1. Introduction
With the growing interest in monoclonal antibodies (mAbs) for therapeutic applications, advances in antibody production have improved drastically over the last decades. Due to the more complex structure of antibodies, their production requires host cells capable of natively folding and modifying the mAb. Modifications include post-translational glycosylation, which is, among other functional properties, critical to reduce their immunogenicity [
1]. For this purpose, mammalian cells fulfil the requirements as appropriate hosts for antibody production [
2,
3]. Advances in transfection protocols and cell engineering have boosted the use of suspension cell lines with the ability to grow at high densities, and increased production yields [
4,
5]. Further within the drug discovery and development process, stable cell lines are generated for the most promising candidate(s), while transient transfection is performed at earlier stages to yield research quantities of mAbs, sufficient for characterization of lead candidates. The accessibility of commercially available transfection reagents with high efficiencies and the use of disposable materials makes transient expression an efficient and cost-effective strategy during early drug discovery [
6]. Human Embryonic Kidney 293 (HEK293) and Chinese Hamster Ovary (CHO) cells are commonly used for transient antibody expression, due to their high expression yields and human-like glycosylation patterns [
1,
7,
8].
The basis of successful antibody production is the correct folding of individual chains, followed by their accurate assembly, resulting in functional heterotetrameric glycoproteins. Misfolded or partially folded antibodies are degraded by the host cell’s intrinsic quality control system, resulting in low production yields. Furthermore, antibodies with undesired folding are not able to effectively engage their target antigen or to mediate effector functions, have unfavourable pharmacokinetics, and tend to aggregate. Besides these biological limitations, purification of antibody products contaminated with aggregated or misfolded mAbs is a major hindrance in the downstream processing of therapeutic molecules and is currently the topic of numerous studies [
9,
10,
11,
12].
Antibody folding starts upon co-translational translocation into the endoplasmic reticulum (ER) [
13]. Following the homodimerization of the two heavy chains (HC), the light chains (LC) are associated and covalently linked via disulphide bonds [
14]. The glycosylation at Asn297 is linked to the CH2 backbone in a co-translational manner [
15]. During translation, chaperones are involved to ensure the correct folding of the individual domains, as well as the final assembly of the tetrameric mAb [
16].
At present, the largest part of transient production of mAbs is carried out using a two-plasmid system, also known as co-transfection, for the expression of
lc and
hc, with each gene driven by its own promoter and transcribed separately [
17,
18]. These are carried out, for the most part, with an equimolar ratio of heavy chain and light chain genes. Nonetheless, contradictory results have been reported using Expi293-F cells. While some publications report that an equimolar gene ratio results in the highest yield of fully assembled IgGs [
8], others have described optimal expression with a 1:2 ratio of heavy and light chain genes, respectively [
19].
A large drawback of the two-plasmid system is the moderate control over the relative expression of
lc and
hc, with fluctuating cell-to-cell transfection efficiencies [
17]. On the other hand, a bidirectional (BiDi) vector with a dual-promoter can ensure the introduction of both
hc and
lc genes into each cell in equal amounts. However, for some applications it could be beneficial to have a stronger expression of one chain over the other. By choosing two suitable promoters controlling the transcription of
hc and
lc, different ratios can be achieved [
20]. While diverse approaches have been developed throughout the years to advance and facilitate recombinant antibody production, the design of expression vectors plays a large role for optimization of expression yields. Recently, Bayat and colleagues (2018) compared the use of three different vector design strategies for the expression of IgG1 antibodies in CHO cells, namely using the conventional two-vector approach with
hc and
lc encoded separately, a bicistronic vector based on internal ribosome entry sites (IRES), and a dual-promoter single vector approach. All vectors were under the control of a human cytomegalovirus (CMV) promoter. Expression analysis revealed that the dual-promoter vector system resulted in the highest mAb yield [
21].
