
New Autoantibody Specificities in Systemic Sclerosis and Very Early Systemic Sclerosis

 ,
,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Studies and Samples
2.2. Antigens
2.3. IFN-α Determination in Sera/Plasma
2.4. ELISA for Anti-CXCL4/CXCL4-L1-Autoantibodies Determination in Sera/Plasma
2.5. Statistical Analyses
3. Results
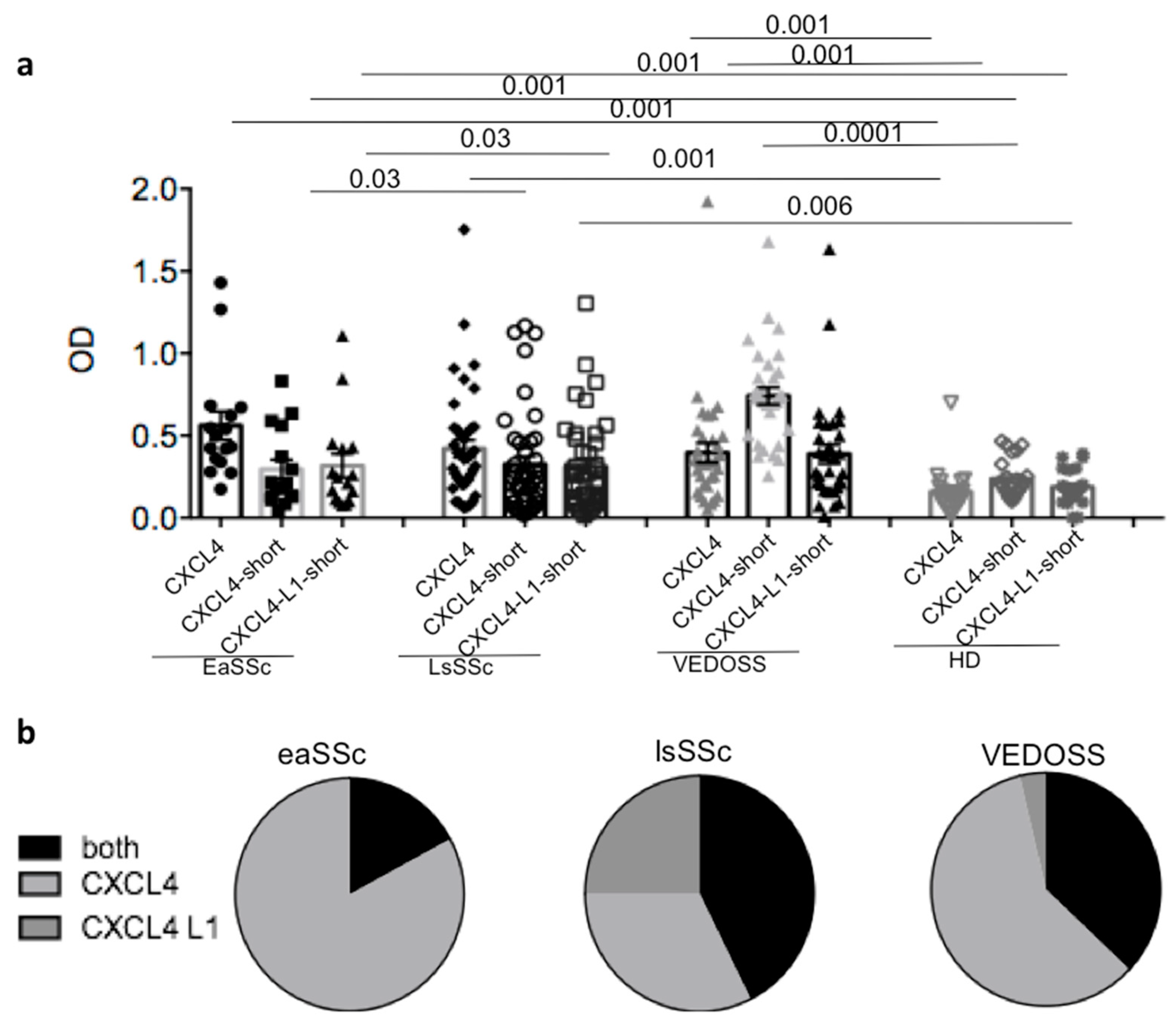
3.1. SSc and VEDOSS Can Share Autoantibody Specificity
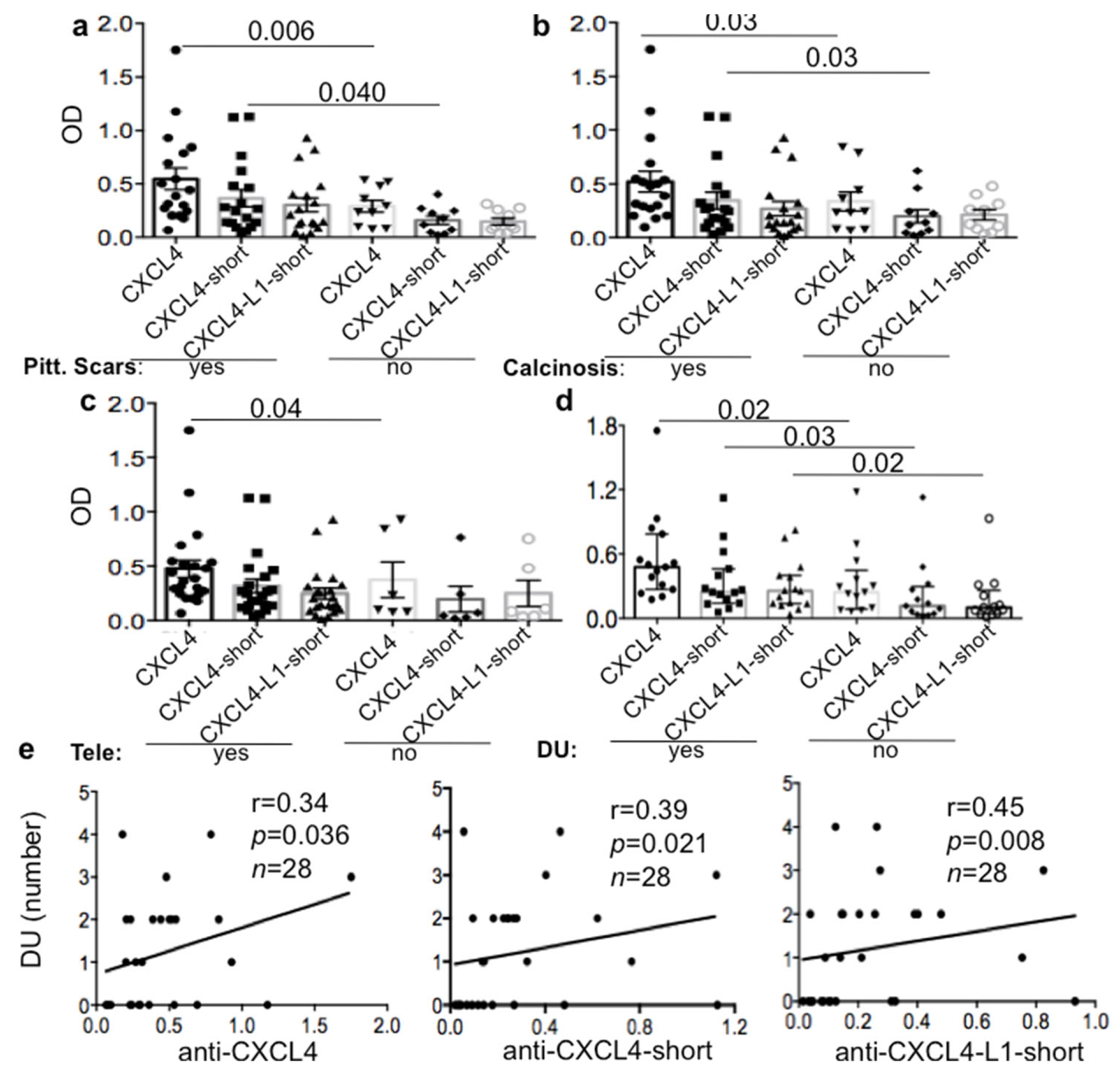
3.2. Anti-CXCL4 Autoantibodies Can Be Associated with Skin Involvement in lsSSc
3.3. Anti-CXCL4/CXCL4-L1 Autoantibodies Correlate with IFN-I in lsSSc but Not in eaSSc/VEDOSS
3.4. IFN-I Expression, but Not Anti-CXCL4/CXCL4-L1 Antibody Reactivity, Differs in VEDOSS SSc-Progressors versus SSc-Non-Progressors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gabrielli, A.; Avvedimento, E.V.; Krieg, T. Scleroderma. N. Engl. J. Med. 2009, 360, 1989–2003. [Google Scholar] [CrossRef]
- Ho, Y.Y.; Lagares, D.; Tager, A.M.; Kapoor, M. Fibrosis: A lethal component of systemic sclerosis. Nat. Rev. Rheumatol. 2014, 10, 390–402. [Google Scholar] [CrossRef]
- Frasca, L.; Lande, R. Toll-like receptors in mediating pathogenesis in systemic sclerosis review. Clin. Exp. Immunol. 2020, 201, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maehara, T.; Kaneko, N.; Perugino, C.A.; Mattoo, H.; Kers, J.; Allard-Chamard, H.; Mahajan, V.S.; Liu, H.; Murphy, J.H.; Ghebremichael, M.; et al. Cytotoxic CD4+ T lymphocytes may induce endothelial cell apoptosis in systemic sclerosis. J. Clin. Investig. 2020, 130, 2451–2464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutolo, M.; Soldano, S.; Smith, V. Pathophysiology of systemic sclerosis: Current understanding and new insights. Expert Rev. Clin. Immunol. 2019, 15, 753–764. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, S. Toll-like receptors in systemic sclerosis: An emerging target. Immunol. Lett. 2018, 195, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Van Bon, L.; Affandi, A.J.; Broen, J.; Christmann, R.B.; Marijnissen, R.J.; Stawski, L.; Farina, G.A.; Stifano, G.; Mathes, A.L.; Cossu, M.; et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N. Engl. J. Med. 2014, 370, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ah Kioon, M.D.; Tripodo, C.; Fernandez, D.; Kirou, K.A.; Spiera, R.F.; Crow, M.K.; Gordon, J.K.; Barrat, F.J. Plasmacytoid dendritic cells promote systemic sclerosis with a key role for TLR8. Sci. Transl. Med. 2018, 423, eaam8458. