
Mammalian Display Platform for the Maturation of Bispecific TCR-Based Molecules

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. PBMC Isolation and Freezing
2.3. Functional Assays
2.4. Stable CHO Cell Line Generation
2.5. TCER Library Generation and Affinity Maturation
2.6. Plasmid Preparation
2.6.1. Vectors
2.6.2. Transformation and Plasmid Isolation
2.6.3. Soluble TCER Expression
2.7. Generation of pHLA Complexes
2.8. Biolayer Interferometry
2.9. Data Analysis
3. Results
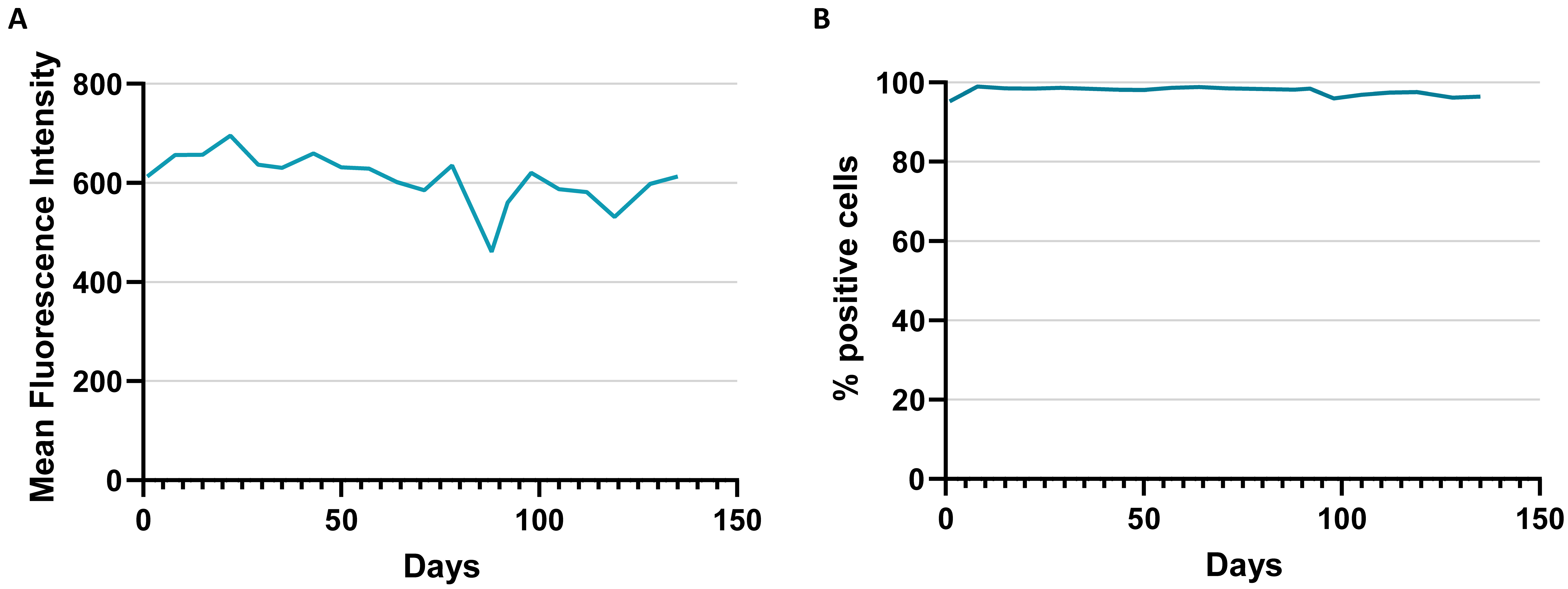
3.1. Generation of High-Expression CHO Cells for Targeted Protein Expression
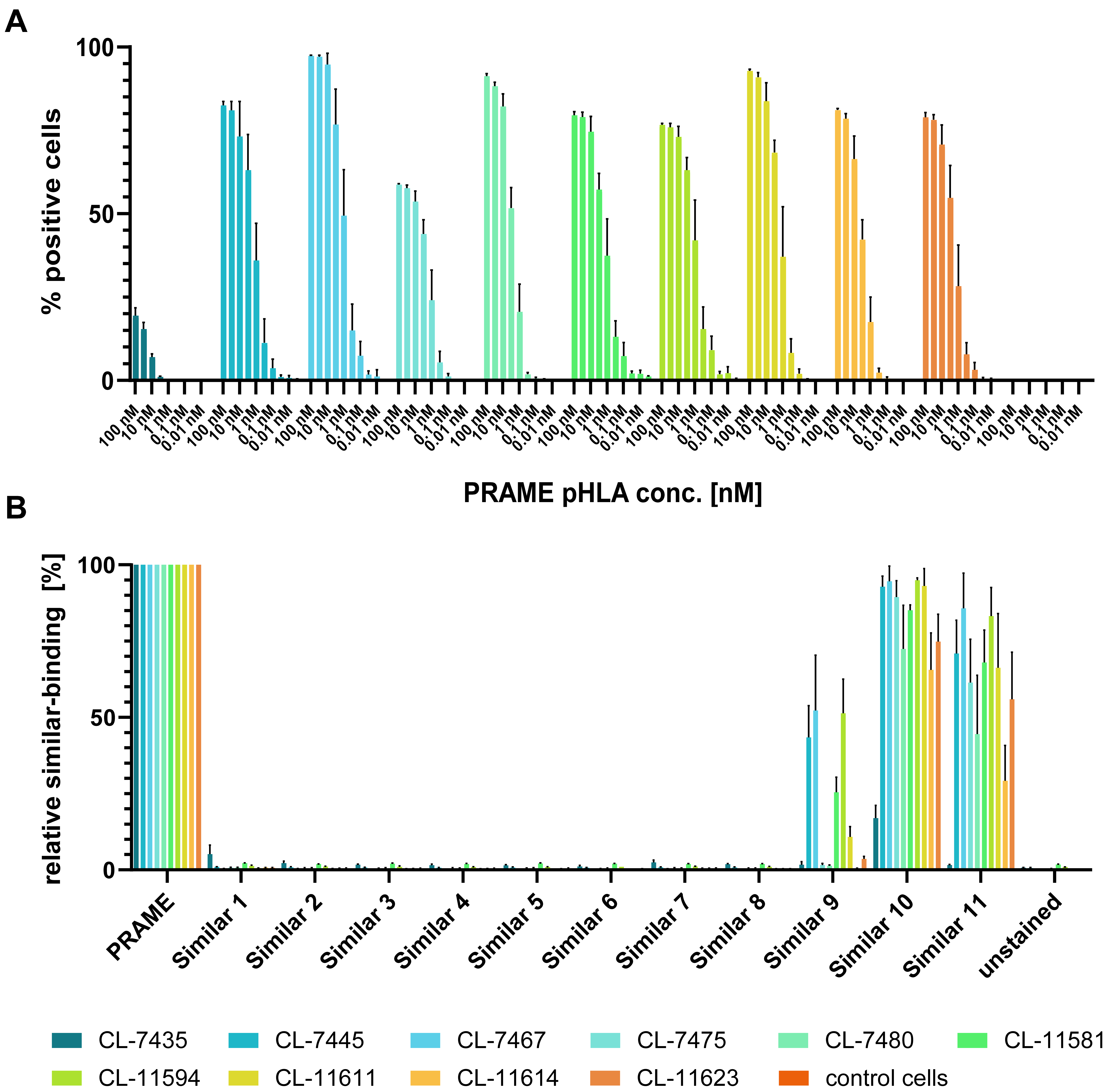
3.2. Generation of PRAME-Specific TCER Library and Selection of Candidates
3.3. Assessment of TCER-Mediated Anti-Tumor Activity
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific Antibodies: A Mechanistic Review of the Pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Staerz, U.D.; Kanagawa, O.; Bevan, M.J. Hybrid Antibodies Can Target Sites for Attack by T Cells. Nature 1985, 314, 628–631. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. FDA Approval of Immunocore’s First-in-Class TCR Therapeutic Broadens Depth of the T Cell Engager Platform. Nat. Rev. Drug Discov. 2022, 21, 170. [Google Scholar] [CrossRef]
- Wang, S.; Chen, K.; Lei, Q.; Ma, P.; Yuan, A.Q.; Zhao, Y.; Jiang, Y.; Fang, H.; Xing, S.; Fang, Y.; et al. The State of the Art of Bispecific Antibodies for Treating Human Malignancies. Embo Mol. Med. 2021, 13, e14291. [Google Scholar] [CrossRef] [PubMed]
- Chandran, S.S.; Klebanoff, C.A. T Cell Receptor-based Cancer Immunotherapy: Emerging Efficacy and Pathways of Resistance. Immunol. Rev. 2019, 290, 127–147. [Google Scholar] [CrossRef]
- Sambi, M.; Bagheri, L.; Szewczuk, M.R. Current Challenges in Cancer Immunotherapy: Multimodal Approaches to Improve Efficacy and Patient Response Rates. J. Oncol. 2019, 2019, 4508794. [Google Scholar] [CrossRef] [Green Version]
- Ventola, C.L. Cancer Immunotherapy, Part 3: Challenges and Future Trends. Pharm. Ther. 2017, 42, 514–521. [Google Scholar]
- Wagner, E.K.; Qerqez, A.N.; Stevens, C.A.; Nguyen, A.W.; Delidakis, G.; Maynard, J.A. Human Cytomegalovirus-Specific T-Cell Receptor Engineered for High Affinity and Soluble Expression Using Mammalian Cell Display. J. Biol. Chem. 2019, 294, 5790–5804. [Google Scholar] [CrossRef] [Green Version]