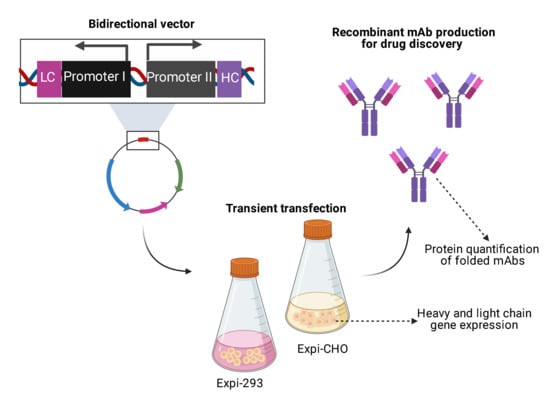
Andersen and co-workers have previously shown the ability of the CMV enhancer to control two core CMV promoters simultaneously, resulting in efficient antibody expression [
22]. With this basis, we sought to investigate different promoters in a bidirectional format to facilitate transient transfection, avoiding co-transfections. We also sought to simplify
hc and
lc gene cloning by establishing a one-step Golden Gate cloning procedure that relies on the simultaneous plasmid incorporation of
hc and
lc genes together with the bidirectional promoter sequence. By analysing
hc and
lc gene expression, as well as through subsequent quantification of fully-folded IgGs, promoter and enhancer element combinations were compared. Here, we show the use of a dual-promoter, single plasmid approach using divergent promoters for the transient expression of two FDA-approved antibodies, Durvalumab and Avelumab, using both Expi293-F and ExpiCHO-S cell systems. This work lays the foundation to facilitate small-scale mAb production in drug discovery programs in a more efficient manner.
2. Materials and Methods
2.1. Plasmids & Cloning of Constructs
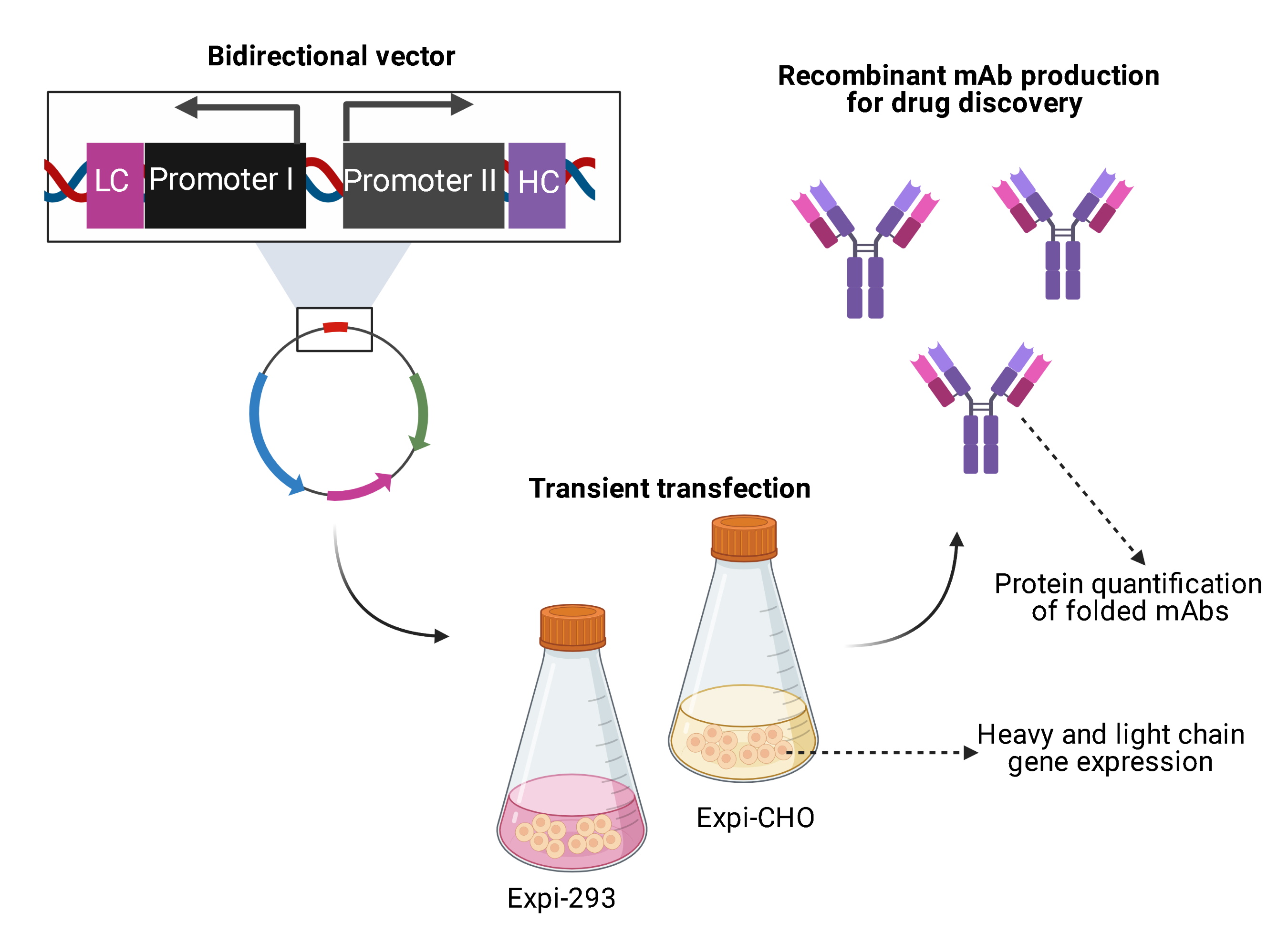
To allow the individual exchange of different variable domains, the utilization of κ and λ isotypes, as well as the usage of different BiDi promoter combinations, the backbone of the mammalian destination (MD) vector was built in a cassette-like manner. For vector amplification in
E. coli, a chloramphenicol resistance was utilized, adjacent to the colE1 and the f1 origins. A stuffer sequence, flanked by
Esp3I restriction sites, was downstream of an inverse-orientated SV40 polyA sequence that was intended to be a terminator signal for the light chain cassette. Upstream of the stuffer sequence, a partial hinge followed by the CH2 and CH3 domains of a human IgG1 were encoded. Again, a SV40 polyA signal sequence served as a terminator signal (
Figure 1A). The plasmid was de novo designed in silico and ordered at GeneArt (Regensburg, Germany).
