sources of dissolved organic carbon during phytoplankton bloom in hypereutrophic Lake Taihu (China). Limnologica 2015, 54, 5–13. [Google Scholar] [CrossRef]
- Chen, F.Z.; Ye, J.L.; Shu, T.T.; Sun, Y.; Li, J. Zooplankton response to the lake restoration in the drinking-water source in Meiliang Bay of subtropical eutrophic Lake Taihu, China. Limnologica 2012, 42, 189–196. [Google Scholar] [CrossRef]
- Ni, J.J.; Yu, Y.H.; Feng, W.S.; Yan, Q.Y.; Pan, G.; Yang, B.; Zhang, X.; Li, X.M. Impacts of algal blooms removal by chitosan-modified soils on zooplankton community in Taihu Lake, China. J. Environ. Sci. 2010, 22, 1500–1507. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, W.W.; Wang, S.H.; Zhang, B.; Hu, J.C. Initial identification of heavy metals contamination in Taihu Lake, a eutrophic lake in China. J. Environ. Sci. 2012, 24, 1539–1548. [Google Scholar] [CrossRef]
- Jin, X.C.; Tu, Q.Y. Criterion of Eutrophiction Survey on Lakes; Environmental Science Press: Beijing, China, 1998. [Google Scholar]
- Hori, T.; Sugiyama, M. Direct spectrophotometric determination of sulphate ion based on the formation of a blue molybdosulphate complex. Analyst 1998, 113, 1639–1642. [Google Scholar] [CrossRef]
- Dennis, K.L.; Wang, Y.; Blatner, N.R.; Wang, S.; Saadalla, A.; Trudeau, E.; Roers, A.; Weaver, C.T.; Lee, J.J.; Gilbert, J.A.; et al. Adenomatous polyps are driven by microbe-instigated focal inflammation and are controlled by IL-10–producing T Cells. Cancer. Res. 2013, 73, 5905–5913. [Google Scholar] [CrossRef] [PubMed]
- Thiago, M.A.; Pereira, R.V.; Caixeta, L.S.; Guard, C.L.; Bicalho, R.C. Microbial diversity in bovine papillomatous digital dermatitis in Holstein dairy cows from upstate New York. FEMS Microbiol. Ecol. 2012, 79, 518–529. [Google Scholar]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Marc, L.; Bjoern, U. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucl. Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.R.; Wang, Q.; Cardenas, E. The Ribosomal Database Project: Improved alignments and new tools for rRNA analysis. Nucleic. Acids. Res. 2009, 37, D141–D145. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 2011, 6, e27310. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sheng, H.F.; He, Y. Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of Illumina Tags. Appl. Environ. Microbiol. 2012, 78, 8264–8271. [Google Scholar] [CrossRef] [PubMed]
- Lepš, J.; Šmilauer, P. Multivariate Analysis of Ecological Data Using CANOCO; Cambridge University Press: New York, NY, USA, 2003. [Google Scholar]
- Chen, C.X.; Jiang, X.; Zhan, Y.Z.; Jin, X.C.; Zhao, Z. Speciation distribution and potential ecological risk assessment of heavy metals in sediments of Taihu Lake. China Environ. Sci. 2011, 31, 1842–1848. [Google Scholar]
- Liu, Y.; Xiao, L. Comprehensive evaluation of ecological risk of heavy metal pollution in the surface sediments of Taihu Lake. Environ. Prot. Sci. 2014, 40, 46–50. [Google Scholar]
- Yang, C.; Wang, P.F.; Liu, J.J.; Wang, C.; Hou, J.; Qian, J. Vertical distribution and migration of heavy metals in sediment cores of Taihu Lake. J. Agro Environ. Sci. 2016, 35, 548–557. [Google Scholar]
- Wang, S.M.; Dou, H.S. Lakes of China; Science Press: Beijing, China, 1998. [Google Scholar]
- Hakanson, L. An ecological risk index for aquatic pollution control a sedimentological approach. Water Res. 1980, 14, 975–1001. [Google Scholar] [CrossRef]