- Harris, D.T.; Kranz, D.M. Adoptive T Cell Therapies: A Comparison of T Cell Receptors and Chimeric Antigen Receptors. Trends Pharmacol. Sci. 2016, 37, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Oates, J.; Hassan, N.J.; Jakobsen, B.K. ImmTACs for Targeted Cancer Therapy: Why, What, How, and Which. Mol. Immunol. 2015, 67, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Lowe, K.L.; Cole, D.; Kenefeck, R.; OKelly, I.; Lepore, M.; Jakobsen, B.K. Novel TCR-Based Biologics: Mobilising T Cells to Warm ‘Cold’ Tumours. Cancer Treat. Rev. 2019, 77, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vafa, O.; Trinklein, N.D. Perspective: Designing T-Cell Engagers With Better Therapeutic Windows. Front. Oncol. 2020, 10, 446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, K.; Boniface, J.; Reay, P.; Schild, H.; Groth, B.F.D.S.; Davis, M. Low Affinity Interaction of Peptide-MHC Complexes with T Cell Receptors. Science 1991, 254, 1788–1791. [Google Scholar] [CrossRef] [PubMed]
- Birch, J.R.; Racher, A.J. Antibody Production. Adv. Drug Deliver Rev. 2006, 58, 671–685. [Google Scholar] [CrossRef] [PubMed]
- Wurm, F.M. Production of Recombinant Protein Therapeutics in Cultivated Mammalian Cells. Nat. Biotechnol. 2004, 22, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Wirth, D.; Gama-Norton, L.; Riemer, P.; Sandhu, U.; Schucht, R.; Hauser, H. Road to Precision: Recombinase-Based Targeting Technologies for Genome Engineering. Curr. Opin. Biotech. 2007, 18, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Puck, T.T.; Cieciura, S.J.; Robinson, A. GENETICS OF SOMATIC MAMMALIAN CELLS. J. Exp. Med. 1958, 108, 945–956. [Google Scholar] [CrossRef] [Green Version]
- Stinson, S.F.; Alley, M.C.; Kopp, W.C.; Fiebig, H.-H.; Mullendore, L.A.; Pittman, A.F.; Kenney, S.; Keller, J.; Boyd, M.R. Morphological and Immunocytochemical Characteristics of Human Tumor Cell Lines for Use in a Disease-Oriented Anticancer Drug Screen. Anticancer Res. 1992, 12, 1035–1054. [Google Scholar]
- Creasey, A.A.; Smith, H.S.; Hackett, A.J.; Fukuyama, K.; Epstein, W.L.; Madin, S.H. Biological Properties of Human Melanoma Cells in Culture. In Vitro 1979, 15, 342–350. [Google Scholar] [CrossRef]