The selected promoter sequences were either ordered as gene strings at Twist Bioscience (EF-1α, minCMV-enh-CMV (GenBank: MK764037) or PCR-amplified from the pTT5 CMV promoter cassette between bases 42–1185 (hereinafter referred to as eCMV) [
23]. To allow for the correct orientation of the promoter sequences, individual primers were used to introduce the respective
BbsI Golden Gate cloning (GGC) signature overhangs. Genes for VH-CH1 and VL-CL of Durvalumab (Imfinzi, κ light chain) and Avelumab (Bavencio, λ light chain) were also ordered as gene strings, already bearing suitable signature sequences for
Esp3I and
BbsI as well as their respective leader sequences. The 200-bp stuffer used between individual promoters consists of the non-functional 3′ coding region of the amp resistance gene for beta-lactamase, followed by ~40 bp of non-coding DNA. Assembly of the MD expression constructs was conducted with 75 ng destination vector and equimolar amounts of the respective fragments, 20 U
BbsI-HF, 10 U
Esp3I, and 200 U T4-DNA ligase (NEB, Frankfurt, Germany) for 30 cycles (1 min; 16 °C; 37 °C). For the reference constructs, VH and VL genes were amplified incorporating
SapI restriction sites and then inserted into a pTT5-derived vector utilizing CH1-CH2-CH3 or κ/λ entry vectors using GGC as described before [
24,
25]. PCR reactions were performed utilizing Q5 polymerase (NEB) according to the manufacturer’s protocol and purified using the Wizard SV Gel and PCR Clean-up System (Promega, Walldorf, Germany). All primers can be found in
Table S1. The DNA sequence for the 2xeCMV BiDi construct can be found in
Sequence S1 in the Supplementary Information.
E. coli XL1-blue were transformed utilizing the Golden Gate reaction mixtures and cultivated on chloramphenicol or ampicillin DYT agar plates for MD or pTT5 constructs, respectively. Resulting colonies were sequenced at MicroSynth SeqLab (Göttingen, Germany), and positive clones were utilized to inoculate 50 mL overnight cultures. Plasmid DNA for transient transfection was isolated using the PureYield Plasmid Midiprep System (Promega, Walldorf, Germany).
2.2. Cell Lines
Expi293-F and ExpiCHO-S cells were obtained from Thermo Fisher Scientific. Cells were incubated at 37 °C, 8% CO2, 110 rpm, and sub-passaged every 3–4 days in their respective expression media, as described in the manufacturer’s protocol (Thermo Fisher Scientific, Schwerte, Germany). Cell count and viability were measured using an automated cell counter (Bio-Rad TC-20) based on trypan blue staining. Cell densities were maintained between 0.3–4 × 106 cells/mL and 0.2–6 × 106 cells/mL for Expi293-F and ExpiCHO-S, respectively.