- Gagen, E.J.; Huber, H.; Meador, T. Novel cultivation-based approach to understanding the miscellaneous crenarchaeotic group (MCG) archaea from sedimentary ecosystems. Appl. Environ. Microbiol. 2013, 79, 6400–6406. [Google Scholar] [CrossRef] [PubMed]
- Kubo, K.; Lloyd, K.G.; Biddle, J.F. Archaea of the Miscellaneous Crenarchaeotal Group are abundant, diverse and widespread in marine sediments. ISME J. 2012, 6, 1949–1965. [Google Scholar] [CrossRef] [PubMed]
- Haller, L.; Tonolla, M.; Zopfi, J.; Peduzzi, R.; Wildi, W.; Poté, J. Composition of bacterial and archaeal communities in freshwater sediments with different contamination levels (Lake Geneva, Switzerland). Water Res. 2011, 45, 1213–1228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Kong, W.; Wu, N. Bacterial diversity and community along the succession of biological soil crusts in the Gurbantunggut Desert, Northern China. J. Basic Microbiol. 2016, 56, 670. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Han, W.Y. Water quality assessment and analysis before and after the decade of the dry period in Lingdingyang Estuary of the Pearl River Mouth. Mar. Environ. Sci. 2001, 20, 28–31. [Google Scholar]
- Chen, Y.W.; Fan, C.X.; Katrin, T. Changes of nutrients and phytoplankton chlorophyll-a in a large shallow lake, Taihu, China: An 8-year investigation. Hydrobiologia 2003, 506, 273–279. [Google Scholar] [CrossRef]
- Pinto, A.B.; Pagnocca, F.C.; Pinheiro, M.A.A.; Fontes, R.F.; de Oliveira, A.J. Heavy metals and TPH effects on microbial abundance and diversity in two estuarine areas of the southern-central coast of São Paulo State, Brazil. Mar. Pollut. Bull. 2015, 96, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.H.A.; Anees, M.; Arshad, M.; Muhammad, Y.S.; Iqbal, M.; Yousaf, S. Effects of illuminance and nutrients on bacterial photo-physiology of hydrocarbon degradation. Sci. Total Environ. 2016, 557, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Q.; Yang, L.Y.; Yin, D.Q. Vertical distribution of physicochemical characteristics and the microbial diversity in different spatial sediments samples in Lake Taihu. Environ. Sci. 2008, 29, 3537–3545. [Google Scholar]
- Freitag, T.E.; Chang, L.; Prosser, J.I. Changes in the community structure and activity of betaproteobacterial ammonia-oxidizing sediment bacteria along a freshwater-marine gradient. Environ. Microbiol. 2006, 8, 684. [Google Scholar] [CrossRef] [PubMed]
- Shao, K.Q.; Gao, G.; Wang, Y.P.; Tang, X.M.; Qin, B.Q. Vertical diversity of sediment bacterial communities in two different trophic states of the eutrophic Lake Taihu, China. J. Environ. Sci. 2013, 25, 1186–1194. [Google Scholar] [CrossRef]
- Feng, B.; Li, X.; Wang, J.; Hu, Z.; Meng, H.; Xiang, L.; Quan, Z. Bacterial diversity of water and sediment in the Changjiang estuary and coastal area of the East China Sea. FEMS Microbiol. Ecol. 2009, 70, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Schmid, E.; de Castro, J.V.; Cardinale, M.; Eberl, L.; Grube, M.; Berg, G.; Riedel, K. Structure and function of the symbiosis partners of the lung lichen (Lobaria pulmonaria L. Hoffm.) analyzed by metaproteomics. Proteomics 2011, 11, 2752–2756. [Google Scholar] [CrossRef] [PubMed]
