
Intracellular Antibodies for Drug Discovery and as Drugs of the Future


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
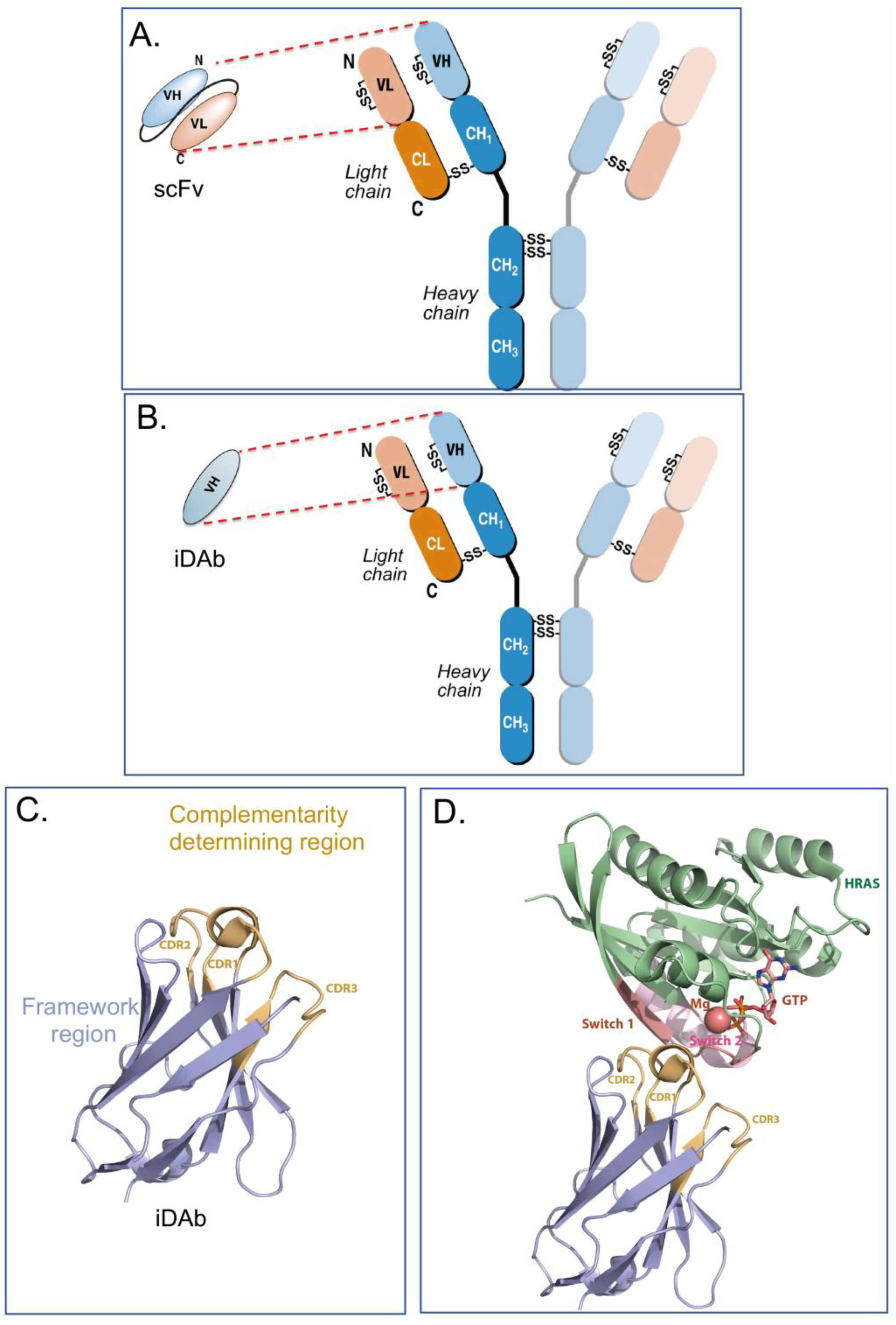
2. Intracellular Antibody Fragments
3. Specialising Intracellular Antibodies by Fusing them with Moieties to Affect Cell Phenotype or Viability

4. Options for Delivery of Intracellular Antibodies to Cells
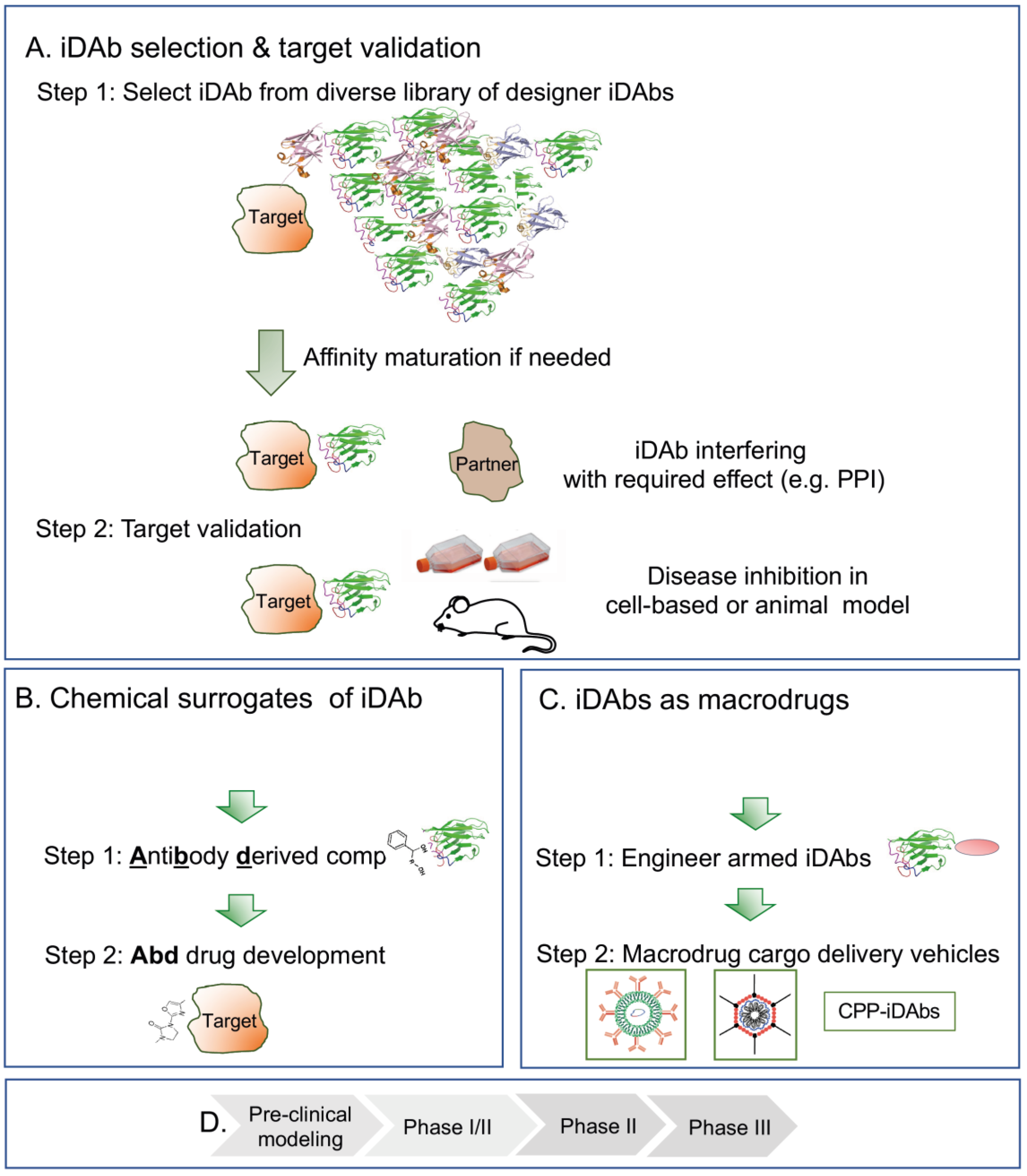
5. Intracellular-Antibody-Derived Compounds: Bridging the Gap between Antibodies and Small Molecules
6. Conclusions
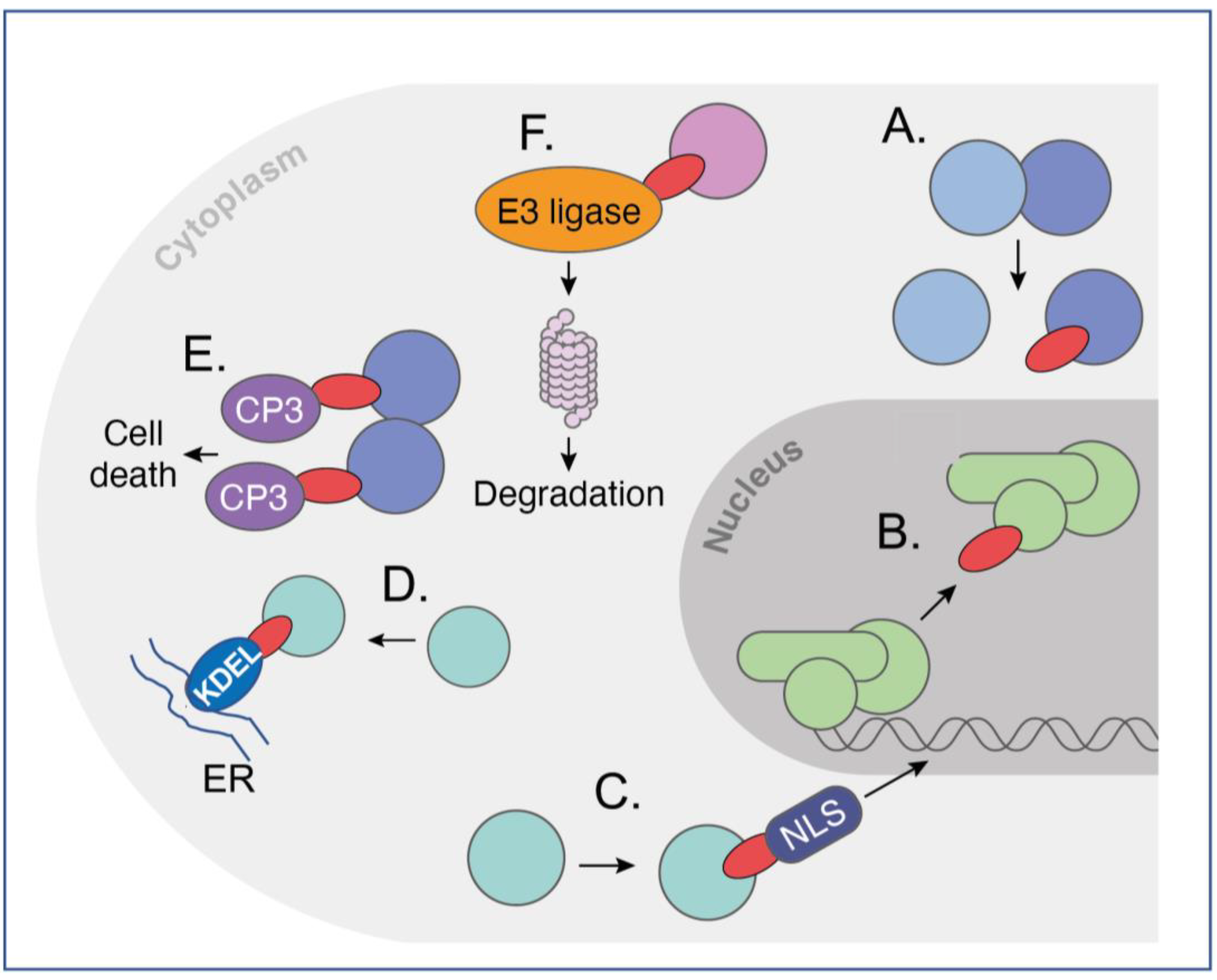