2.3. 24-Well Transfection
For gene expression and protein quantification, small-scale transfections using Axygen 24-well deep-well plates (Corning, New York, NY, USA) were performed. One day prior to transfection, cells were seeded into wells at a final cell density of 1.8 × 106 or 3 × 106 viable cells/mL in 2.5 mL expression medium for Expi293-F or ExpiCHO-S, respectively, and incubated under shaking conditions in a humified atmosphere at 37 °C, 8% CO2, 225 rpm. The following day, the cell density was adjusted to 3 × 106 or 6 × 106 viable cells/mL in 2.5 mL expression medium for Expi293-F or ExpiCHO-S, respectively. DNA:Expifectamine complexes were incubated at room temperature with either 3 µg BiDi plasmid or 2 µg heavy and 2 µg light chain plasmid for co-transfections (two-plasmid reference) for 20 or 1 min for Expi293-F or ExpiCHO-S, respectively, before adding dropwise to the cells. Feeding procedures were carried out according to manufacturer’s instructions. For gene expression analysis, cells were harvested 3 days post-transfection, while protein quantification was carried out 6 days post-transfection.
2.4. RNA Isolation
Three days post-transfection, Expi293-F or ExpiCHO-S cells were harvested by centrifugation and cell pellets were processed through a QIAshredder column (QIAGEN, Hilden, Germany). Total RNA extraction was carried out using RNeasy Mini Kit (QIAGEN) following the manufacturer’s instructions. RNA concentration was determined spectroscopically using a NanoDrop One (Thermo Fisher), ensuring pure RNA was isolated with a A260/280 ratio of 2.0.
2.5. Gene Expression Analysis by Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
Expression levels of heavy and light chain (both κ and λ) were analysed using 100 ng RNA per well in Hard-Shell
® 96-well PCR plates (Bio-Rad, Hercules, CA, USA) and iTaq Universal SYBR Green One-step Kit (Bio-Rad) with designed SYBR Green primers (Sigma Aldrich, Munich, Germany) using a CFX96 qPCR instrument (Bio-Rad). Relative expression levels were analysed using the integrated software from Bio-Rad (CFX Manager, Hercules, CA, USA) and normalized to housekeeping genes GAPDH and RPLP0 (IDT, Coralville, IA, USA). The primers used can be found in
Table S2.
2.6. Protein Purification
To purify the antibodies from small-scale transfections, cells were harvested by centrifugation and cell culture supernatants were purified using Protein A HP SpinTrap columns (Cytiva, Freiburg im Breisgau, Germany) following the manufacturer’s protocol. Antibodies were eluted in 0.1 M glycine-HCl, pH 2.7. Protein concentration was determined using a NanoDrop One (Thermo Fisher) using the corresponding molecular weights and extinction coefficients.
2.7. Protein Quantification and Affinity Determination Using Biolayer Interferometry (BLI)
Six days post-transfection, cells were harvested by centrifugation and the cell culture supernatants were sterile-filtered (0.45 µm). BLI experiments were performed on an Octet Red96 (FortéBio, Fremont, CA, USA). Using Protein A biosensors (Sartorius, Göttingen, Germany) for quantification, mAb concentration was measured from the cell culture supernatants. An in-house produced mAb was used as a standard within the range of 3.13–400 µg/mL.
For affinity determination, anti-human Fab-CH1 2nd generation (FAB2G) biosensors (Sartorius) were used. Purified antibodies were loaded onto the tips at 10 µg/mL until a layer thickness of 1 nm was reached. Association was measured using a serial dilution of His-PD-L1-TwinStrep (produced in-house). Kinetics were determined using Savitzky-Golay filtering and a 1:1 Langmuir binding model.
3. Results
To produce full-length antibodies in a bidirectional manner, we first designed the respective vector in silico. This vector encoded the fragment crystallizable (Fc) region of an IgG1, which is the most common isotype found in therapeutic antibodies [
26]. In order to allow the flexible use for a variety of binders, the fragment antigen binding (Fab), which can be of the κ or λ isotype, was not encoded on the plasmid. Instead, a stuffer sequence, flanked by
Esp3I sites was incorporated (
Figure 1A).
As a reference antibody to establish different promoter combinations, the Fab of Durvalumab, an FDA-approved anti-PD-L1 antibody of the κ type, was chosen [
27]. PCR amplicons of VH-CH1 and VL-CL were generated introducing
Esp3I and
BbsI restriction sites. By utilization of a BiDi promoter system, flanked by
BbsI sites, a Golden Gate reaction resulted in a re-circularized vector (
Figure 1B,C). In this process, the stuffer that was included in the parental MD vector was replaced by a CL-VL-PromoterI-Stuffer-PromoterII-VH-CH1 sequence. Owing to this cloning strategy, a straightforward exchange of different Fabs and different BiDi promoters is feasible. The resulting vector exhibited the functional ORFs for the heavy and the light chains, resulting in the production of full-length antibodies (
Figure 1D).
