Simulating the Fate and Transport of Coal Seam Gas Chemicals in Variably-Saturated Soils Using HYDRUS




Abstract
:1. Introduction
2. Theory
2.1. Variably-Saturated Water Flow
2.2. Solute Transport Applications of the Standard HYDRUS Model
2.3. Solute Transport Applications of the UnsatChem Module
2.4. Solute Transport Applications of the HP1 Module
3. Applications
3.1. Standard HYDRUS-1D: Fate of Hydraulic Fracturing Chemicals During Natural Attenuation
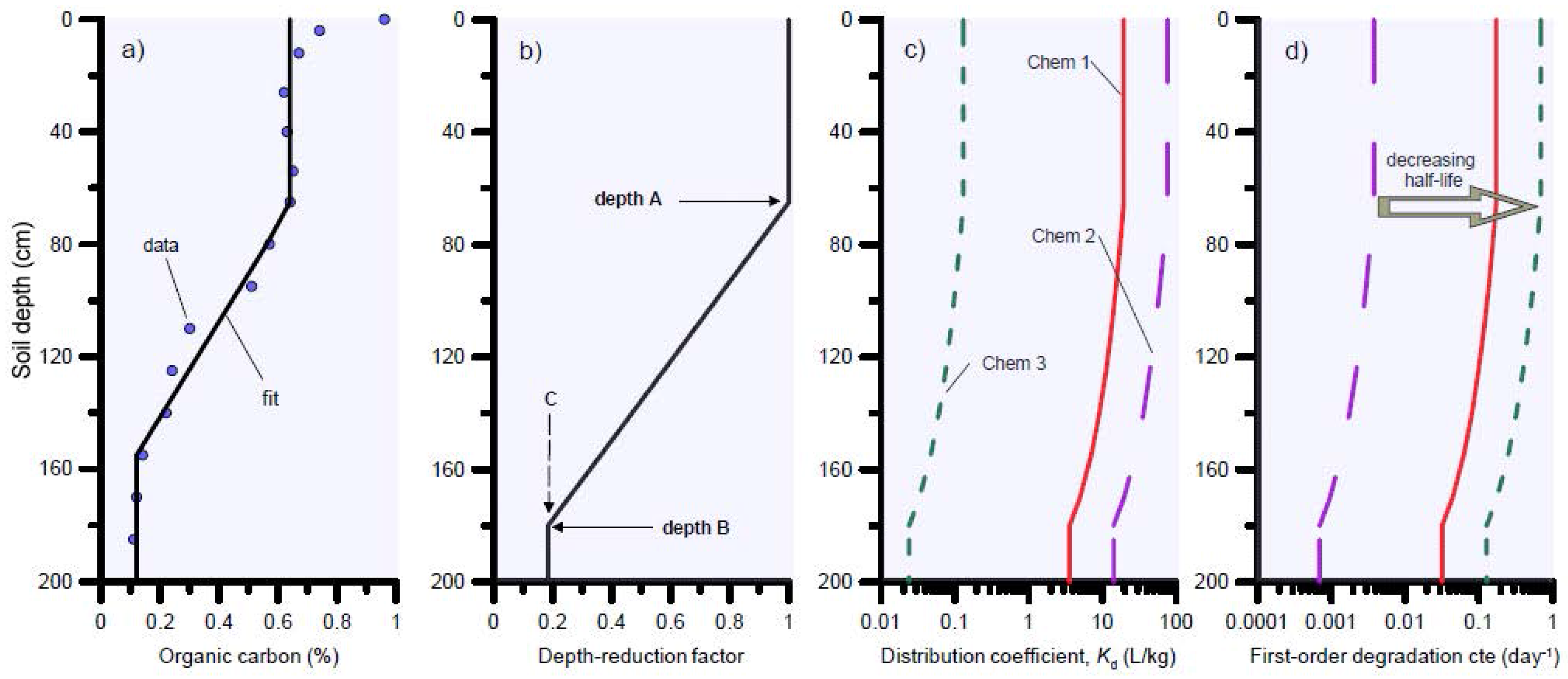
3.1.1. Problem Definition
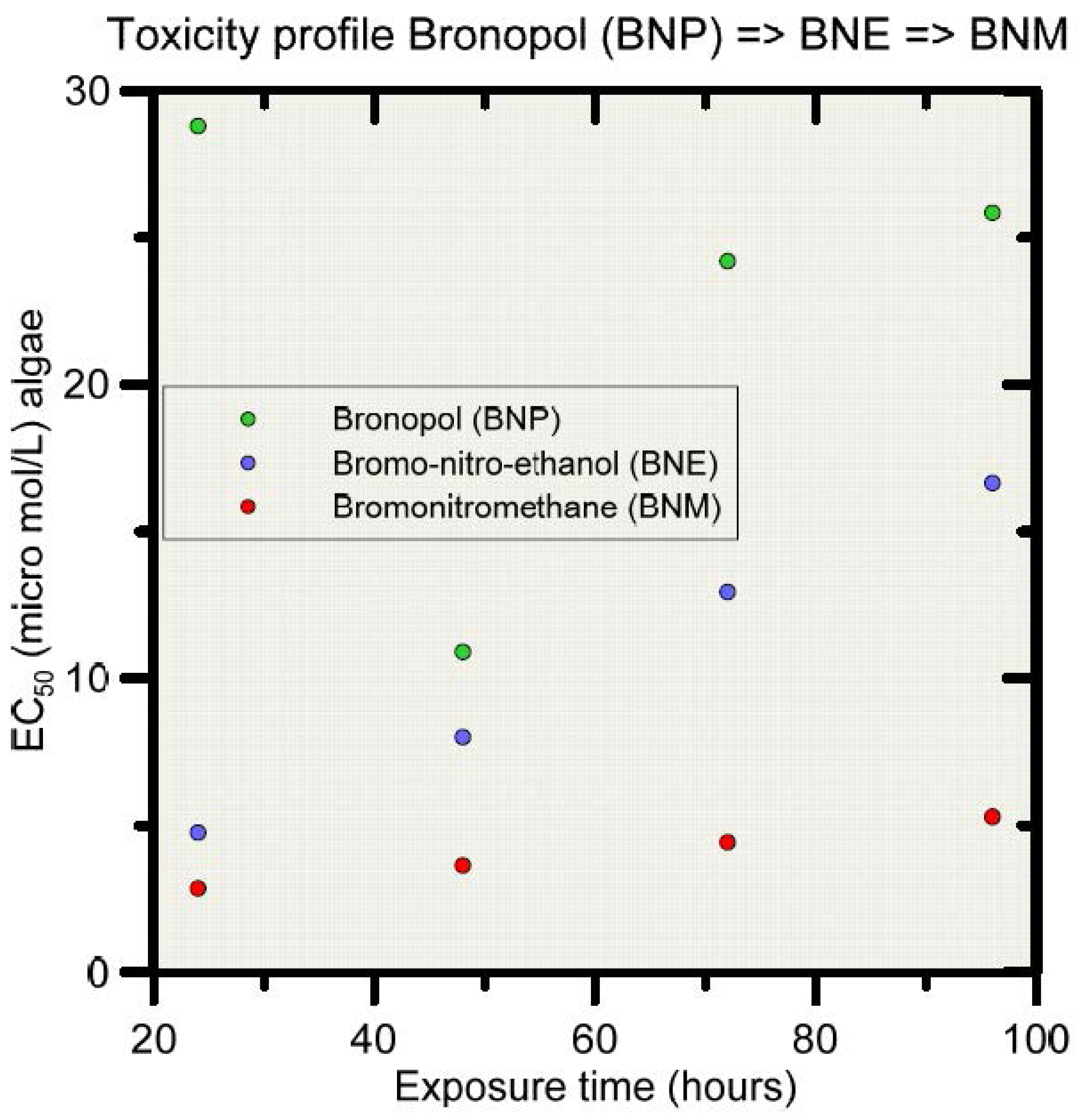
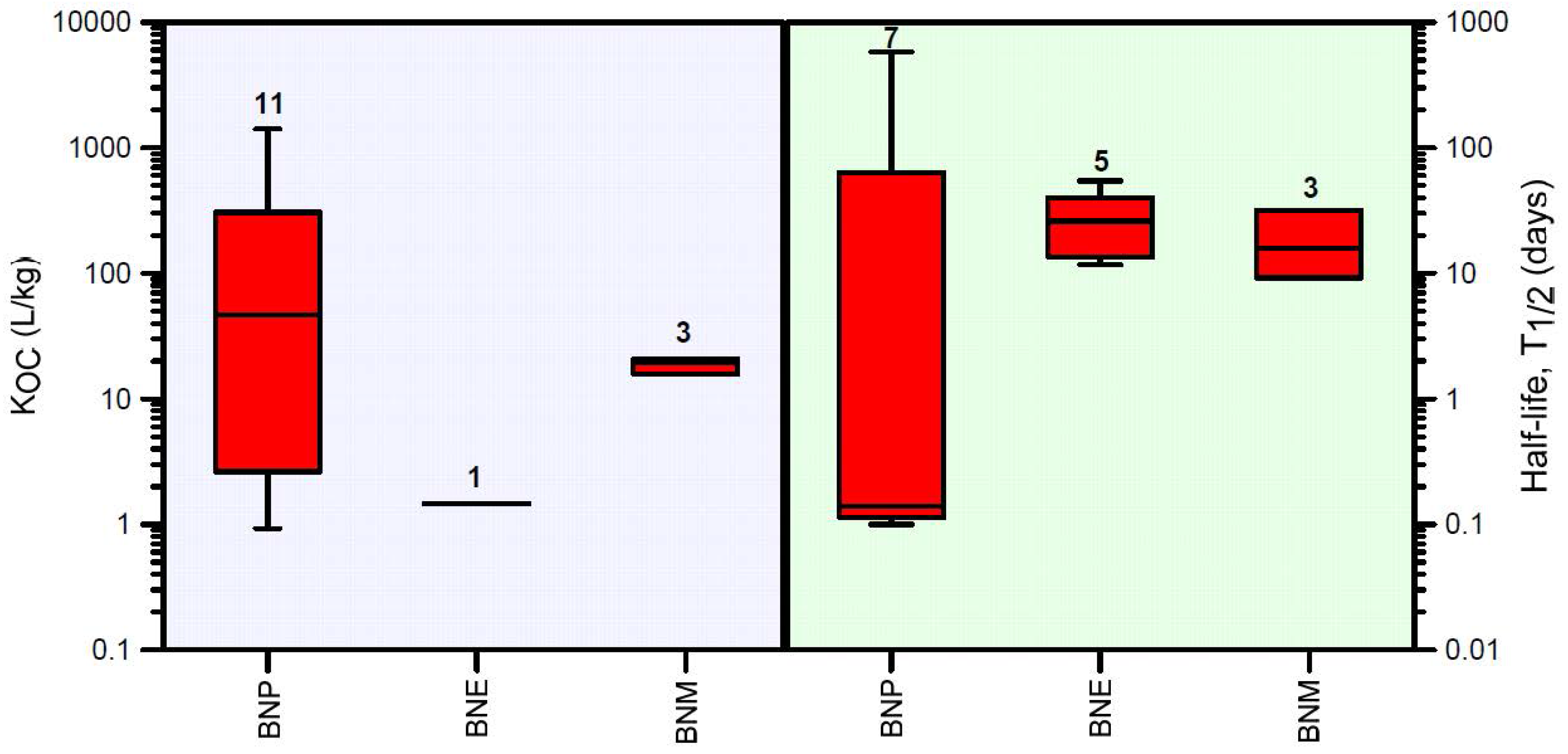
3.1.2. Biocide Transformation Pathways and Natural Soil Attenuation Parameters
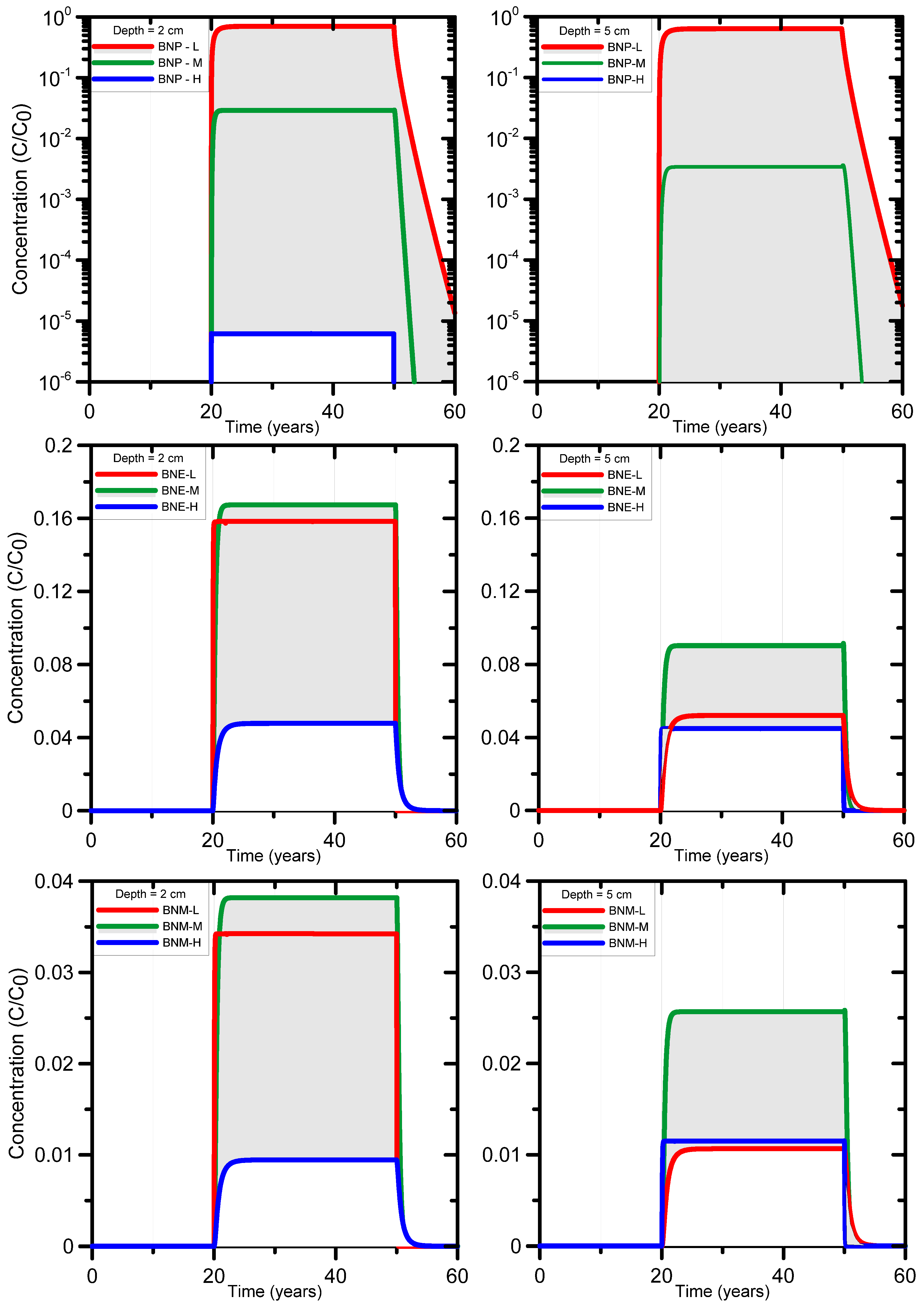
3.1.3. HYDRUS-1D Simulation Results for the Bronopol Degradation Chain
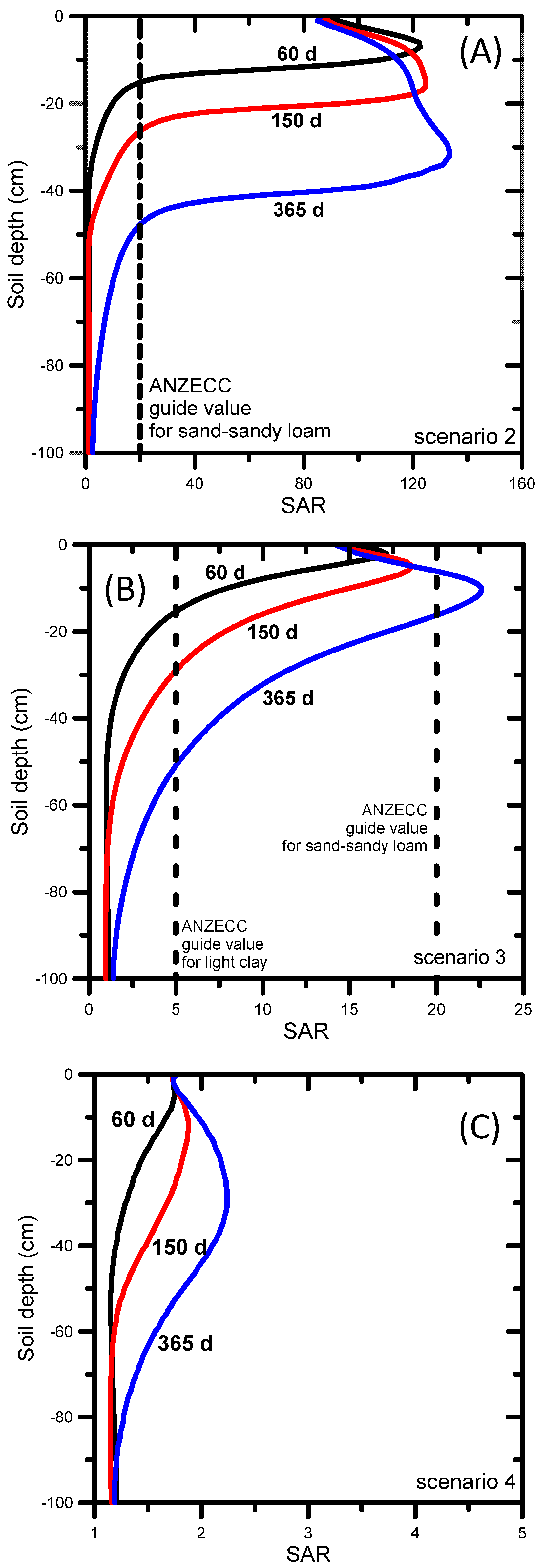
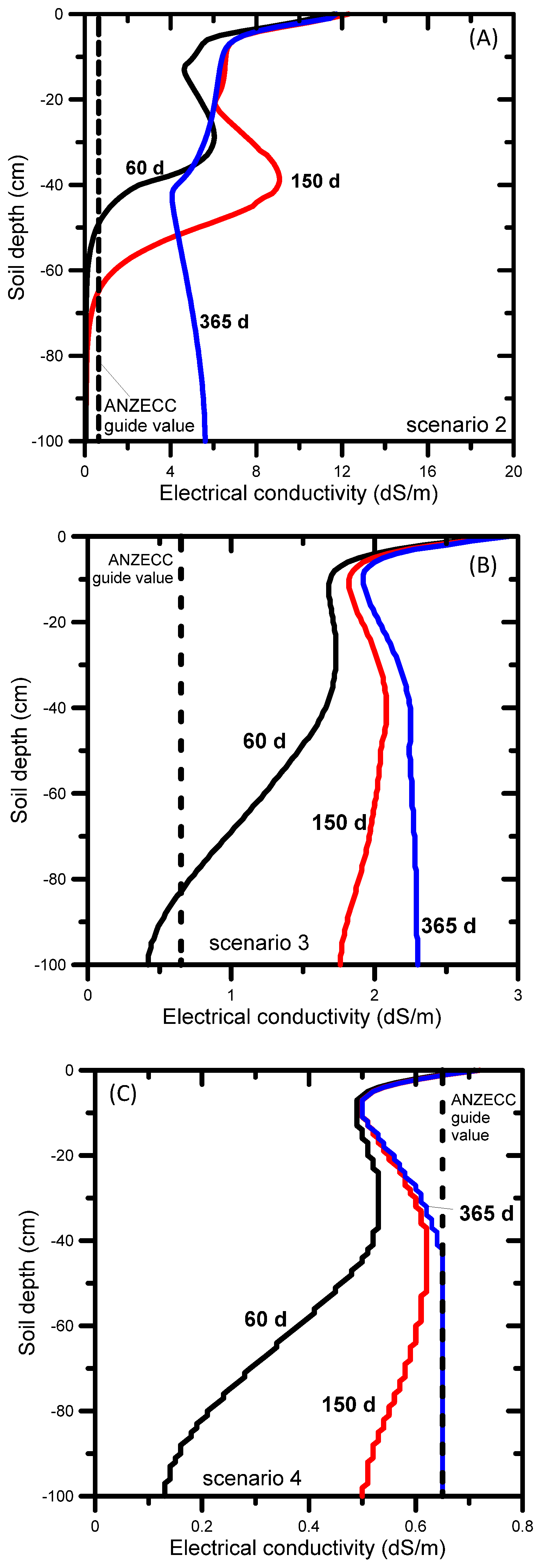
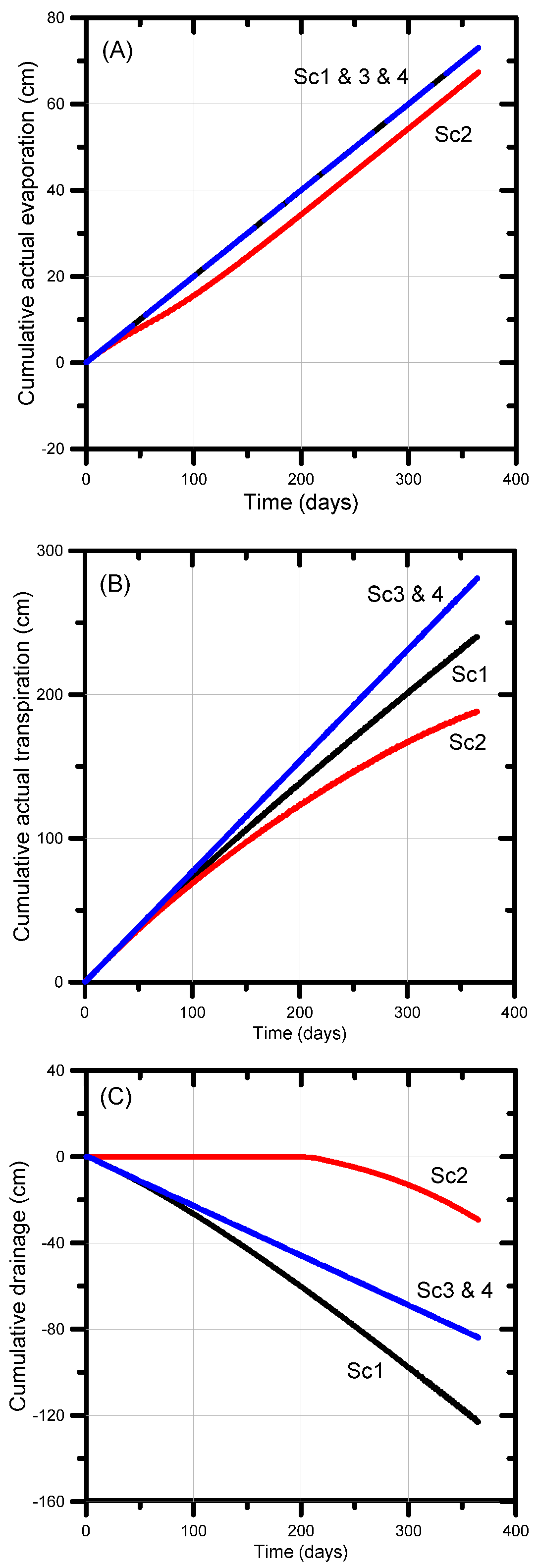
3.2. UNSATCHEM: Optimizing Coal Seam Gas Produced Water for Sustainable Irrigation
3.2.1. Problem Definition
3.2.2. Soil Hydraulic and Chemical Model for Major Ion Transport
3.2.3. UNSATCHEM Simulation Results
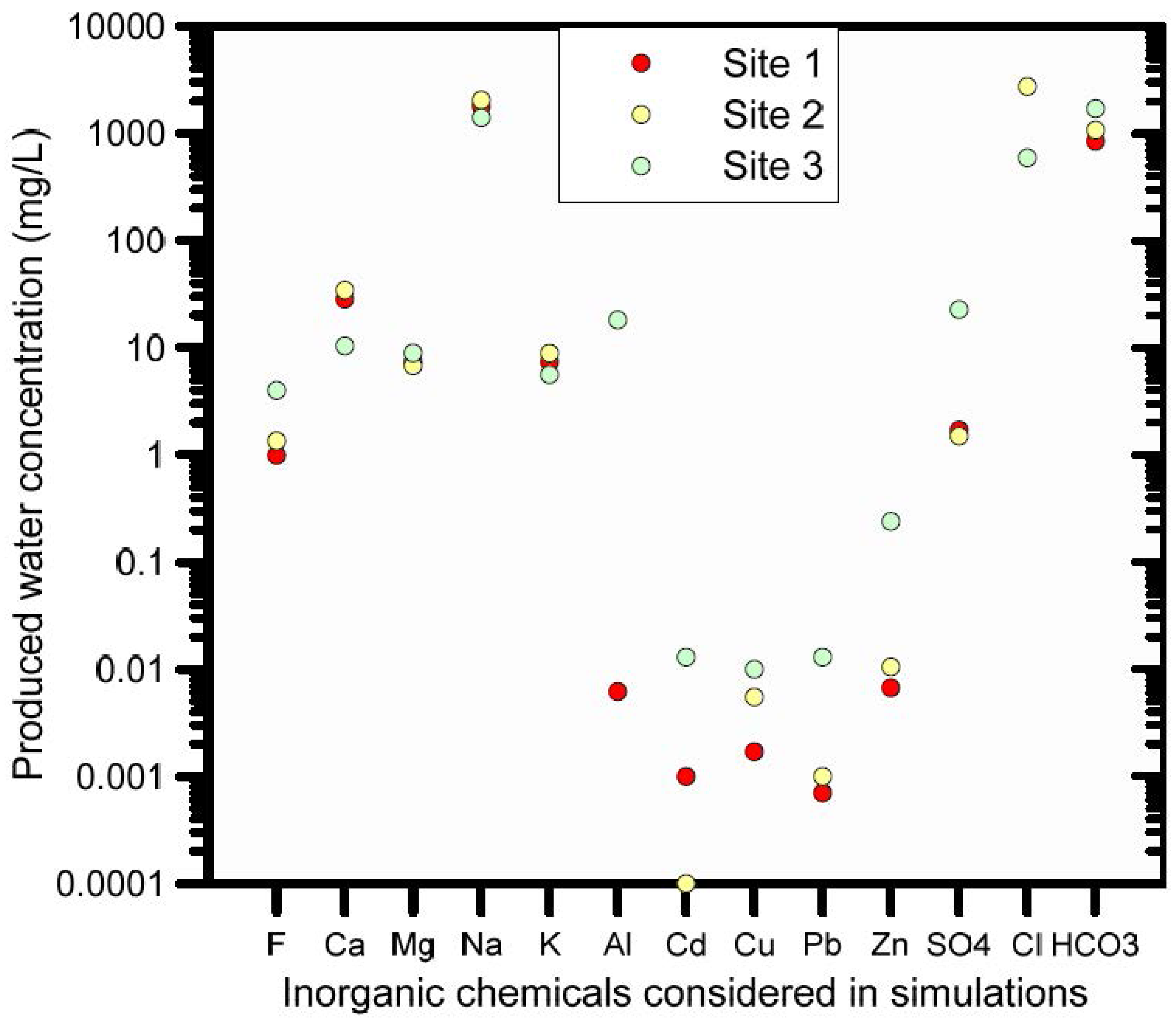
3.3. HP1: Trace Metal Transport in Soil Leached with Coal Seam Gas Produced Water
3.3.1. Problem Definition
3.3.2. Soil Geochemical Model for Trace Metal Transport
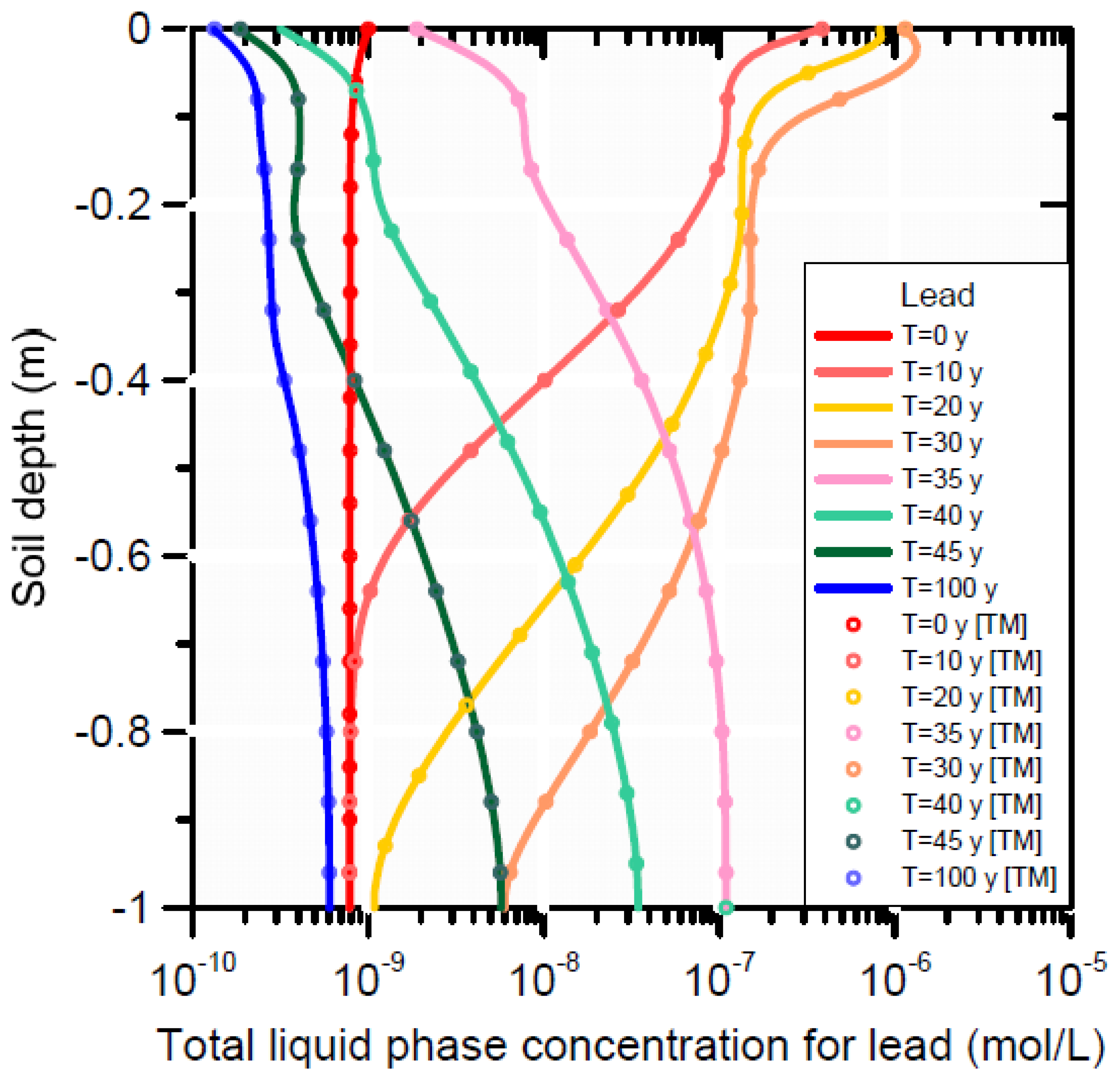
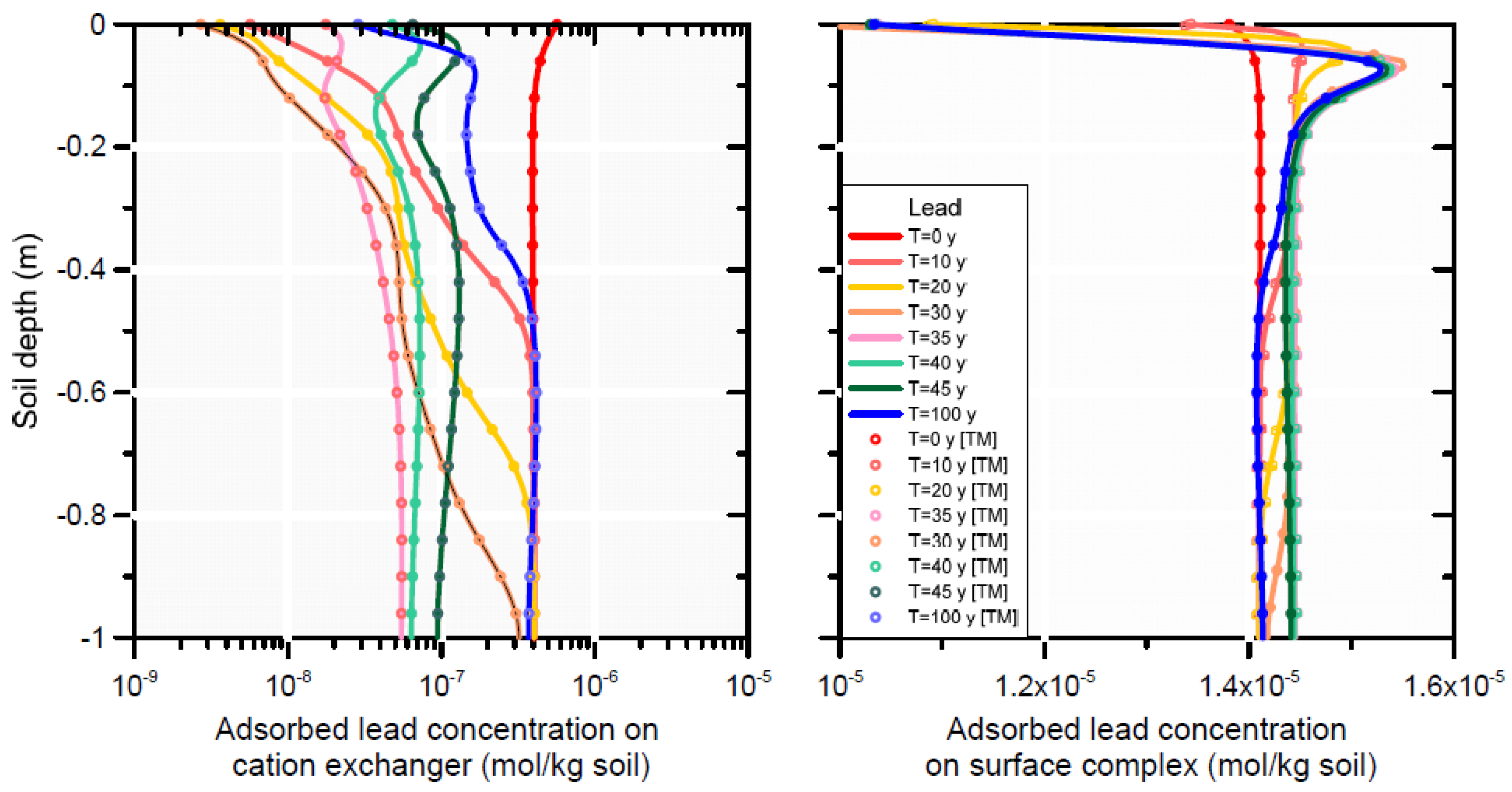
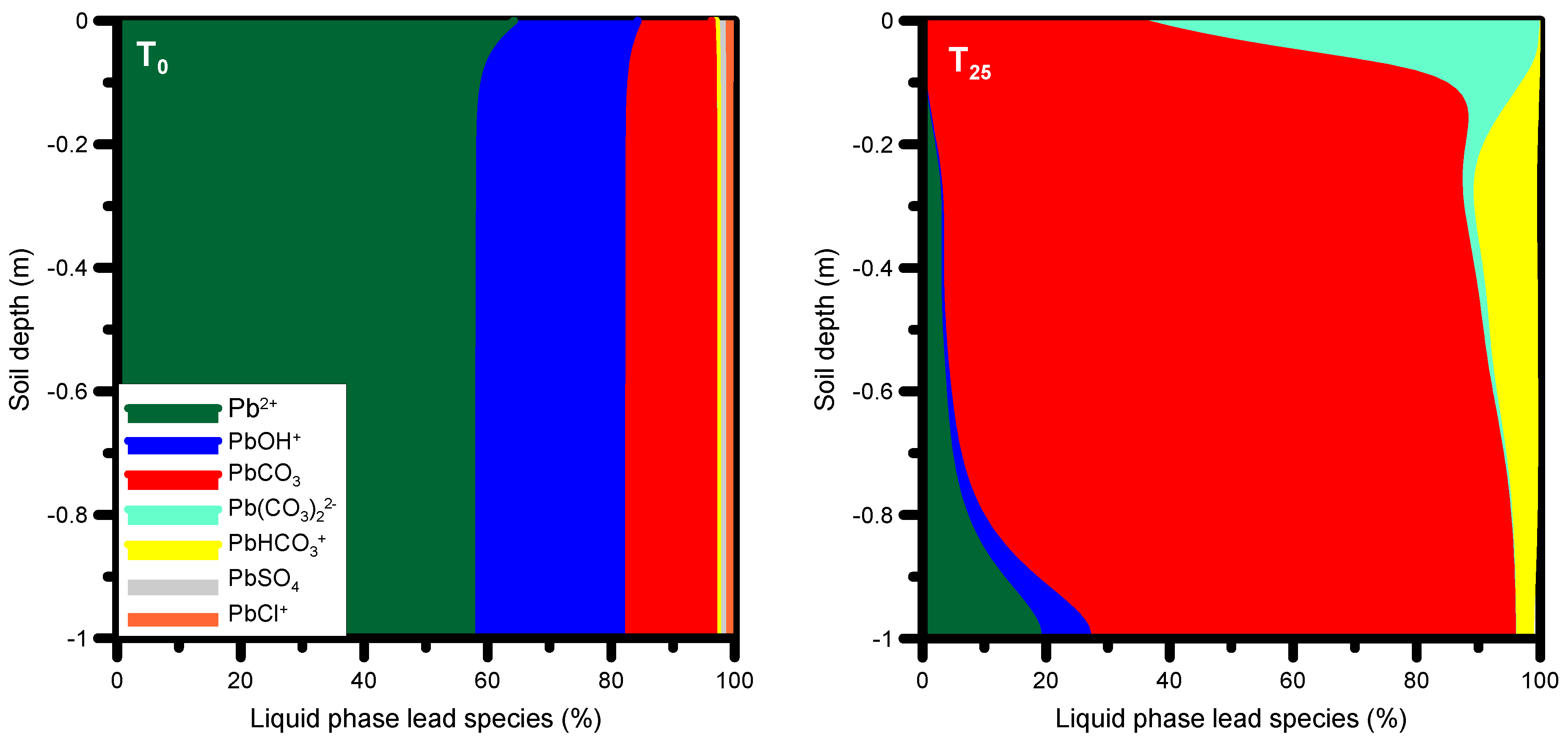
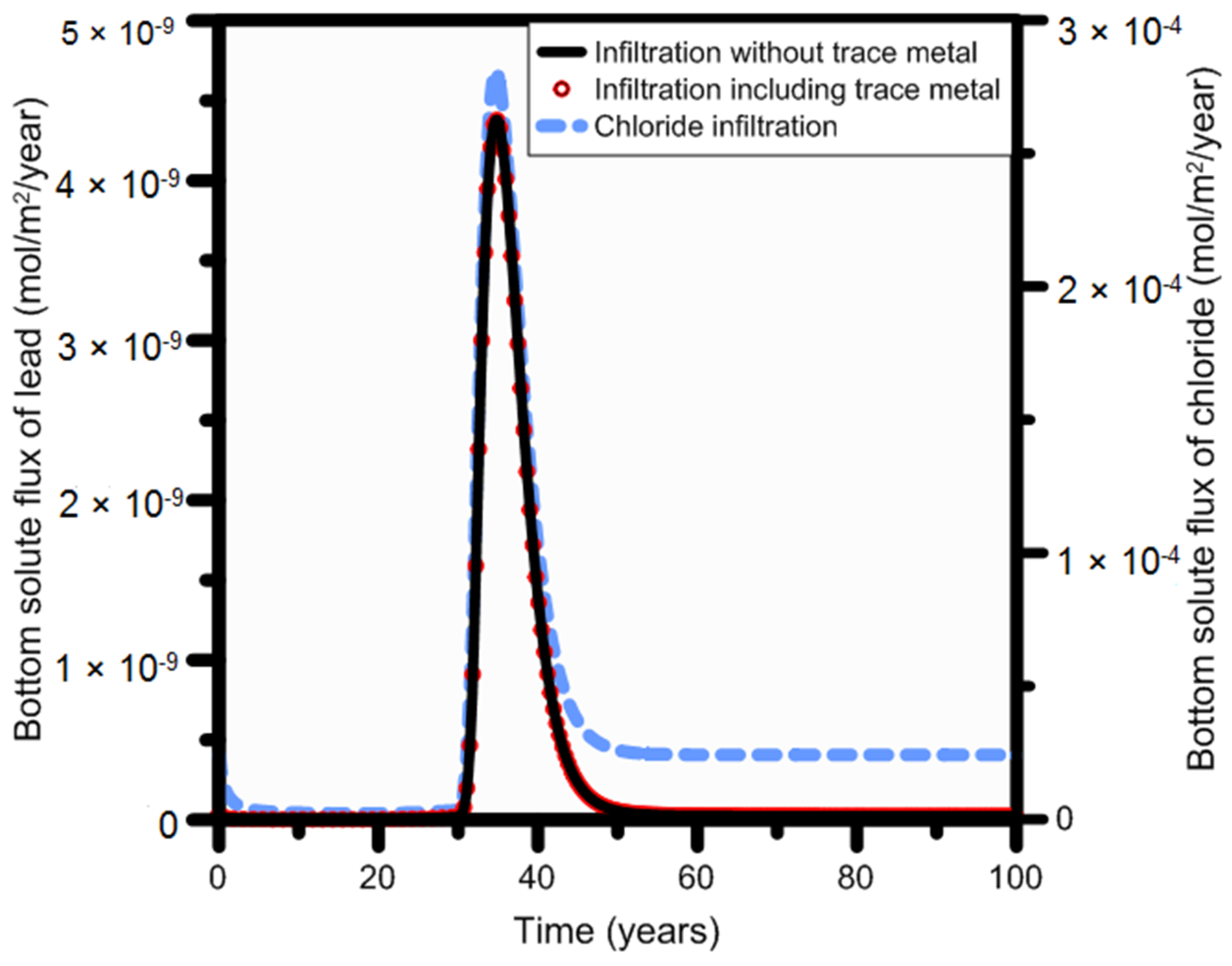
3.3.3. HP1 Simulation Results
4. Summary and Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Singh, G.; Kaur, G.; Williard, K.; Schoonover, J.; Kang, J. Monitoring of Water and Solute Transport in the Vadose Zone: A Review. Vadose Zone J. 2017. [Google Scholar] [CrossRef]
- US EPA. Hydraulic Fracturing for Oil and Gas: Impacts from the Hydraulic Fracturing Water Cycle on Drinking Water Resources in the United States; Office of Research and Development: Washington, DC, USA, 2016; EPA/600/R-16/236Fa.
- Christensen, T.H.; Kjeldsen, P.; Bjerg, P.L.; Jensen, D.L.; Christensen, J.B.; Baun, A.; Albrechtsen, H.-J.; Heron, G. Biogeochemistry of landfill leachate plumes. Appl. Geochem. 2001, 16, 659–718. [Google Scholar] [CrossRef]
- Akcil, A.; Koldas, S. Acid mine drainage (AMD): Causes, treatment and case studies. J. Clean. Prod. 2006, 14, 1139–1145. [Google Scholar] [CrossRef]
- Fredrickson, J.K.; Zachara, J.M.; Balkwill, D.L.; Kennedy, D.; Li, S.-M.W.; Kostandarithes, H.M.; Daly, M.J.; Romine, M.F.; Brockman, F.J. Geomicrobiology of High-Level Nuclear Waste-Contaminated Vadose Sediments at the Hanford Site, Washington State. Appl. Environm. Microbiol. 2004, 70, 4230–4241. [Google Scholar] [CrossRef] [PubMed]
- Vandenhove, H.; Sweeck, L.; Mallants, D.; Vanmarcke, H.; Aitkulov, A.; Sadyrov, O.; Savosin, M.; Tolongutov, B.; Mirzachev, M.; Clerc, J.J.; et al. Assessment of radiation exposure in the uranium and milling area of Mailuu Suu, Kyrgyzstan. J. Environ. Radioact. 2006, 88, 118–139. [Google Scholar] [CrossRef] [PubMed]
- Lapworth, D.J.; Baran, N.; Stuart, M.E.; Ward, R.S. Emerging organic contaminants in groundwater: A review of sources, fate and occurrence. Environ. Pollut. 2012, 163, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Mallants, D.; Diels, L.; Bastians, L.; Vos, J.; Moors, H.; Wang, L.; Maes, N.; Vandenhove, H. Removal of Uranium and Arsenic from Groundwater Using Six Different Reactive Materials: Assessment of Removal Efficiency. In Uranium in the Aquatic Environment; Merkel, B.J., Planer-Friedrich, B., Wolkersdolfer, C., Eds.; Springer: Berlin, Germany, 2002; pp. 561–568. [Google Scholar]