- Grube, M.; Koeberl, M.; Lackner, S.; Berg, C.; Berg, G. Host-parasite interaction and microbiome response: Effects of fungal infections on the bacterial community of the Alpine lichen Solorina crocea. FEMS Microbiol. Ecol. 2012, 82, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Lertsethtakarn, P.; Ottemann, K.M.; Hendrixson, D.R. Motility and chemotaxis in campylobacter and helicobacter. Annu. Rev. Microbiol. 2011, 65, 389–410. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Takaki, Y.; Shimamura, S.; Reysenbach, A.L.; Takai, K.; Horikoshi, K. Deep-sea vent epsilon-proteobacterial genomes provide insights into emergence of pathogens. Proc. Natl. Acad. Sci. USA 2007, 104, 12146–12150. [Google Scholar] [CrossRef] [PubMed]
- Grube, M.; Cernava, T.; Soh, J.; Fuchs, S.; Aschenbrenner, I.; Lassek, C.; Wegner, U.; Becher, D.; Riedel, K.; Sensen, C.W.; et al. Exploring functional contexts of symbiotic sustain within lichen associated bacteria by comparative omics. ISME J. 2015, 9, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Yang, J.S.; Qu, J.H.; Li, H.F.; Liu, W.J.; Li, B.Z.; Wang, E.T.; Yuan, H.L. Sediment prokaryote communities in different sites of eutrophic Lake Taihu and their interactions with environmental factors. World J. Microbiol. Biotechnol. 2015, 31, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, H.; Sekiguchi, Y.; Hanada, S.; Nakamura, K.; Nomura, N.; Matsumura, M.; Kamagata, Y. Comparative analysis of bacterial diversity in freshwater sediment of a shallow eutrophic lake by molecular and improved cultivation-based techniques. Appl. Environ. Microbiol. 2005, 71, 2162–2169. [Google Scholar] [CrossRef] [PubMed]
- Borin, S.; Brusetti, L.; Mapelli, F. Sulfur cycling and methanogenesis primarily drive microbial colonization of the highly sulfidic urania deep hypersaline basin. Proc. Natl. Acad. Sci. USA 2009, 106, 9151–9156. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Qi, R.; Yang, M. Bacterial community characteristics under long-term antibiotic selection pressures. Water Res. 2011, 45, 6063–6073. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, M.; Xia, Y. Pyrosequencing analysis of bacterial diversity in 14 wastewater treatment systems in china. Appl. Environ. Microbiol. 2012, 78, 7042. [Google Scholar] [CrossRef] [PubMed]
- Takai, K.; Suzuki, M.; Nakagawa, S.; Miyazaki, M.; Suzuki, Y.; Inagaki, F.; Horikoshi, K. Sulfurimonas paralvinellae sp. nov., a novel mesophilic, hydrogen- and sulfur-oxidizing chemolithoautotroph within the Epsilonproteobacteria isolated from a deep-sea hydrothermal vent polychaete nest, reclassification of Thiomicrospira denitrificans as Sulfurimonas denitrificans comb. nov. and emended description of the genus Sulfurimonas. Int. J. Syst. Evol. Microbiol. 2006, 56, 1725–1733. [Google Scholar] [PubMed]
- Kodama, Y.; Watanabe, K. Sulfuricurvum kujiense gen. nov., sp. nov., a facultatively anaerobic, chemolithoautotrophic, sulfur oxidizing bacterium isolated from an underground crude-oil storage cavity. Int. J. Syst. Evol. Microbiol. 2004, 54, 2297–2300. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, J.; Lapidus, A.; Copeland, A.; Del Rio, T.G.; Nolan, M.; Lucas, S.; Chen, F.; Tice, H.; Cheng, J.F.; Saunders, E.; et al. Complete genome sequence of Sulfurospirillum deleyianum type strain (5175T). Stand. Genomic Sci. 2010, 2, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Kodama, Y.; Ha, L.T.; Watanabe, K. Sulfurospirillum cavolei sp. nov., a facultatively anaerobic sulfur-reducing bacterium isolated from an underground crude oil storage cavity. Int. J. Syst. Evolut. Microbiol. 2007, 57, 827–831. [Google Scholar] [CrossRef] [PubMed]