3.1. Cloning Promoter Combinations
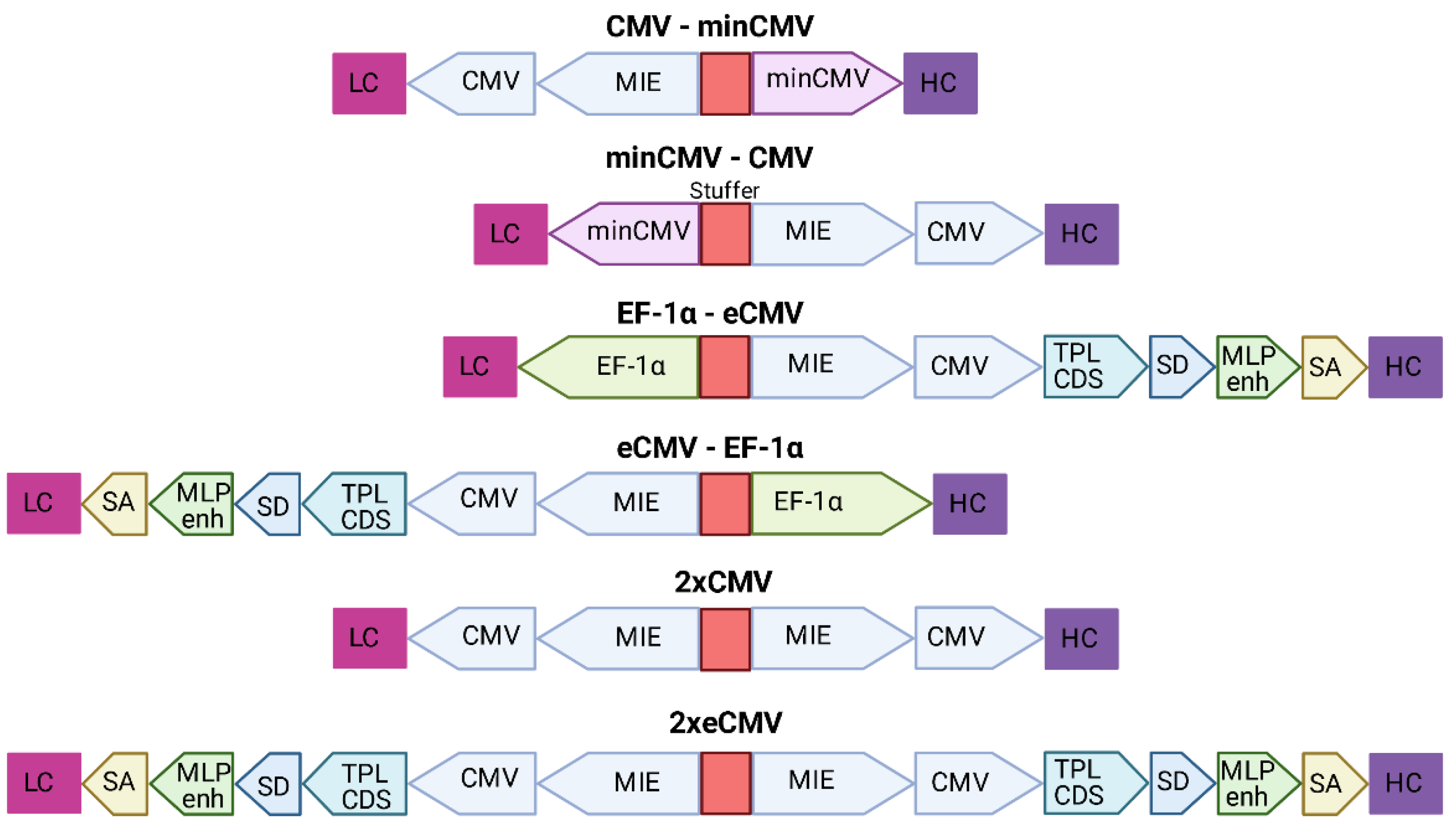
Based on the findings from Andersen et al. [
22] that a single enhancer adjacent to a bidirectional CMV promoter allows for efficient antibody production using a single expression plasmid, six different bidirectional promoter complexes were generated comprising not only the minCMV-CMV cassette, but also combinations of other strong promoters including their individual enhancer elements, such as the CMV with its major immediate early enhancer (MIE), the optimized CMV cassette from pTT5 (denoted as eCMV in this study), and the human translation elongation factor 1 alpha (EF-1α) promoter, that were selected based on their capability to produce fully-folded IgG molecules.