- Giard, D.J.; Aaronson, S.A.; Todaro, G.J.; Arnstein, P.; Kersey, J.H.; Dosik, H.; Parks, W.P. In Vitro Cultivation of Human Tumors: Establishment of Cell Lines Derived From a Series of Solid Tumors. J. Natl. Cancer Inst. 1973, 51, 1417–1423. [Google Scholar] [CrossRef] [PubMed]
- Uozumi, K.; Otsuka, M.; Ohno, N.; Moriya, T.; Suzuki, S.; Shimotakahara, S.; Matsumura, I.; Hanada, S.; Ari, T. Establishment and Characterization of a New Human Megakaryoblastic Cell Line (SET-2) That Spontaneously Matures to Megakaryocytes and Produces Platelet-like Particles. Leukemia 2000, 14, 142–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garboczi, D.N.; Hung, D.T.; Wiley, D.C. HLA-A2-Peptide Complexes: Refolding and Crystallization of Molecules Expressed in Escherichia Coli and Complexed with Single Antigenic Peptides. Proc. Natl. Acad. Sci. USA 1992, 89, 3429–3433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodenko, B.; Toebes, M.; Hadrup, S.R.; van Esch, W.J.E.; Molenaar, A.M.; Schumacher, T.N.M.; Ovaa, H. Generation of Peptide–MHC Class I Complexes through UV-Mediated Ligand Exchange. Nat. Protoc. 2006, 1, 1120–1132. [Google Scholar] [CrossRef]
- Beerli, R.R.; Bauer, M.; Buser, R.B.; Gwerder, M.; Muntwiler, S.; Maurer, P.; Saudan, P.; Bachmann, M.F. Isolation of Human Monoclonal Antibodies by Mammalian Cell Display. Proc. Natl. Acad. Sci. USA 2008, 105, 14336–14341. [Google Scholar] [CrossRef] [Green Version]
- Breous-Nystrom, E.; Schultze, K.; Meier, M.; Flueck, L.; Holzer, C.; Boll, M.; Seibert, V.; Schuster, A.; Blanusa, M.; Schaefer, V.; et al. Retrocyte Display® Technology: Generation and Screening of a High Diversity Cellular Antibody Library. Methods 2014, 65, 57–67. [Google Scholar] [CrossRef]
- Waldmeier, L.; Hellmann, I.; Gutknecht, C.K.; Wolter, F.I.; Cook, S.C.; Reddy, S.T.; Grawunder, U.; Beerli, R.R. Transpo-MAb Display: Transposition-Mediated B Cell Display and Functional Screening of Full-Length IgG Antibody Libraries. Mabs 2016, 8, 726–740. [Google Scholar] [CrossRef] [Green Version]
- Valldorf, B.; Hinz, S.C.; Russo, G.; Pekar, L.; Mohr, L.; Klemm, J.; Doerner, A.; Krah, S.; Hust, M.; Zielonka, S. Antibody Display Technologies: Selecting the Cream of the Crop. Biol. Chem. 2021, 403, 455–477. [Google Scholar] [CrossRef]
- Parthiban, K.; Perera, R.L.; Sattar, M.; Huang, Y.; Mayle, S.; Masters, E.; Griffiths, D.; Surade, S.; Leah, R.; Dyson, M.R.; et al. A Comprehensive Search of Functional Sequence Space Using Large Mammalian Display Libraries Created by Gene Editing. Mabs 2019, 11, 884–898. [Google Scholar] [CrossRef] [Green Version]