- Scherer, M.M.; Richter, S.; Valentine, R.L.; Alvarez, P.J.J. Chemistry and microbiology of permeable reactive barriers for in situ groundwater clean up. Crit. Rev. Environ. Sci. Technol. 2000, 30, 363–411. [Google Scholar] [CrossRef]
- Šimůnek, J.; Jacques, D.; Langergraber, G.; Bradford, S.A.; Šejna, M.; van Genuchten, M.T. Numerical Modelling of Contaminant Transport Using HYDRUS and Its Specialized Modules, Invited paper for the Special Issue “Water Management in Changing Environment”. J. Indian Inst. Sci. 2013, 93, 265–284. [Google Scholar]
- Šimůnek, J.; van Genuchten, M.T.; Šejna, M. Recent developments and applications of the HYDRUS computer software packages. Vadose Zone J. 2016, 15, 25. [Google Scholar] [CrossRef]
- Šimůnek, J.; Bradford, S. Vadose Zone Modelling: Introduction and importance. Vadose Zone J. 2008, 7, 581–586. [Google Scholar] [CrossRef]
- Seuntjens, P.; Mallants, D.; Šimůnek, J.; Patyn, J.; Jacques, D. Sensitivity analysis of physical and chemical properties affecting field-scale cadmium transport in a heterogeneous soil profile. J. Hydrol. 2002, 264, 185–200. [Google Scholar] [CrossRef]
- Jacques, D.; Šimůnek, J.; Mallants, D.; van Genuchten, M.T. Modelling coupled hydrologic and chemical processes: Long-term Uranium transport following Phosphorus fertilization. Vadose Zone J. 2008, 7, 698–711. [Google Scholar] [CrossRef]
- Mallants, D.; Jacques, D.; Zeevaert, T. Modelling 226Ra, 222Rn, and 210Pb Migration in a Proposed Surface Repository of Very Low-Level Long-Lived Radioactive Waste. In Proceedings of the ICEM 2003, the 9th International Conference on Radioactive Waste Management and Environmental Remediation, Oxford, UK, 21–25 September 2003; icem03-4632. p. 8. [Google Scholar]
- Li, Y.; Šimůnek, J.; Zhang, Z.; Jing, L.; Ni, L. Evaluation of nitrogen balance in a direct-seeded-rice field experiment using Hydrus-1D. Agric. Water Manag. 2015, 148, 213–222. [Google Scholar] [CrossRef]
- Papiernik, S.K.; Yates, S.R.; Koskinen, W.C.; Barber, B. Processes affecting the dissipation of the herbicide isoxaflutole and its diketonitrile metabolite in agricultural soils under field conditions. J. Agric. Food Chem. 2007, 55, 8630–8639. [Google Scholar] [CrossRef] [PubMed]
- Schaerlaekens, J.; Mallants, D.; Šimůnek, J.; van Genuchten, M.T.; Feyen, J. Numerical simulation of transport and sequential biodegradation of chlorinated aliphatic hydrocarbons using CHAIN_2D. Hydrol. Proc. 1999, 13, 2847–2859. [Google Scholar] [CrossRef]
- Fan, Z.; Casey, F.X.M.; Hakk, H.; Larsen, G.L. Discerning and modelling the fate and transport of testosterone in undisturbed soil. J. Environ. Qual. 2007, 36, 864–873. [Google Scholar] [CrossRef] [PubMed]
- Unold, M.; Šimůnek, J.; Kasteel, R.; Groeneweg, J.; Vereecken, H. Transport of manure-based applied sulfadiazine and its main transformation products in soil columns. Vadose Zone J. 2009, 8, 677–689. [Google Scholar] [CrossRef]
- Šimůnek, J.; Jacques, D.; van Genuchten, M.T.; Mallants, D. Multicomponent geochemical transport modelling using Hydrus-1D and HP1. J. Am. Water Resour. Assoc. 2006, 42, 1537–1547. [Google Scholar] [CrossRef]
- Šimůnek, J.; Suarez, D.L. Two-dimensional transport model for variably saturated porous media with major ion chemistry. Water Resour. Res. 1994, 30, 1115–1133. [Google Scholar] [CrossRef]