- Muyzer, G.; Stams, A. The ecology and biotechnology of sulphate-reducing bacteria. Nat. Rev. Microbiol. 2008, 6, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Abell, G.C.; Bowman, J.P. Ecological and biogeographic relationships of class Flavobacteria in the Southern Ocean. FEMS Microbiol. Ecol. 2005, 51, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X; Tian, J.; Liu, C.; Chen, L. Composition and dynamics of microbial community in a zeolite biofilter-membrane bioreactor treating coking wastewater. Appl. Microbiol. Biotechnol. 2013, 97, 8767–8775. [Google Scholar] [CrossRef] [PubMed]
- Bonmati, A.; Sotres, A.; Mu, Y.; Rozendal, R.; Rabaey, K. Oxalate degradation in a bioelectrochemical system: Reactor performance and microbial community characterization. Bioresour. Technol. 2013, 143, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, S.; Kawanabe, A.; Ito, H.; Kandori, H.; Kogure, K. Diversity and functional analysis of proteorhodopsin in marine flavobacteria. Environ. Microbiol. 2012, 14, 1240–1248. [Google Scholar] [CrossRef] [PubMed]
- Jian, H.Q.; Hong, L.Y.; Wang, E.T.; Li, C.; Huang, H.Z. Bacterial diversity in sediments of the eutrophic Guanting Reservoir, China, estimated by analyses of 16S rDNA sequence. Biodivers. Conserv. 2008, 17, 1667–1683. [Google Scholar]
- Bauld, J.; Brock, T.D. Ecological studies of Chloroflexis, a gliding photosynthetic bacterium. Arch. Microbiol. 1973, 92, 267–284. [Google Scholar] [CrossRef]
- Liu, K.; Chunbo, H.; Jiao, J.J.; Jidong, G. Bacterial Investigation of Ammonium-Rich Sediment in the Pearl River Delta, China; American Geophysical Union: Washington, DC, USA, 2011. [Google Scholar]
- Johnson, D.B.; Hallberg, K.B. Carbon, iron and sulfur metabolism in acidophilic micro-organisms. Adv. Microb. Physiol. 2009, 54, 201–255. [Google Scholar]
- Falagán, C.; Sánchez-España, J.; Johnson, D.B. New insights into the biogeochemistry of extremely acidic environments revealed by a combined cultivation-based and culture independent study of two stratifi ed pit lakes. FEMS Microbiol. Ecol. 2014, 87, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Mota, C.; Head, M.A.; Ridenoure, J.A.; Cheng, J.J. Effects of aeration cycles on nitrifying bacterial populations and nitrogen removal in intermittently aerated reactors. Appl. Environ. Microbiol. 2005, 71, 8565–8572. [Google Scholar] [CrossRef] [PubMed]
- Altmann, D.; Stief, P.; Amann, R.; de Beer, D.; Schramm, A. In situ distribution and activity of nitrifying bacteria in freshwater sediment. Environ. Microbiol. 2003, 5, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.D.; Huang, Y.; Gao, H. Effect of heavy metal of A/O biological nitrogen removal system. J. Shenyang Arch. Civ. Eng. Univ. 2003, 19, 154–156. [Google Scholar]
- Ceven, F.; Semerci, N.; Geyik, A.G. Inhibition of respiration and distribution of Cd, Pb, Hg, Ag and Cr species in a nitrifying sludge. J. Hazard. Mater. 2010, 178, 619–627. [Google Scholar]