- Parola, C.; Neumeier, D.; Friedensohn, S.; Csepregi, L.; Tacchio, M.D.; Mason, D.M.; Reddy, S.T. Antibody Discovery and Engineering by Enhanced CRISPR-Cas9 Integration of Variable Gene Cassette Libraries in Mammalian Cells. Mabs 2019, 11, 1367–1380. [Google Scholar] [CrossRef]
- Phan, Q.V.; Contzen, J.; Seemann, P.; Gossen, M. Site-Specific Chromosomal Gene Insertion: Flp Recombinase versus Cas9 Nuclease. Sci. Rep. 2017, 7, 17771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggen, D.H.; Chervin, A.S.; Insaidoo, F.K.; Piepenbrink, K.H.; Baker, B.M.; Kranz, D.M. Identification and Engineering of Human Variable Regions That Allow Expression of Stable Single-Chain T Cell Receptors. Protein Eng. Des. Sel. 2011, 24, 361–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieke, M.C.; Shusta, E.V.; Boder, E.T.; Teyton, L.; Wittrup, K.D.; Kranz, D.M. Selection of Functional T Cell Receptor Mutants from a Yeast Surface-Display Library. Proc. Natl. Acad. Sci. USA 1999, 96, 5651–5656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shusta, E.V.; Kieke, M.C.; Parke, E.; Kranz, D.M.; Wittrup, K.D. Yeast Polypeptide Fusion Surface Display Levels Predict Thermal Stability and Soluble Secretion Efficiency11Edited by J. A. Wells. J. Mol. Biol. 1999, 292, 949–956. [Google Scholar] [CrossRef]
- Chen, C.; Li, N.; Zhao, Y.; Hang, H. Coupling Recombinase-mediated Cassette Exchange with Somatic Hypermutation for Antibody Affinity Maturation in CHO Cells. Biotechnol. Bioeng. 2016, 113, 39–51. [Google Scholar] [CrossRef]
- Steinwand, M.; Droste, P.; Frenzel, A.; Hust, M.; Dübel, S.; Schirrmann, T. The Influence of Antibody Fragment Format on Phage Display Based Affinity Maturation of IgG. Mabs 2013, 6, 204–218. [Google Scholar] [CrossRef] [Green Version]
- Robertson, N.; Lopez-Anton, N.; Gurjar, S.A.; Khalique, H.; Khalaf, Z.; Clerkin, S.; Leydon, V.R.; Parker-Manuel, R.; Raeside, A.; Payne, T.; et al. Development of a Novel Mammalian Display System for Selection of Antibodies against Membrane Proteins. J. Biol. Chem. 2020, 295, 18436–18448. [Google Scholar] [CrossRef]