- Šimůnek, J.; Suarez, D.L. Sodic soil reclamation using multicomponent transport modelling. J. Irrig. Drain. Eng. 1997, 123, 367–376. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. User’s Guide to PHREEQ C (Version 2)—A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport and Inverse Geochemical Calculations. In Water-Resources Investigations; Report 99-4259; U.S. Geological Survey: Earth Science Information Center: Denver, CO, USA, 1999; p. 312. [Google Scholar]
- Jacques, D.; Šimůnek, J.; Mallants, D.; van Genuchten, M.T. Modelling coupled water flow, solute transport and geochemical reactions affecting heavy metal migration in a podzol soil. Geoderma 2008, 145, 449–461. [Google Scholar] [CrossRef]
- Langergraber, G.; Šimůnek, J. Modelling variably-saturated water flow and multi-component reactive transport in constructed wetlands. Vadose Zone J. 2005, 4, 924–938. [Google Scholar] [CrossRef]
- Langergraber, G.; Šimůnek, J. Reactive transport modelling of subsurface flow constructed wetlands. Vadose Zone J. 2012, 11. [Google Scholar] [CrossRef]
- Langergraber, G.; Rousseau, D.; García, J.; Mena, J. CWM1—A general model to describe biokinetic processes in subsurface flow constructed wetlands. Water Sci. Technol. 2009, 59, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Šimůnek, J.; Changming, H.; Pang, L.; Bradford, S.A. Colloid-facilitated transport in variably-saturated porous media: Numerical model and experimental verification. Vadose Zone J. 2006, 5, 1035–1047. [Google Scholar] [CrossRef]
- Pang, L.; Šimůnek, J. Evaluation of bacteria-facilitated cadmium transport in gravel columns using the HYDRUS colloid-facilitated solute transport model. Water Resour. Res. 2006, 42, W12S10. [Google Scholar] [CrossRef]
- Zhang, M.; Engelhardt, I.; Šimůnek, J.; Bradford, S.A.; Kasel, D.; Berns, A.E.; Vereecken, H.; Klumpp, E. Co-transport of chlordecone and sulfadiazine in the presence of functionalized multi-walled carbon nanotubes in soils. Environ. Pollut. 2017, 221, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Spurlock, F.; Šimůnek, J.; Johnson, B.; Tuli, A. Sensitivity analysis of vadose zone fumigant transport and volatilization. Vadose Zone J. 2013, 12, 12. [Google Scholar] [CrossRef]
- Spurlock, F.; Johnson, B.; Tuli, A.; Gao, S.; Tao, J.; Sartori, F.; Qin, R.; Sullivan, D.; Stanghellini, M.; Ajwa, H. Simulation of fumigant transport and volatilization from tarped broadcast applications. Vadose Zone J. 2013, 12, 10. [Google Scholar] [CrossRef]
- Brooks, R.H.; Corey, A.T. Hydraulic Properties of Porous Media. In Hydrology Paper No. 3; Colorado State University: Fort Collins, CO, USA, 1964. [Google Scholar]
- van Genuchten, M.T. A closed-form equation for predicting the hydraulic conductivity of unsaturated soils. Soil Sci. Soc. Am. J. 1980, 44, 892–898. [Google Scholar] [CrossRef]
- Vogel, T.; Císlerová, M. On the reliability of unsaturated hydraulic conductivity calculated from the moisture retention curve. Transp. Porous Med. 1988, 3, 1–15. [Google Scholar] [CrossRef]
- Kosugi, K. Lognormal distribution model for unsaturated soil hydraulic properties. Water Resour. Res. 1996, 32, 2697–2703. [Google Scholar] [CrossRef]
- Durner, W. Hydraulic conductivity estimation for soils with heterogeneous pore structure. Water Resour. Res. 1994, 30, 211–233. [Google Scholar] [CrossRef]
- Šimůnek, J.; Hopmans, J.W. Modelling compensated root water and nutrient uptake. Ecol. Model. 2009, 220, 505–521. [Google Scholar] [CrossRef]
- Šimůnek, J.; Jarvis, N.J.; van Genuchten, M.T.; Gärdenäs, A. Review and comparison of models for describing non-equilibrium and preferential flow and transport in the vadose zone. J. Hydrol. 2003, 272, 14–35. [Google Scholar] [CrossRef]