- Liu, S.; Shen, L.D.; Lou, L.P.; Tian, G.M.; Zheng, P.; Hu, B.L. Spatial distribution and factors shaping the niche segregation of ammonia-oxidizing microorganisms in the Qiantang River, China. Appl. Environ. Microbiol. 2013, 79, 4065–4071. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.H.; Lin, G.H.; Gao, G.; Qin, B.Q.; Zhang, J.S.; Zhao, G.P.; Zhou, Z.H.; Shen, J.H. Bacterial and archaeal assemblages in sediments of a large shallow freshwater lake Lake Taihu as revealed by denaturing gradient gel electrophoresis. J. Appl. Microbiol. 2009, 106, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Dorador, C.; Vila, I.; Remonsellez, F.; Imhoff, J.F.; Witzel, K.-P. Unique clusters of archaea in salar de huasco, an athalassohaline evaporitic basin of the Chilean Altiplano. FEMS Microbiol. Ecol. 2010, 73, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Ciobanu, M.C.; Rabineau, M.; Droz, L. Sedimentological imprint on subseafloor microbialcommunities in Western Mediterranean Sea Quaternary sediments. Biogeosciences 2012, 9, 3491–3512. [Google Scholar] [CrossRef]
- Jung, M.Y.; Park, S.J.; Kim, S.J.; Kim, J.G.; Sinninghe Damsté, J.S.; Jeon, C.O.; Rhee, S.K. A mesophilic, autotrophic, ammonia-oxidizing archaeon of thaumarchaeal group I.1a cultivated from a deep oligotrophic soil horizon. Appl. Environ. Microbiol. 2014, 80, 3645–3655. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, M.; Schwab, C.; Jensen, B.B.; Engberg, R.M.; Spang, A.; Canibe, N.; Højberg, O.; Milinovich, G.; Fragner, L.; Schleper, C.; et al. Methylotrophic methanogenic Thermoplasmata implicated in reduced methane emissions from bovine rumen. Nat. Commun. 2013, 4, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Zhang, Y.; Yang, W.; Xu, H. Microbial community characterization of an UASB treating increased organic loading rates of vitamin C biosynthesis wastewater. Water Sci. Technol. 2012, 65, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.S.; Tian, J.Q.; Jiang, N.; Guo, X.P.; Wang, Y.F.; Dong, X.Z. Methanogen community in Zoige wetland of Tibetan plateau and phenotypic characterization of adominant uncultured methanogen cluster ZC-1. Environ. Microbiol. 2008, 10, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Gorra, R.; Webster, G.; Martin, M.; Celi, L.; Mapelli, F.; Weightman, A.J. Dynamic microbial community associated with iron–arsenic co-precipitation products from a groundwater storage system in Bangladesh. Microb. Ecol. 2012, 64, 171–186. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Creek Name | Length (m) | Width (m) | Depth (m) | Sediment (m) | SD (m) | Flow (m3/s) |
|---|---|---|---|---|---|---|
| Sewage creek | 150 | 8.4 | 0.68 | 0.10 | 0.56 | 0.026 |
| Industry creek | 560 | 6.7 | 0.72 | 0.10 | 0.24 | 0.026 |
| Target Gene | Primer Sequence (5′-3′) | Thermal Programme | Reference |
|---|---|---|---|
| Archaeal 16S rRNA | Arch519F: CAGCCGCCGCGGTAA Arch 915R: GTGCTCCCCCGCCAATTCCT | 5 min at 98 °C, 28 cycles at 98 °C for 30 s, 55 °C for 45 s, 72 °C for 1 min, and a final extension at 72 °C for 5 min. | [30] |
| Bacterial 16S rRNA | 338F: ACTCCTACGGGAGGCAGCA 806R: GGACTACHVGGGTWTCTAAT | 5 min at 95 °C, 30 cycles of 95 °C for 1 min, 55 °C for 1 min, 72 °C for 1 min, and a final extension at 72 °C for 7 min | [31] |
| Sample Site | DO | T | pH | Chla | TP | TOC | COD | NH4+-N | NO3−-N | TN | SO42− | Cl− |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sewage Creek | ||||||||||||
| D1 | 5.66 | 19.6 | 7.13 | 0.075 | 0.49 | 22.2 | 22.3 | 2.28 | 3.36 | 7.14 | 66.7 | 166.1 |