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Peck, A.; Santer, D.; Patole, P.; Schwartz, S.M.; Molitor, J.A.; Arnett, F.C.; Elkon, K.B. Induction of interferon-alpha by scleroderma sera containing autoantibodies to topoisomerase I: Association of higher interferon-alpha activity with lung fibrosis. Arthritis Rheum. 2008, 58, 2163–2173. [Google Scholar] [CrossRef]
- Eloranta, M.L.; Franck-Larsson, K.; Lövgren, T.; Kalamajski, S.; Rönnblom, A.; Rubin, K.; Alm, G.V.; Rönnblom, L. Type I interferon system activation and association with disease manifestations in systemic sclerosis. Ann. Rheum Dis. 2010, 69, 1396–1402. [Google Scholar] [CrossRef]
- Brkic, Z.; van Bon, L.; Cossu, M.; van Helden-Meeuwsen, C.G.; Vonk, M.C.; Knaapen, H.; van den Berg, W.; Dalm, V.A. The interferon type I signature is present in systemic sclerosis before overt fibrosis and might contribute to its pathogenesis through high BAFF gene expression and high collagen synthesis. Ann. Rheum. Dis. 2016, 75, 1567–1573. [Google Scholar] [CrossRef]
- Lande, R.; Lee, E.Y.; Palazzo, R.; Marinari, B.; Pietraforte, I.; Santos, G.S.; Mattenberger, Y.; Spadaro, F.; Stefanantoni, K.; Iannace, N.; et al. CXCL4 assembles DNA into liquid crystalline complexes to amplify TLR9-mediated interferon-alpha production in systemic sclerosis. Nat. Commun. 2019, 10, 1731–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persson, E.K.; Verstraete, K.; Heyndrickx, I.; Gevaert, E.; Aegerter, H.; Percier, J.M.; Deswarte, K.; Verschueren, K.H.G.; Dansercoer, A.; Gras, D.; et al. Protein crystallization promotes type 2 immunity and is reversible by antibody treatment. Science 2019, 364, eaaw4295. [Google Scholar] [CrossRef]
- Arepally, G.M.; Cines, D.B. Pathogenesis of heparin-induced thrombocytopenia. Transl. Res. 2020, 225, 131–140. [Google Scholar] [CrossRef]
- Lande, R.; Mennella, A.; Palazzo, R.; Pietraforte, I.; Stefanantoni, K.; Iannace, N.; Butera, A.; Boirivant, M.; Pica, R.; Conrad, C.; et al. Anti-CXCL4 antibody reactivity is present in Systemic Sclerosis (SSc) and correlates with the SSc Type I Interferon signature. Int. J. Mol. Sci. 2020, 21, 5102. [Google Scholar] [CrossRef]