The MIE-CMV promoter, being one of the most commonly used in mammalian expression vectors, is designated as a strong driver for recombinant protein expression [
28,
29]. As shown before by Andersen and colleagues [
22], the MIE enhancer is also capable of facilitating elevated expression levels in the divergently oriented minCMV promoter, although to a lesser extent. This was based on the presumption that the formation of the large transcription complex might be sterically hindered. As a reference, we selected a similar bidirectional promoter setup lacking the unique sequence upstream of the enhancer. Additionally, we went for a mirror-symmetric approach comprised of two individual CMV promoters, each having adjacent MIE enhancers that were separated by a 200-bp-stuffer. The eCMV cassette comprises—besides the MIE enhancer and core promoter—several additional regulatory elements that have been described to increase expression levels. These elements include the non-coding adenoviral tripartite leader sequence (TPL), the adenovirus major late promoter enhancer (MLP enh.), as well as distinct splicing sites allowing for prolonged mRNA stability [
30,
31]. Furthermore, we also utilized the strong human translation elongation factor 1 alpha promoter (EF-1α) that has proven to be advantageous over the CMV promoter in some cell types and in the expression of distinct proteins of interests [
32,
33].
Based on the modular setup of our MD vector and utilization of GGC, we generated different combinations of the aforementioned promoters to analyse for highest full-length antibody expression levels and product yield of Durvalumab. A schematic overview of the BiDi combinations is depicted in
Figure 2.
3.2. Gene Expression Analysis in Mammalian Cells
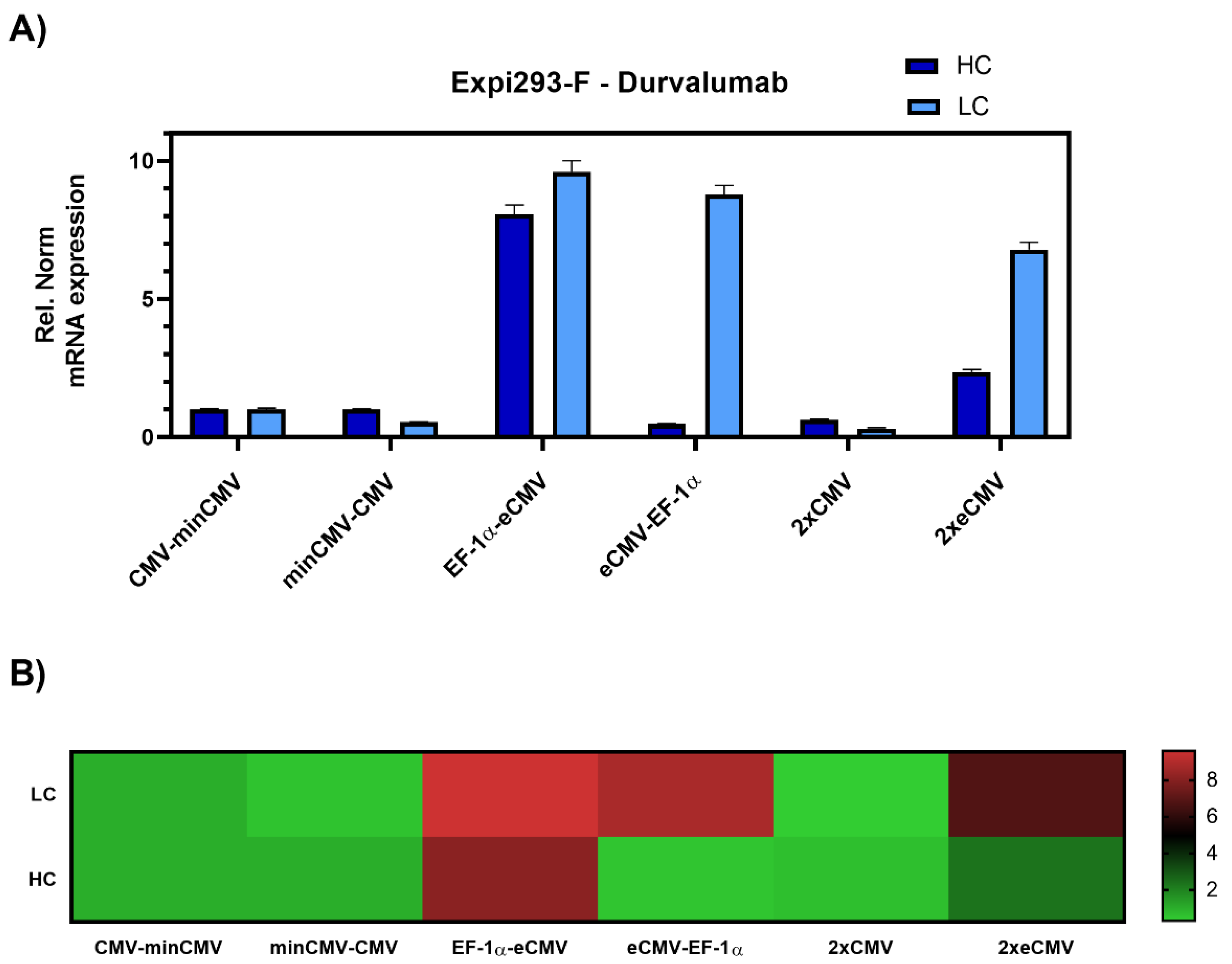
Transient transfections of all six promoter combinations were performed in 24-well plates using Expi293-F cells, an established cell line for transient antibody production. Three days post-transfection, cells were harvested, and RNA was isolated. Relative gene expression of
hc and
lc mRNA levels was measured by RT-qPCR (
Figure 3).