- Saito, H.; Šimůnek, J.; Mohanty, B. Numerical analyses of coupled water, vapor, and heat transport in the vadose zone. Vadose Zone J. 2006, 5, 784–800. [Google Scholar] [CrossRef]
- Pontedeiro, E.M.; van Genuchten, M.T.; Cotta, R.M.; Šimůnek, J. The effects of preferential flow and soil texture on risk assessments of a NORM waste disposal site. J. Hazard. Mater. 2010, 174, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Hanson, B.R.; Šimůnek, J.; Hopmans, J.W. Numerical modelling of urea-ammonium-nitrate fertigation under micro-irrigation. Agric. Water Manag. 2006, 86, 102–113. [Google Scholar] [CrossRef]
- Ramos, T.B.; Šimůnek, J.; Gonçalves, M.C.; Martins, J.C.; Prazeres, A.; Pereira, L.S. Two-dimensional modelling of water and nitrogen fate from sweet sorghum irrigated with fresh and blended saline waters. Agric. Water Manag. 2012, 111, 87–104. [Google Scholar] [CrossRef]
- Casey, F.X.M.; Šimůnek, J. Inverse analyses of the transport of chlorinated hydrocarbons subject to sequential transformation reactions. J. Environ. Qual. 2001, 30, 1354–1360. [Google Scholar] [CrossRef] [PubMed]
- Casey, F.X.M.; Larsen, G.L.; Hakk, H.; Šimůnek, J. Fate and transport of 17β-Estradiol in soil-water systems. Environ. Sci. Technol. 2003, 37, 2400–2409. [Google Scholar] [CrossRef] [PubMed]
- Casey, F.X.M.; Larsen, G.L.; Hakk, H.; Šimůnek, J. Fate and transport of testosterone in agriculturally significant soils. Environ. Sci. Technol. 2004, 38, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Casey, F.X.M.; Šimůnek, J.; Lee, J.; Larsen, G.L.; Hakk, H. Sorption, mobility, and transformation of estrogenic hormones in natural soil. J. Environ. Qual. 2005, 34, 1372–1379. [Google Scholar] [CrossRef] [PubMed]
- Das, B.S.; Lee, L.S.; Rao, P.S.C.; Hultgren, R.P. Sorption and degradation of steroid hormones in soils during transport: Column studies and model evaluation. Environ. Sci. Technol. 2004, 38, 1460–1470. [Google Scholar] [CrossRef] [PubMed]
- Wehrhan, A.; Kasteel, R.; Šimůnek, J.; Groeneweg, J.; Vereecken, H. Transport of sulfadiazine in soil columns—experiments and modelling approaches. J. Contam. Hydrol. 2007, 89, 107–135. [Google Scholar] [CrossRef] [PubMed]
- Dontsova, K.M.; Yost, S.L.; Šimůnek, J.; Pennington, J.C.; Williford, C. Dissolution and transport of TNT, RDX, and Composition B in saturated soil columns. J. Environ. Qual. 2006, 35, 2043–2054. [Google Scholar] [CrossRef] [PubMed]
- Dontsova, K.M.; Pennington, J.C.; Hayes, C.; Šimůnek, J.; Williford, C.W. Dissolution and transport of 2,4-DNT and 2,6-DNT from M1 propellant in soil. Chemosphere 2009, 77, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Šimůnek, J.; van Genuchten, M.T. Modeling Nonequilibrium Flow and Transport Processes Using HYDRUS. Vadose Zone J. 2008, 7, 782–797. [Google Scholar] [CrossRef]
- Šimůnek, J.; Valocchi, A.J. Geochemical Transport. In Methods of Soil Analysis, Part 1, Physical Methods, Chapter 6.9, 3rd ed.; Dane, J.H., Topp, G.C., Eds.; SSSA: Madison, WI, USA, 2002; pp. 1511–1536. [Google Scholar]
- Kaledhonkar, M.J.; Keshari, A.K. Modelling the effects of saline water use in agriculture. Irrig. Drain. 2006, 55, 177–190. [Google Scholar] [CrossRef]
- Gonçalves, M.C.; Šimůnek, J.; Ramos, T.B.; Martins, J.C.; Neves, M.J.; Pires, F.P. Multicomponent solute transport in soil lysimeters irrigated with waters of different quality. Water Resour. Res. 2006, 42, W08401. [Google Scholar] [CrossRef]