- Bowers, P.M.; Horlick, R.A.; Neben, T.Y.; Toobian, R.M.; Tomlinson, G.L.; Dalton, J.L.; Jones, H.A.; Chen, A.; Altobell, L.; Zhang, X.; et al. Coupling Mammalian Cell Surface Display with Somatic Hypermutation for the Discovery and Maturation of Human Antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 20455–20460. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Qiu, J.; Chen, C.; Liu, C.; Liu, Y.; An, L.; Jia, J.; Tang, J.; Wu, L.; Hang, H. Affinity Maturation of Anti-TNF-Alpha ScFv with Somatic Hypermutation in Non-B Cells. Protein Cell 2012, 3, 460–469. [Google Scholar] [CrossRef] [Green Version]
- Cameron, B.J.; Gerry, A.B.; Dukes, J.; Harper, J.V.; Kannan, V.; Bianchi, F.C.; Grand, F.; Brewer, J.E.; Gupta, M.; Plesa, G.; et al. Identification of a Titin-Derived HLA-A1-Presented Peptide as a Cross-Reactive Target for Engineered MAGE A3-Directed T Cells. Sci. Transl. Med. 2013, 5, 197ra103. [Google Scholar] [CrossRef]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular Toxicity and Titin Cross-Reactivity of Affinity-Enhanced T Cells in Myeloma and Melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence at Peptide Position | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
| PRAME | S | L | L | Q | H | L | I | G | L |
| Similar 1 | . | x | . | . | x | . | . | x | x |
| Similar 2 | . | . | x | x | . | . | x | . | . |
| Similar 3 | . | . | x | . | . | . | x | x | x |
| Similar 4 | . | . | . | x | x | . | . | x | . |
| Similar 5 | . | . | . | x | . | . | x | x | . |
| Similar 6 | . | . | . | . | . | x | . | x | . |
| Similar 7 | . | . | . | . | x | x | x | x | . |
| Similar 8 | . | . | x | . | x | . | x | . | x |
| Similar 9 | . | x | . | x | x | . | . | . | x |
| Similar 10 | x | . | . | x | . | x | . | . | . |
| Similar 11 | x | x | x | x | . | . | . | x | x |
| CDRα1 | CDRα2 | CDRα3 | CDRβ1 | CDRβ2 | CDRβ3 |
|---|---|---|---|---|---|
| DRGSQS | YSNGDKE | DNAHGGM | SGHRS | EHGLER | CASSPWDSPNVQY |
| DRGSQL | YQEGDKE | DNDQGGI | EGHRA | FSETQR | CASSPWDSPNEQY |
| YQTGDKE | DNDVGGI | PGHKA | IHGEER | ||
| YQAGDKE | DNEQGGM | PGHRA | IHGQER | ||
| YPQGDKK | DNKAGGI | PGHRS | IHGVER | ||
| YSQGDKE | DNPAGGI | QGHRA | VHGAER | ||
| DNPRGGM | VHGEER | ||||
| DNPVGGP | VHGIER | ||||
| ENKPGGP | VHGKER | ||||
| GNAQGGM | VHGLER | ||||
| GNDLGGI | VHGMER | ||||
| NNPSGGM | VHGNER | ||||
| PNPPGGK | VHGQER | ||||
| PNTHGGP | VHGRER | ||||
| SNFGNEK | VHGVER | ||||
| TNIAGGS | VHGYAR |
| Clone | CDRα1 | CDRα2 | CDRα3 | CDRβ1 | CDRβ2 | CDRβ3 |
|---|---|---|---|---|---|---|