The data was set relative to the CMV-minCMV construct and normalised to housekeeping genes. Looking at relative mRNA levels, both variants with minCMV and CMV, independent of the promoter orientation, did not yield high mRNA expression for either lc or hc. Remarkably, the combinations with the EF-1α promoter and the enhanced CMV cassette (eCMV) showed significant differences in expression levels depending on their orientation. Steering lc expression with the EF-1α promoter (EF-1α-eCMV) resulted in a 9.5-fold upregulation in lc and 8-fold upregulation in hc mRNA levels. Conversely, having the more potent eCMV in the light chain direction and EF-1α in the heavy chain direction (eCMV-EF-1α) led to a 9-fold lc upregulation, and low relative hc expression levels. Based on these results, mirrored constructs containing both promoter and enhancer cassettes in both directions were tested, one with the traditional CMV promoter (2xCMV), and the other with the eCMV cassette (2xeCMV). Interestingly, the 2xCMV construct did not result in increased lc or hc mRNA levels compared to the bidirectional construct containing two identical eCMV promoters or constructs containing two different promoters. The BiDi combination of two mirrored eCMV promoter cassettes (2xeCMV) yielded in a 7-fold upregulation of lc mRNA and a modest upregulation in hc levels.
As promoter strength may vary depending on the cell line, particularly for the CMV promoter [
33], ExpiCHO-S cells were also investigated. Gene expression analysis resulted in similar results as in Expi293-F cells, with the 2xeCMV complex showing the strongest upregulation in both
hc and
lc mRNA levels compared to the other promoter combination, namely a modest 10-fold upregulation of
hc, and a 20-fold upregulation of
lc mRNA (
Figure 4).
3.3. Protein Yield Determination via BLI
As mRNA transcript levels do not indicate successful secretion of fully functional recombinant antibodies, protein quantification studies were performed. Expi293-F cells were transiently transfected and the amount of secreted therapeutic antibody Durvalumab [
27] was quantified by biolayer interferometry (BLI) using sterile-filtered cell culture supernatants six days post-transfection. The mAb concentrations for the different constructs were interpolated from a standard curve generated using an in-house produced mAb. In line with the gene expression analysis, protein quantification resulted in a clear ranking of the different promoter combinations (
Figure 5).
From the bidirectional combinations tested with transient transfections, the 2xeCMV BiDi construct showed the highest mAb concentration with 353 µg/mL (
Figure 5). Interestingly, mRNA expression was higher for EF-1α-eCMV compared to 2xeCMV for both
hc and
lc, but antibody yield was significantly reduced in Expi293-F cells (
Figure 3). This may likely be due to the fact that EF-1α-eCMV displayed similar
hc and
lc mRNA expression patterns, while 2xeCMV revealed a higher accumulation of the
lc compared to the
hc mRNA. It is well known that the expression of excess light chain over heavy chain is often beneficial for antibody production [
34,
35,
36,
37]. These findings corroborate that 2xeCMV resulted in the most promising bidirectional promoter combination, particularly in view of the fact that the usage of this promoter combination resulted, both in Expi293-F and ExpiCHO-S cell lines, in significantly enhanced mRNA synthesis with an excess of
lc over
hc.