- Kaledhonkar, M.J.; Keshari, A.K.S.; van der Zee, E.A.T.M. Relative sensitivity of ESP profile to spatial and temporal variability in cation exchange capacity and pore water velocity under simulated field conditions. Agric. Water Manag. 2006, 83, 58–68. [Google Scholar] [CrossRef]
- Kaledhonkar, M.J.; Sharma, D.R.; Tyagi, N.K.; Kumar, A.; van der Zee, S.E.A.T.M. Modelling for conjunctive use irrigation planning in sodic groundwater areas. Agric. Water Manag. 2012, 107, 14–22. [Google Scholar] [CrossRef]
- Skaggs, T.H.; Shouse, P.J.; Poss, J.A. Irrigation of forage crops with saline drainage waters: 2. Modelling drainage and root water uptake. Vadose Zone J. 2006, 5, 824–837. [Google Scholar] [CrossRef]
- Schoups, G.; Hopmans, J.W.; Tanji, K.K. Evaluation of model complexity and space–time resolution on the prediction of long-term soil salinity dynamics, western San Joaquin Valley, California. Hydrol. Process. 2006, 20, 2647–2668. [Google Scholar] [CrossRef]
- Corwin, D.L.; Rhoades, J.D.; Šimůnek, J. Leaching requirement for soil salinity control: Steady-state vs. transient-state models. Agric. Water Manag. 2007, 90, 165–180. [Google Scholar] [CrossRef]
- Ramos, T.B.; Šimůnek, J.; Gonçalves, M.C.; Martins, J.C.; Prazeres, A.; Castanheira, N.L.; Pereira, L.S. Field evaluation of a multicomponent solute transport model in soils irrigated with saline waters. J. Hydrol. 2011, 407, 129–144. [Google Scholar] [CrossRef]
- Rasouli, F.; Pouya, A.K.; Šimůnek, J. Modelling the effects of saline water use in wheat-cultivated lands using the UNSATCHEM model. Irrig. Sci. 2013, 31, 1009–1024. [Google Scholar] [CrossRef]
- Jakubowski, R.; Haws, N.; Ellerbrock, D.; Murtagh, J.; MacFarlane, D. Development of a management tool to support the beneficial use of treated coal seam gas water for irrigation in Eastern Australia. Mine Water Environ. 2014, 33, 133–145. [Google Scholar] [CrossRef]
- Steefel, C.I.; Appelo, C.A.J.; Arora, B.; Jacques, D.; Kalbacher, T.; Kolditz, O.; Lagneau, V.; Lichtner, P.C.; Mayer, K.U.; Meeussen, J.C.L.; et al. Reactive transport codes for subsurface environmental simulation. Comput. Geosci. 2015, 19, 445–447. [Google Scholar] [CrossRef]
- Mayer, K.U.; Alt-Epping, P.; Jacques, D.; Arora, B.; Steefel, C.I. Benchmark problems for reactive transport modeling of the generation and attenuation of acid rock drainage. Comput. Geosci. 2015, 19, 599–611. [Google Scholar] [CrossRef]
- Xie, M.; Mayer, K.U.; Claret, F.; Alt-Epping, P.; Jacques, D.; Steefel, C.; Chiaberge, C.; Simunek, J. Implementation and evaluation of permeability-porosity and tortuosity-porosity relationships linked to mineral dissolution-precipitation. Comput. Geosci. 2015, 19, 655–671. [Google Scholar] [CrossRef]
- Jacques, D.; Šimůnek, J.; Mallants, D.; van Genuchten, M.T. The HPx reactive transport models: Summary of recent developments and applications. In Proceedings of the 4th International Conference “HYDRUS Software Applications to Subsurface Flow and Contaminant Transport Problems”, Prague, Czech Republic, 21–22 March 2013; Šimůnek, J., van Genuchten, M.T., Kodešová, R., Eds.; Czech University of Life Sciences: Prague, Czech Republic; pp. 7–16, ISBN 978-80-213-2380-3. [Google Scholar]
- Leterme, B.; Blanc, P.; Jacques, D. A reactive transport model for mercury fate in soil—application to different anthropogenic pollution sources. Environ. Sci. Pollut. R. 2014, 21, 12279–12293. [Google Scholar] [CrossRef] [PubMed]
- Leterme, B.; Jacques, D. A reactive transport model for mercury fate in contaminated soil-sensitivity analysis. Environ. Sci. Pollut. R. 2015, 22, 16830–16842. [Google Scholar] [CrossRef] [PubMed]