 ) inside cells that have differ-ent functionalities, ranging from iDAb protein (A,B) to those fused to effector moieties (warheads) that invoke a cellular property for the effectiveness of the iDAb (C–F). In (A): an iDAb acts as an in-hibitor of a protein–protein interaction; in (B): an iDAb acts as an inhibitor of a transcription factor interaction with DNA (this could also be applied to protein-RNA interaction); in (C): an iDAb fused to an NLS relocates its protein target from the cytoplasm to nucleus; in (D): an iDAb fused with a endoplasmic reticulum signal sequence (KDEL) sequesters a target protein in the endoplasmic re-ticulum; in (E): two iDAbs linked to procaspase 3 (CP3) bind to a target protein and cause auto-cleavage of procaspase 3 to active caspase 3, thereby initiating programmed cell death in an anti-gen-dependent fashion, as may occur to target chromosomal translocation fusion proteins; in (F): iDAbs are turned into biodegraders to induce targeted protein degradation, by fusing an iDAb to an E3 ligase, allowing a binary complex to form with the target protein, resulting in ubiquitination of the protein and proteasomal degradation. These various functionalities can also be applied with the use of DARPins, monobodies and affimers. This figure is adapted from a previous publication [62]. The figure was created by Claudia Stocker, Vivid Biology.