- Vandercappellen, J.; Van Damme, J.; Struyf, S. The role of the CXC chemokines platelet factor-4 (CXCL4/PF-4) and its variant (CXCL4L1/PF-4var) in inflammation, angiogenesis and cancer. Cytokine Growth Factor Rev. 2011, 22, 1–18. [Google Scholar] [CrossRef] [PubMed]
- von Hundelshausen, P.; Petersen, F.; Brandt, E. Platelet-derived chemokines in vascular biology. Thromb. Haemost. 2007, 97, 704–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsouras, M.D.; Sikara, M.P.; Grika, E.P.; Moutsopoulos, H.M.; Tzioufas, A.G.; Vlachoyiannopoulos, P.G. Elevated expression of platelet-derived chemokines in patients with antiphospholipid syndrome. J. Autoimmun. 2015, 65, 30–37. [Google Scholar] [CrossRef]
- Baroni, G.; Banzato, A.; Bison, E.; Denas, G.; Zoppellaro, G.; Pengo, V. The role of platelets in antiphospholipid syndrome. Platelets 2017, 28, 762–766. [Google Scholar] [CrossRef]
- Ramirez, G.A.; Franchini, S.; Rovere-Querini, P.; Sabbadini, M.G.; Manfredi, A.A.; Maugeri, N. The role of platelets in the pathogenesis of systemic sclerosis. Front. Immunol. 2012, 3, 160. [Google Scholar] [CrossRef] [Green Version]
- Bournia, V.; Patsouras, M.; Vlachoyiannis, N.; Tzioufas, A.; Sfikakis, P.; Vlachoyiannopoulos, P. CXCL4-L1 Levels Are Elevated in Systemic Sclerosis Patients and Correlate with Pulmonary Arterial Hypertension and Capillaroscopic Indices of Vascular Damage. Available online: https://acrabstracts.org/abstract/cxcl4-l1-levels-are-elevated-in-systemic-sclerosis-patients-and-correlate-with-pulmonary-arterial-hypertension-and-capillaroscopic-indices-of-vascular-damage/ (accessed on 26 March 2021).
- Bellando-Randone, S.; Matucci-Cerinic, M. From Raynaud’s Phenomenon to Very Early Diagnosis of Systemic Sclerosis—The VEDOSS approach. Curr. Rheumatol. Rev. 2013, 9, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Valentini, G.; Pope, J.E. Undifferentiated connective tissue disease at risk for systemic sclerosis: Which patients might be labeled prescleroderma? Autoimmun. Rev. 2020, 19, 10265–10275. [Google Scholar] [CrossRef]