3.4. Correlation of mRNA and Protein Levels
After performing both mRNA and protein studies, the correlation of the cycle threshold (Ct) values from RT-qPCR and the mAb concentrations from protein quantification in Expi293-F was determined (
Figure 6A). Overall, a Pearson’s coefficient of −0.6407 was calculated, indicating, as expected, a negative correlation between Ct values and mAb concentration. Looking at the respective Ct values of either the
hc or
lc for 2xeCMV, one can observe they have a low Ct value and resulted in the largest mAb yield. While some claim that abundance of
hc may hinder productivity, it appears that the excess of
lc allowing for correct mAb folding and assembly is sufficient for higher mAb yields. On the contrary, the Ct values of eCMV-EF-1α
lc is similar to that of 2xeCMV, with the only difference being in the
hc expression, ultimately resulting in much lower yields. Thus, this further shows the importance of a fine-tuned
hc and
lc expression, substantiating the potential of 2xeCMV for a BiDi antibody production system.
3.5. Antibody—And Light Chain-Independence
To ensure the promoter combination used for our BiDi technology was not antibody- or light chain isotype-dependent, the FDA-approved anti-PD-L1 antibody Avelumab of the λ isotype was also tested [
38]. As 70% of approved antibodies belong to the κ type [
39,
40], only the most promising 2xeCMV construct was used to validate the established system for a λ-based mAb, compared to the conventional co-transfection approach. For co-transfection, Expi293-F cells were transfected using a 1:1 ratio of HC and LC DNA, with each plasmid carrying the same promoter and enhancer cassette as the bidirectional vector, namely eCMV. As can be appreciated from
Figure 6B, there was no significant variation in antibody yield between transfections with BiDi and the two-plasmid reference for either Durvalumab or Avelumab. Similarly, no variation was observed in ExpiCHO-S production (data not shown). Kinetics determination of both antibodies produced with either the two-plasmid reference or our established 2xeCMV BiDi plasmid bound to its target PD-L1 with comparable affinities (
Table 1,
Figures S1 and S2). Additionally, SDS-PAGE analysis under reducing conditions resulted in the expected heavy and light chains bands (
Figure S3).
Thus, this indicated the established system is compatible with different binders and can be employed for both κ- and λ-light chain isotypes.
4. Discussion
This work describes the generation of a bidirectional vector construct for recombinant antibody expression using two eCMV promoter cassettes, controlling the expression of lc and hc individually in each direction. By performing a thorough analysis of different bidirectional promoter combinations with varying lengths and strengths, the 2xeCMV combination showed the most promising hc and lc mRNA synthesis in two regularly used mammalian cell lines, and, more importantly, the highest yields after protein quantification comparable to those using the conventional two-plasmid system. Other BiDi constructs showed potentially promising gene expression profiles, such as EF-1α-eCMV combination, with high relative mRNA levels of both hc and lc mRNA. Nonetheless, having higher levels of hc mRNA in the cells appear to curb productivity of fully folded IgG formation, resulting in drastically decreased antibody yields compared to both the two-plasmid reference and the 2xeCMV BiDi vector.
While using a bidirectional approach takes away some flexibility in terms of being able to alter the
hc:lc ratios during co-transfection, the overexpression of both genes and, especially the excess
lc expression, results in sufficient material for antibody hit screening. For convenience, we established a one-step cloning procedure for simultaneous plasmid incorporation of the heavy and light chain encoding segments obviating the need for generating two separate plasmids. Not only does this approach lower plasmid preparation efforts, but it also increases handling for transfection of numerous mAbs during screening and characterization as only a single plasmid is required. The use of two FDA-approved antibodies with either a κ- or λ-light chain shows there is no antibody- or light chain-dependence using this system, indicating that it can be implemented ubiquitously. Further options remain to increase the yields of IgG molecules, such as optimization of the stuffer region between the two eCMV promoter cassettes, potentially reducing any steric hinderance and increasing transcription efficiency [
22,
41].
In conclusion, this work displays the benefits of using a one-plasmid bidirectional system with 2xeCMV promoters for fully folded IgGs within drug discovery. In terms of practicality, handling of a single plasmid for antibody production may be superior to the conventional way. Moreover, yields of fully folded IgGs are comparable between the two systems. Future directions for this technology go beyond recombinant production of classical antibody formats, as reduction of the number of plasmid constructs could also be considered feasible for the expression of bispecific antibodies and other antibody formats in the frame of antibody drug discovery.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}