- Bessinger, B.A.; Marks, C.D. Treatment of Mercury-Contaminated Soils With Activated Carbon: A Laboratory, Field, and Modelling Study. Remediat. J. 2010, 21, 115–135. [Google Scholar] [CrossRef]
- Thaysen, E.M.; Jacques, D.; Jessen, S.; Andersen, C.E.; Laloy, E.; Ambus, P.; Postma, D.; Jakobsen, I. Inorganic carbon fluxes across the vadose zone of planted and unplanted soil mesocosms. Biogeosciences 2014, 11, 7179–7192. [Google Scholar] [CrossRef]
- Thaysen, E.M.; Jessen, S.; Postma, D.; Jakobsen, R.; Jacques, D.; Ambus, P.; Laloy, E.; Jakobsen, I. Effects of lime and concrete waste of vadose zone carbon cycling. Vadose Zone J. 2014, 13, 11. [Google Scholar] [CrossRef]
- Zhou, D.; Thiele-Bruhn, S.; Arenz-Leufen, M.G.; Jacques, D.; Lichtner, P.; Engelhardt, I. Impact of manure-related DOM on sulfonamide transport in arable soils. J. Contam. Hydrol. 2016, 192, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Makselon, J.; Zhou, D.; Engelhardt, I.; Jacques, D.; Klumpp, E. Experimental and Numerical Investigations of Silver Nanoparticle Transport under Variable Flow and Ionic Strength in Soil. Environ. Sci. Technol. 2017, 51, 2096–2104. [Google Scholar] [CrossRef] [PubMed]
- US Congress. Chemicals Used in Hydraulic Fracturing; United States House of Representatives—Committee on Energy and Commerce: Washington, DC, USA, 2011; p. 29.
- Kahrilas, G.A.; Blotevogel, J.; Stewart, P.S.; Borch, T. Biocides in Hydraulic Fracturing Fluids: A Critical Review of Their Usage, Mobility, Degradation, and Toxicity. Environ. Sci. Technol. 2014, 49, 16–32. [Google Scholar] [CrossRef] [PubMed]
- QGC. Approval Conditions 49 to 52: Stage 2 CSG Water Monitoring and Management Plan; 2nd Revision to 23 April 2012 Submission; 13.0 Well Stimulation; QGC: Brisbane, Australia, 2012. [Google Scholar]
- Madsen, T.; Buchardt Boyd, H.; Nylén, D.; Pedersen, A.R.; Petersen, G.I.; Simonsen, F. Environmental and Health Assessment of Substances in Household Detergents and Cosmetic Detergent Products; Environmental Project No. 615; Danish Environmenal Protection Agency: Copenhagen, Denmark, 2001. [Google Scholar]
- US EPA. OPP Pesticide Ecotoxicity Database. Available online: http://www.ipmcenters.org/ecotox/ (accessed on 23 June 2016).
- US EPA. Registration Eligibility Decision (RED) Bronopol; United States Environmental Protection Agency, Office of Prevention, Pesticides and Toxic Substances (EPA-738-R-95-034), U.S. Government Printing Office: Washington, DC, USA, 1995.
- Swenberg, J.A.; Kerns, W.D.; Mitchell, R.I.; Gralla, E.J.; Pavkov, K.L. Induction of squamous cell carcinomas of the rat nasal cavity by inhalation exposure to formaldehyde vapor. Cancer Res. 1980, 40, 3398–3402. [Google Scholar] [PubMed]
- Dunnett, P.C.; Telling, G.M. Study of the fate of bronopol and the effects of antioxidants on N-nitrosamine formation in shampoos and skin creams. Int. J. Cosmet. Sci. 1984, 6, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Challis, B.C.; Yousaf, T.I. Facile formation of N-nitrosamines from bromonitromethane and secondary amines. J. Chem. Soc. Chem. Commun. 1990, 22, 1598–1599. [Google Scholar] [CrossRef]
- Loeppky, R.N. Nitrosamine and N-Nitroso Compound Chemistry and Biochemistry. In ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1994. [Google Scholar]
- Douglass, M.L.; Kabacoff, B.L.; Anderson, G.A.; Chent, M.C. The chemistry of nitrosamine formation, inhibition and destruction. J. Cosmet. Sci. 1978, 29, 581–606. [Google Scholar]