| Parental TCR | DRGSQS | YSNGDKE | SNFGNEK | SGHRS | FSETGR | CASSPWDSPNEQY |
| CL-7435 | DRGSQS | YQAGDKE | GNDLGGI | SGHRS | VHGEER | CASSPWDSPNEQY |
| CL-7445 | DRGSQS | YSNGDKE | DNPRGGM | QGHRA | VHGEER | CASSPWDSPNEQY |
| CL-7467 | DRGSQS | YQAGDKE | GNAQGGM | PGHRA | VHGEER | CASSPWDSPNVQY |
| CL-7475 | DRGSQS | YPQGDKK | DNPAGGI | SGHRS | VHGEER | CASSPWDSPNVQY |
| CL-7480 | DRGSQS | YQEGDKE | SNFGNEK | PGHRA | VHGEER | CASSPWDSPNEQY |
| CL-11581 | DRGSQS | YSQGDKE | DNPRGGM | PGHRS | VHGEER | CASSPWDSPNVQY |
| CL-11594 | DRGSQS | YSNGDKE | DNEQGGM | PGHRS | VHGEER | CASSPWDSPNVQY |
| CL-11611 | DRGSQS | YQEGDKE | NNPSGGM | QGHRA | VHGEER | CASSPWDSPNEQY |
| CL-11614 | DRGSQS | YSQGDKE | DNPAGGI | SGHRS | VHGEER | CASSPWDSPNEQY |
| CL-11623 | DRGSQS | YSQGDKE | NNPSGGM | PGHRS | VHGEER | CASSPWDSPNVQY |
| Clone | KD [nM] |
|---|---|
| CL-7467 | 3.4 |
| CL-7445 | 3.7 |
| CL-11581 | 5.2 |
| CL-11594 | 6.1 |
| CL-11623 | 6.6 |
| CL-11611 | 12.0 |
| CL-7435 | 16.5 |
| CL-11614 | 17.8 |
| CL-7480 | 24.5 |
| CL-7475 | 37.4 |
| Parental TCR | 1230.0 |
| Name | EC50 UACC-257 [pM] | EC50 HS695T [pM] |
|---|---|---|
| CL-7435 | 1136 | 1380 |
| CL-7445 | 115 | 644 |
| CL-7467 | 61 | 172 |
| CL-7475 | 1864 | 1556 |
| CL-7480 | 730 | 1849 |
| CL-11581 | 310 | 587 |
| CL-11594 | 450 | 808 |
| CL-11611 | 656 | 984 |
| CL-11614 | 914 | 1424 |
| CL-11623 | 136 | 849 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dilchert, J.; Hofmann, M.; Unverdorben, F.; Kontermann, R.; Bunk, S. Mammalian Display Platform for the Maturation of Bispecific TCR-Based Molecules. Antibodies 2022, 11, 34. https://doi.org/10.3390/antib11020034
Dilchert J, Hofmann M, Unverdorben F, Kontermann R, Bunk S. Mammalian Display Platform for the Maturation of Bispecific TCR-Based Molecules. Antibodies. 2022; 11(2):34. https://doi.org/10.3390/antib11020034
Chicago/Turabian StyleDilchert, Janine, Martin Hofmann, Felix Unverdorben, Roland Kontermann, and Sebastian Bunk. 2022. "Mammalian Display Platform for the Maturation of Bispecific TCR-Based Molecules" Antibodies 11, no. 2: 34. https://doi.org/10.3390/antib11020034
APA StyleDilchert, J., Hofmann, M., Unverdorben, F., Kontermann, R., & Bunk, S. (2022). Mammalian Display Platform for the Maturation of Bispecific TCR-Based Molecules. Antibodies, 11(2), 34. https://doi.org/10.3390/antib11020034