- Melsens, K.F.; De Keyser, S.; Decuman, Y.; Piette, E.; Vandecasteele, E.; Smith, V. Disease activity indices in systemic sclerosis: A systematic literature review. Clin. Exp. Rheumatol. 2016, 34 (Suppl. 100), 186–192. [Google Scholar]
- Hagedorn, M.; Zilberberg, L.; Lozano, R.M.; Cuevas, P.; Canron, X.; Redondo-Horcajo, M.; Gimenez-Gallego, G.; Bikfalvi, A. A short peptide domain of platelet factor 4 blocks angiogenic key events induced by FGF-2. FASEB J. 2001, 15, 550–552. [Google Scholar] [CrossRef]
- Valenzuela, A.; Song, P.; Chung, L. Calcinosis in scleroderma. Curr. Opin. Rheumatol. 2018, 30, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Herrick, A.L.; Shukla, R.; Watson, R.E.B. Frontiers in translational systemic sclerosis research: A focus on the unmet ’cutaneous’ clinical needs (Viewpoint). Exp. Dermatol. 2020, 29, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Barsotti, S.; Venturini, V.; Di Battista, M.; Janowska, A.; Dini, V.; Della Rossa, A.; Mosca, M. The impact of skin calcinosis on digital ulcers in patients with SSc: Clinical and prognostic stratification using the “wound bed score”. Int. Wound J. 2020, 17, 1783–1790. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.-H.; Chen, Y.-P.; Liu, J.-S.; Dubrac, A.; Quemener, C.; Prats, H.; Bikfalvi, A.; Wu, W.-G.; Sue, S.-C. Alternative C-terminal helix orientation alters chemokine function: Structure of the anti-angiogenic chemokine, CXCL4L1. J. Biol. Chem. 2013, 288, 13522–13533. [Google Scholar] [CrossRef] [Green Version]
- Valentini, G.; Riccardi, A.; Vettori, S.; Irace, R.; Iudici, M.; Tolone, S.; Docimo, L.; Bocchino, M. CXCL4 in undifferentiated connective tissue disease at risk for systemic sclerosis (SSc) (previously referred to as very early SSc). Clin. Exp. Med. 2017, 17, 411–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skaug, B.; Assassi, S. Type I interferon dysregulation in Systemic Sclerosis. Cytokine 2020, 132, 154635. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Main Clinical, Demographic and Laboratory Parameters | SSc (n = 42) | VEDOSS1 (n = 31) (Discovery Cohort) | VEDOSS2 (n = 48) (Replication Cohort) | p Values VEDOSS1 vs. VEDOSS2 | HD (n = 25) |
|---|---|---|---|---|---|
| Age, mean (range): years | 52.5 (32–71) | 50 (26–61) | 47 (25–70) | ns | 48 (29–57) |
| Sex (M/F): | 1/41 | 0/31 | 2/46 | ns | 10/15 |