- Cui, N.; Zhang, X.; Xie, Q.; Want, S.; Chen, J.; Huang, L.; Qiao, X.; Li, X.; Cai, X. Toxicity profile of labile preservative bronopol in water: The role of more persistent and toxic transformation products. Environ. Pollut. 2011, 159, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Scow, K.M.; Johnson, C.R. Effect of sorption on biodegradation of soil pollutants. Adv. Agron. 1997, 58, 1–56. [Google Scholar] [CrossRef]
- Chiou, C.T. Roles of Organic Matter, Minerals, and Moisture in Sorption of Non-Ionic Compounds and Pesticides by Soil. In Humic Substances in Soil and Crop Sciences; Selected Readings; MacCarthy, P., Clapp, C.E., Malcolm, R.L., Bloom, P.R., Eds.; SSSA, ASA: Madison, WI, USA, 1991; pp. 111–160. [Google Scholar]
- Delle Site, A.D. Factors Affecting Sorption of Organic Compounds in Natural Sorbent Water Systems and Sorption Coefficients for Selected Pollutants. A Review. J. Phys. Chem. Ref. Data 2001, 30, 187–436. [Google Scholar] [CrossRef]
- Ringrose-Voase, A.J.; Young, R.R.; Paydar, Z.; Huth, N.I.; Bernardi, A.L.; Cresswell, H.P.; Keating, B.A.; Scott, J.F.; Stauffacher, M.; Banks, R.G.; et al. Deep Drainage under Different Land Uses in the Liverpool Plains Catchment; Report 3, Agricultural Resource Management Report Series; NSW Agriculture: Canberra, Australia, 2003. [Google Scholar]
- Bennett, S. Key Spatiotemporal Factors Determining Uncertainty of Deep Drainage in a Semi-Arid Area. Ph.D. Thesis, The University of New South Wales, New South Wales, Australia, 2012. [Google Scholar]
- ANZECC. Australian and New Zealand Guidelines for Fresh and Marine Water Quality; Volume 1: The Guidelines (Chapters 1–7); Australian and New Zealand Environment and Conservation Council: Canberra, Australia, 2000. [Google Scholar]
- Stearns, M.; Tindall, J.; Cronin, G.; Friedel, M.; Bergquist, E. Effects of coal-bed methane discharge waters on the vegetation and soil ecosystem in Powder River Basin, Wyoming. Water Air Soil Pollut. 2005, 168, 33–57. [Google Scholar] [CrossRef]
- Beletse, Y.G.; Annandale, J.G.; Steyn, J.M.; Hll, I.; Aken, M.E. Can crops be irrigated with sodium bicarbonate rich CBM deep aquifer water? Theoretical and field evaluation. Ecol. Eng. 2008, 33, 26–36. [Google Scholar] [CrossRef]
- Nghiem, L.D.; Ren, T.; Aziz, N.; Porter, I.; Regmi, G. Treatment of coal seam gas co-produced water for beneficial use in Australia: A review of best practices. Desal. Water Treat. 2011, 32, 316–323. [Google Scholar] [CrossRef]
- Mace, J.E.; Amrhein, C. Leaching and reclamation of a soil irrigated with moderate SAR waters. Soil Sci. Soc. Am. J. 2001, 65, 199–204. [Google Scholar] [CrossRef]
- Vance, G.F.; King, L.A.; Ganjegunte, G.K. Soil and plant responses from land application of saline-sodic waters: Implications of management. J. Environ. Qual. 2008, 37, S139–S148. [Google Scholar] [CrossRef] [PubMed]
- Crosbie, R.; Morrow, D.; Cresswell, R.; Leaney, F.; Lamontagne, S.; Lefournour, M. New Insights to the Chemical and Isotopic Composition of Rainfall across Australia; Water for a Healthy Country Flagship Report Series; CSIRO: Canberra, Australia, 2012; ISSN 1835-095X. [Google Scholar]
- Ringrose-Voase, A.J. Water Balance and Deep Drainage under Irrigated Cotton. In WaterPak; Dugdale, H., Harris, G., Neilsen, J., Richards, D., Roth, G., Williams, D., Eds.; Cotton Research and Development Corporation: New South Wales, Australia, 2004; pp. 17–28. [Google Scholar]
- Bureau of Meteorology (BOM). Recent Evapotranspiration. 2014. Available online: http://www.bom.gov.au/watl/eto/ (accessed on 2 June 2015).