| D2 | 5.68 | 19.8 | 7.14 | 0.023 | 0.18 | 16.9 | 23.0 | 1.05 | 1.37 | 4.51 | 59.2 | 133.5 |
| D3 | 6.97 | 20.2 | 7.26 | 0.020 | 0.15 | 17.4 | 21.7 | 0.68 | 1.21 | 2.52 | 55.6 | 123.7 |
| Mean | 6.10 | 19.9 | 7.17 | 0.039 | 0.27 | 18.8 | 22.3 | 1.33 | 1.98 | 4.72 | 60.5 | 141.1 |
| Industry Creek | ||||||||||||
| J1 | 7.27 | 19.8 | 7.44 | 0.003 | 0.19 | 14.7 | 6.33 | 0.83 | 1.60 | 3.36 | 176.8 | 131.2 |
| J2 | 6.47 | 19.4 | 7.35 | 0.003 | 0.15 | 15.1 | 5.55 | 1.06 | 1.38 | 3.66 | 129.6 | 100.5 |
| J3 | 8.06 | 19.6 | 7.65 | 0.006 | 0.10 | 14.6 | 4.79 | 0.71 | 1.53 | 3.24 | 66.5 | 60.7 |
| Mean | 7.26 | 19.6 | 7.48 | 0.004 | 0.15 | 14.8 | 5.56 | 0.86 | 1.50 | 3.42 | 124.3 | 97.4 |
| Sample Site | Cr | Ni | Cu | As | Cd | Zn | Pb |
|---|---|---|---|---|---|---|---|
| Sewage Creek | |||||||
| D1 | 44.17 | 30.83 | 38.12 | 9.04 | 0.62 | 131.78 | 102.58 |
| D2 | 41.46 | 24.78 | 29.51 | 8.22 | 1.06 | 116.21 | 53.13 |
| D3 | 40.45 | 24.89 | 30.98 | 7.35 | 0.91 | 137.92 | 10.25 |
| Mean | 42.02 | 26.83 | 32.87 | 8.20 | 0.86 | 128.63 | 55.32 |
| Industry Creek | |||||||
| J1 | 45.82 | 29.68 | 51.13 | 12.79 | 1.55 | 197.35 | 92.51 |
| J2 | 45.47 | 25.82 | 47.82 | 10.46 | 0.38 | 115.14 | 89.32 |
| J3 | 41.99 | 23.10 | 21.75 | 10.14 | 1.21 | 70.70 | 52.96 |
| Mean | 44.42 | 26.20 | 40.23 | 11.13 | 1.05 | 127.73 | 78.26 |
| Sample site | TSI | Level | RI | Level |
|---|---|---|---|---|
| Sewage Creek | ||||
| D1 | 76 | Hyper | 189 | Middle |
| D2 | 69 | Middle | 277 | Middle |
| D3 | 68 | Middle | 234 | Middle |
| Industry Creek | ||||
| J1 | 67 | Middle | 409 | High |
| J2 | 63 | Middle | 364 | High |
| J3 | 57 | Light | 310 | High |
| Sample | Reads | OTUs | Ace | Chao1 | Coverage | Shannon |
|---|---|---|---|---|---|---|
| Bacterial 16S rRNA | ||||||
| D1 | 34,091 | 1207 | 1302 | 1302 | 0.99 | 5.12 |
| D2 | 21,115 | 1144 | 1248 | 1275 | 0.99 | 5.91 |
| D3 | 34,233 | 1332 | 1382 | 1391 | 0.99 | 5.96 |
| J1 | 47,498 | 1192 | 1301 | 1320 | 0.99 | 5.32 |
| J2 | 31,031 | 1328 | 1402 | 1421 | 0.99 | 5.85 |
| J3 | 18,265 | 1115 | 1256 | 1273 | 0.98 | 5.75 |
| Archaeal 16S rRNA | ||||||
| D1 | 32,255 | 690 | 748 | 733 | 0.98 | 4.27 |
| D2 | 61,300 | 652 | 735 | 750 | 0.99 | 3.79 |
| D3 | 36,363 | 590 | 721 | 703 | 0.99 | 3.86 |
| J1 | 46,435 | 585 | 614 | 608 | 0.99 | 3.54 |
| J2 | 39,875 | 523 | 679 | 694 | 0.98 | 3.53 |
| J3 | 52,445 | 547 | 619 | 615 | 0.99 | 3.63 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.; Jiang, X.; Wang, J.; Wang, K.; Zheng, B. Effect of Sewage and Industrial Effluents on Bacterial and Archaeal Communities of Creek Sediments in the Taihu Basin. Water 2017, 9, 373. https://doi.org/10.3390/w9060373
Li D, Jiang X, Wang J, Wang K, Zheng B. Effect of Sewage and Industrial Effluents on Bacterial and Archaeal Communities of Creek Sediments in the Taihu Basin. Water. 2017; 9(6):373. https://doi.org/10.3390/w9060373
Chicago/Turabian StyleLi, Da, Xia Jiang, Jinzhi Wang, Kun Wang, and Binghui Zheng. 2017. "Effect of Sewage and Industrial Effluents on Bacterial and Archaeal Communities of Creek Sediments in the Taihu Basin" Water 9, no. 6: 373. https://doi.org/10.3390/w9060373
APA StyleLi, D., Jiang, X., Wang, J., Wang, K., & Zheng, B. (2017). Effect of Sewage and Industrial Effluents on Bacterial and Archaeal Communities of Creek Sediments in the Taihu Basin. Water, 9(6), 373. https://doi.org/10.3390/w9060373