| Disease duration from 1st visit (months)(range) | 74.4 (12–252) | 135.6 (36–504) | 120 (36–500) | ns | N/A |
| SSc Form (limited/diffuse) | 1/41 | N/A | N/A | N/A | N/A |
| Ea lim/ea diffuse | 0/14 | N/A | N/A | N/A | N/A |
| mRSS (mean, range) | 16.6 (6–36) | N/A | N/A | N/A | N/A |
| ACA positivity | 5% | 64% | 61% | ns | N/A |
| ATA positivity | 71% | 23% | 5% | p = 0.04 | N/A |
| aRNAP3 positivity | 14% | 3% | - | N/A | N/A |
| Calcinosis | 50% | 0% | 2.5% | ns | N/A |
| Pitting scars | 60% | 0% | - | N/A | N/A |
| Raynaud Phenomenon | 93% | 100% | 97% | ns | N/A |
| DU | 50% | 0% | 0% | ns | N/A |
| Teleangectasia | 71% | 0% | 5% | ns | N/A |
| Pulm Art. Hypertension | 25% | 0% | 8% | ns | N/A |
| Lung fibrosis (%) | 33% | 0% | 0% | ns | N/A |
| DLCO (%) (mean) | 65.7% | 85.6% | 85.5% | ns | N/A |
| DLCO < 80% | 88% | 38% | 26% | p = 0.001 | N/A |
| Gastroint. Involv. | 0% | 0% | 0% | ns | N/A |
| Synovitis | 0% | 0% | 0% | ns | N/A |
| Sclerodactilia | 69% | - | - | N/A | N/A |
| DMARDs | 99% | 16% | 18% | ns | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lande, R.; Palazzo, R.; Mennella, A.; Pietraforte, I.; Cadar, M.; Stefanantoni, K.; Conrad, C.; Riccieri, V.; Frasca, L. New Autoantibody Specificities in Systemic Sclerosis and Very Early Systemic Sclerosis. Antibodies 2021, 10, 12. https://doi.org/10.3390/antib10020012
Lande R, Palazzo R, Mennella A, Pietraforte I, Cadar M, Stefanantoni K, Conrad C, Riccieri V, Frasca L. New Autoantibody Specificities in Systemic Sclerosis and Very Early Systemic Sclerosis. Antibodies. 2021; 10(2):12. https://doi.org/10.3390/antib10020012
Chicago/Turabian StyleLande, Roberto, Raffaella Palazzo, Anna Mennella, Immacolata Pietraforte, Marius Cadar, Katia Stefanantoni, Curdin Conrad, Valeria Riccieri, and Loredana Frasca. 2021. "New Autoantibody Specificities in Systemic Sclerosis and Very Early Systemic Sclerosis" Antibodies 10, no. 2: 12. https://doi.org/10.3390/antib10020012
APA StyleLande, R., Palazzo, R., Mennella, A., Pietraforte, I., Cadar, M., Stefanantoni, K., Conrad, C., Riccieri, V., & Frasca, L. (2021). New Autoantibody Specificities in Systemic Sclerosis and Very Early Systemic Sclerosis. Antibodies, 10(2), 12. https://doi.org/10.3390/antib10020012