- Appelo, C.A.J.; Postma, D. Geochemistry, Groundwater, and Pollution, 2nd ed.; Balkema, A.A., Ed.; CRC Press: Rotterdam, The Netherlands, 2005. [Google Scholar]
- McNeal, B.L. Prediction of the effect of mixed-salt solutions on soil hydraulic conductivity. Soil Sci. Soc. Am. Proc. 1968, 32, 190–193. [Google Scholar] [CrossRef]
- McNeal, B.L. Soil Salts and Their Effects on Water Movement. In Drainage for Agriculture; Agronomy No. 17; van Schilfgaarde, J., Ed.; American Society of Agronomy: Madison, WI, USA, 1974. [Google Scholar]
- Biggs, A.; Witheyman, S.L.; Williams, K.M.; Cupples, N.; de Voil, C.A.; Power, R.E.; Stone, B.J. Assessing the Salinity Impacts of Coal Seam Gas Water on landscapes and Surface Streams. In Final Report of Activity 3 of the Healthy HeadWaters Coal Seam Gas Water Feasibility Study; Department of Natural Resources and Mines: Toowoomba, Australia, 2012. [Google Scholar]
- De Caritat, P.; Lech, M.E. Thomson Region Geochmical Survey, Northwestern New South Wales ; CRC LEME Open File Report 145; CRC LEME: Bentley, WA, USA, 2007. [Google Scholar]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical | Mean R | Min R | Max R | N | Mean T1/2 | Min T1/2 | Max T1/2 | N |
|---|---|---|---|---|---|---|---|---|
| Bronopol (BNP) | 187 | 2 | 1170 | 11 | 87 | 0.1 | 548 | 7 |
| 2-bromo-2-nitroethanol (BNE) | 2 | 2 | 2 | 1 | 28 | 11 | 51 | 5 |
| Bromonitromethane (BNM) | 17 | 14 | 18 | 3 | 18 | 8.7 | 30 | 3 |
| Units | Alkalinity | Cl | SO4 | Ca | K | Mg | Na | ECT | SAR |
|---|---|---|---|---|---|---|---|---|---|
| Untreated produced water | |||||||||
| mg L−1 | 1706 | 593 | 22.8 | 10.4 | 5.6 | 8.9 | 1406 | - | - |
| meq L−1 | 41.9 | 16.7 | 0.47 | 0.52 | 0.14 | 0.74 | 61.1 | - | 77 |
| ECi (µS cm−1) @ | 1245 | 1278 | 38 | 31 | 11 | 39 | 3064 | 5705 | - |
| %ECi @ | 21.8 | 22.4 | 0.7 | 0.5 | 0.2 | 0.7 | 53.7 | 100% | - |
| Amended produced water (3:1 ratio surface water:produced water) | |||||||||
| mg L−1 | 183 | 164 | 337 | 10.3 | 34.8 | 6.5 | 184 | - | - |
| meq L−1 | 4.5 | 4.6 | 7.0 | 0.51 | 0.89 | 0.53 | 8.0 | - | 11 |
| ECi (µS cm−1) @ | 134 | 353 | 561 | 31 | 65 | 28 | 401 | 1573 | - |
| %ECi @ | 8.5 | 22.5 | 35.7 | 1.9 | 4.2 | 1.8 | 25.5 | 100% | - |
| RO-Treated produced water | |||||||||
| mg L−1 | 25 | 67 | 1 | 10.33 | 0.43 | 7.6 | 28.7 | - | - |
| meq L−1 | 0.61 | 1.89 | 0.02 | 0.52 | 0.011 | 0.62 | 1.25 | - | 1.6 |
| ECi (µS cm−1) @ | 18.2 | 144.3 | 1.67 | 30.7 | 0.81 | 33 | 62.5 | 291 | - |
| %ECi @ | 6.3 | 49.6 | 0.6 | 10.5 | 0.3 | 11.3 | 21.5 | 100% | - |
| Treated/rain water* | 417 | 103 | 3 | 43 | 4 | 95 | 60 | ||
| Molar conductivity Ʌi for ions as trace concentration in water at 25 °C | |||||||||
| S cm2 mol−1 | 44.5 | 76.35 | 160 | 119 | 73.5 | 106 | 50.1 | ||
| Ca2+ | Mg2+ | Na+ | K+ | Total Sorption Capacity | Units |
|---|---|---|---|---|---|
| 0.1745 | 0.0891 | 0.0026 | 0.0129 | 0.279 | [eq kg−1] = [mol(+) kg−1] |
| 0.0875 | 0.0445 | 0.0026 | 0.0129 | 0.147 | [mol kg−1] |
| 0.044 | 0.022 | 0.0013 | 0.0065 | 0.074 | [mol kg−1] 50% |
| Cation exchange capacity | 0.184 | (mole charge dm−3 soil) | |||
| Surface complexation capacity | 0.00826 | (mole charge dm−3 soil) | |||
| Aqueous Phase Solution Species | ||
|---|---|---|
| Element | Reaction | logk |
| Pb | Pb2+ + H2O = PbOH++ H+ | −7.71 |
| Pb2+ + 2 H2O = Pb(OH)2 + 2 H+ | −17.12 | |
| Pb2+ + 3 H2O = Pb(OH)3− + 3 H+ | −28.06 | |
| Pb2+ + 4 H2O = Pb(OH)42− + 4 H+ | −39.7 | |
| 2 Pb2+ + H2O = Pb2OH3+ + H+ | −6.36 | |
| Pb2+ + Cl− = PbCl+ | 1.6 | |
| Pb2+ + 2 Cl− = PbCl2 | 1.8 | |
| Pb2+ + 3 Cl− = PbCl3− | 1.7 | |
| Pb2+ + 4 Cl− = PbCl42− | 1.38 | |
| Pb2+ + CO32− = PbCO3 | 7.24 | |
| Pb2+ + 2 CO32−= Pb(CO3)22− | 10.64 | |
| Pb2+ + HCO3− = PbHCO3+ | 2.9 | |
| Pb2+ + SO42−= PbSO4 | 2.75 | |
| Pb2+ + 2 SO42− = Pb(SO4)22− | 3.47 | |
| Pb2+ + NO3− = PbNO3 | 1.17 | |
| Pb2+ + F− = PbF+ | 1.25 | |
| Pb2+ + 2 F− = PbF2 | 2.56 | |
| Cation Exchange | ||
|---|---|---|
| Element | Reaction | logk |
| Cd | Cd2+ + 2X− = CdX2 | 0.8 |
| Cu | Cu2+ + 2X− = CuX2 | 0.6 |
| Pb | Pb2+ + 2X− = PbX2 | 1.05 |
| UO2 | UO22+ + 2X− = UO2X2 | 0.8 |
| Zn | Zn2+ + 2X−= ZnX2 | 0.8 |
| Surface Complexation | ||
|---|---|---|
| Element | Reaction | logk |
| Cd | Hfo_wOH + Cd2+ = Hfo_wOCd++ H+ | −2.91 |
| Cu | Hfo_wOH + Cu2+ = Hfo_wOCu+ + H+ | 0.6 |
| Pb | Hfo_wOH + Pb2+ = Hfo_wOPb+ + H+ | 0.3 |
| UO2 | Hfo_wOH + UO22+ = Hfo_wOUO2+ + H+ | 2.8 |
| Zn | Hfo_wOH + Zn2+ = Hfo_wOZn+ + H+ | −1.99 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mallants, D.; Šimůnek, J.; Genuchten, M.T.v.; Jacques, D. Simulating the Fate and Transport of Coal Seam Gas Chemicals in Variably-Saturated Soils Using HYDRUS. Water 2017, 9, 385. https://doi.org/10.3390/w9060385
Mallants D, Šimůnek J, Genuchten MTv, Jacques D. Simulating the Fate and Transport of Coal Seam Gas Chemicals in Variably-Saturated Soils Using HYDRUS. Water. 2017; 9(6):385. https://doi.org/10.3390/w9060385
Chicago/Turabian StyleMallants, Dirk, Jirka Šimůnek, Martinus Th. van Genuchten, and Diederik Jacques. 2017. "Simulating the Fate and Transport of Coal Seam Gas Chemicals in Variably-Saturated Soils Using HYDRUS" Water 9, no. 6: 385. https://doi.org/10.3390/w9060385
APA StyleMallants, D., Šimůnek, J., Genuchten, M. T. v., & Jacques, D. (2017). Simulating the Fate and Transport of Coal Seam Gas Chemicals in Variably-Saturated Soils Using HYDRUS. Water, 9(6), 385. https://doi.org/10.3390/w9060385