) inside cells that have differ-ent functionalities, ranging from iDAb protein (A,B) to those fused to effector moieties (warheads) that invoke a cellular property for the effectiveness of the iDAb (C–F). In (A): an iDAb acts as an in-hibitor of a protein–protein interaction; in (B): an iDAb acts as an inhibitor of a transcription factor interaction with DNA (this could also be applied to protein-RNA interaction); in (C): an iDAb fused to an NLS relocates its protein target from the cytoplasm to nucleus; in (D): an iDAb fused with a endoplasmic reticulum signal sequence (KDEL) sequesters a target protein in the endoplasmic re-ticulum; in (E): two iDAbs linked to procaspase 3 (CP3) bind to a target protein and cause auto-cleavage of procaspase 3 to active caspase 3, thereby initiating programmed cell death in an anti-gen-dependent fashion, as may occur to target chromosomal translocation fusion proteins; in (F): iDAbs are turned into biodegraders to induce targeted protein degradation, by fusing an iDAb to an E3 ligase, allowing a binary complex to form with the target protein, resulting in ubiquitination of the protein and proteasomal degradation. These various functionalities can also be applied with the use of DARPins, monobodies and affimers. This figure is adapted from a previous publication [62]. The figure was created by Claudia Stocker, Vivid Biology.
) inside cells that have differ-ent functionalities, ranging from iDAb protein (A,B) to those fused to effector moieties (warheads) that invoke a cellular property for the effectiveness of the iDAb (C–F). In (A): an iDAb acts as an in-hibitor of a protein–protein interaction; in (B): an iDAb acts as an inhibitor of a transcription factor interaction with DNA (this could also be applied to protein-RNA interaction); in (C): an iDAb fused to an NLS relocates its protein target from the cytoplasm to nucleus; in (D): an iDAb fused with a endoplasmic reticulum signal sequence (KDEL) sequesters a target protein in the endoplasmic re-ticulum; in (E): two iDAbs linked to procaspase 3 (CP3) bind to a target protein and cause auto-cleavage of procaspase 3 to active caspase 3, thereby initiating programmed cell death in an anti-gen-dependent fashion, as may occur to target chromosomal translocation fusion proteins; in (F): iDAbs are turned into biodegraders to induce targeted protein degradation, by fusing an iDAb to an E3 ligase, allowing a binary complex to form with the target protein, resulting in ubiquitination of the protein and proteasomal degradation. These various functionalities can also be applied with the use of DARPins, monobodies and affimers. This figure is adapted from a previous publication [62]. The figure was created by Claudia Stocker, Vivid Biology.
) inside cells that have differ-ent functionalities, ranging from iDAb protein (A,B) to those fused to effector moieties (warheads) that invoke a cellular property for the effectiveness of the iDAb (C–F). In (A): an iDAb acts as an in-hibitor of a protein–protein interaction; in (B): an iDAb acts as an inhibitor of a transcription factor interaction with DNA (this could also be applied to protein-RNA interaction); in (C): an iDAb fused to an NLS relocates its protein target from the cytoplasm to nucleus; in (D): an iDAb fused with a endoplasmic reticulum signal sequence (KDEL) sequesters a target protein in the endoplasmic re-ticulum; in (E): two iDAbs linked to procaspase 3 (CP3) bind to a target protein and cause auto-cleavage of procaspase 3 to active caspase 3, thereby initiating programmed cell death in an anti-gen-dependent fashion, as may occur to target chromosomal translocation fusion proteins; in (F): iDAbs are turned into biodegraders to induce targeted protein degradation, by fusing an iDAb to an E3 ligase, allowing a binary complex to form with the target protein, resulting in ubiquitination of the protein and proteasomal degradation. These various functionalities can also be applied with the use of DARPins, monobodies and affimers. This figure is adapted from a previous publication [62]. The figure was created by Claudia Stocker, Vivid Biology.

Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviation/Term | Definition |
| AIDA | Antibody–antigen interaction-dependent apoptosis |
| Abd technology | Antibody-derived compound technology |
| Affimer | Small non-antibody proteins that bind antigens |
| Biodegrader | An intracellular antibody engineered as a fusion with an E3 ligase for targeted protein degradation of a target protein |
| CDR | Antibody-complementarity-determining regions |
| CPPs | Cell-penetrating peptides |
| CP3 | Caspase 3 |
| DARPins | Designed ankyrin repeat proteins |
| EBV | Epstein–Barr virus |
| epitope | Antigen-binding amino acids that interact with antibody paratope |
| Rule of 5 | Empirical rules for parameters of drug-like properties of compounds. See [53] |
| H and L chain | Antibody heavy and light chains |
| HIV | Human immunodeficiency virus |
| iDAb | Intracellular domain antibody |
| IgG | Immunoglobulin G-class antibody |
| LNP | Lipid nanoparticle |
| macrodrug | An intracellular antibody used as a drug per se |
| monobody | Synthetic proteins based on fibronectin type III domain |
| nanobody | Camelid VHH fragments |
| NLS | Nuclear localisation signal |
| paratope | Antibody-binding amino acids that interact with antigenic epitope |
| PPI | Protein–protein interaction |
| PROTAC | Proteolysis-targeting chimeric |
| scFv | Single-chain fragment variable |
| SIT technology | Suicide (or silencing) intrabody technology |
| V region | Antibody variable region |
| VNAR | Shark heavy-chain V region |
| warhead | Generic term for effector region fused to intracellular antibodies instead of the Fc region of immunoglobulin |
References
- Carlson, J.R. A new means of inducibly inactivating a cellular protein. Mol. Cell Biol. 1988, 8, 2638–2646. [Google Scholar] [CrossRef]
- Biocca, S.; Neuberger, M.S.; Cattaneo, A. Expression and targeting of intracellular antibodies in mammalian cells. EMBO J. 1990, 9, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Marasco, W.A.; Haseltine, W.A.; Chen, S.Y. Design, intracellular expression, and activity of a human anti-human immunodeficiency virus type 1 gp120 single-chain antibody. Proc. Natl. Acad. Sci. USA 1993, 90, 7889–7893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Rabbitts, T.H. Functional intracellular antibody fragments do not require invariant intra-domain disulfide bonds. J. Mol. Biol. 2008, 376, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Ward, E.S.; Gussow, D.; Griffiths, A.D.; Jones, P.T.; Winter, G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli. Nature 1989, 341, 544–546. [Google Scholar] [CrossRef]
- Tanaka, T.; Lobato, M.N.; Rabbitts, T.H. Single domain intracellular antibodies: A minimal fragment for direct in vivo selection of antigen-specific intrabodies. J. Mol. Biol. 2003, 331, 1109–1120. [Google Scholar] [CrossRef]
- Tanaka, T.; Rabbitts, T.H. Protocol for the selection of single-domain antibody fragments by third generation intracellular antibody capture. Nat. Protoc. 2010, 5, 67–92. [Google Scholar] [CrossRef]
- Hamers-Casterman, C.; Atarhouch, T.; Muyldermans, S.; Robinson, G.; Hamers, C.; Songa, E.B.; Bendahman, N.; Hamers, R. Naturally occurring antibodies devoid of light chains. Nature 1993, 363, 446–448. [Google Scholar] [CrossRef]
- Helma, J.; Cardoso, M.C.; Muyldermans, S.; Leonhardt, H. Nanobodies and recombinant binders in cell biology. J. Cell Biol. 2015, 209, 633–644. [Google Scholar] [CrossRef] [Green Version]
- Roux, K.H.; Greenberg, A.S.; Greene, L.; Strelets, L.; Avila, D.; McKinney, E.C.; Flajnik, M.F. Structural analysis of the nurse shark (new) antigen receptor (NAR): Molecular convergence of NAR and unusual mammalian immunoglobulins. Proc. Natl. Acad. Sci. USA 1998, 95, 11804–11809. [Google Scholar] [CrossRef] [Green Version]
- Hantschel, O.; Biancalana, M.; Koide, S. Monobodies as enabling tools for structural and mechanistic biology. Curr. Opin. Struct. Biol. 2020, 60, 167–174. [Google Scholar] [CrossRef]
- Pluckthun, A. Designed ankyrin repeat proteins (DARPins): Binding proteins for research, diagnostics, and therapy. Annu. Rev. Pharm. Toxicol. 2015, 55, 489–511. [Google Scholar] [CrossRef]
- Tiede, C.; Bedford, R.; Heseltine, S.J.; Smith, G.; Wijetunga, I.; Ross, R.; AlQallaf, D.; Roberts, A.P.; Balls, A.; Curd, A.; et al. Affimer proteins are versatile and renewable affinity reagents. Elife 2017, 6, 24903. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Li, H.C.; Tanaka, T.; Rabbitts, T.H. Selection of human single domain antibodies recognizing the CMYC protein using enhanced intracellular antibody capture. J. Immunol. Methods 2015, 426, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Chothia, C.; Novotny, J.; Bruccoleri, R.; Karplus, M. Domain association in immunoglobulin molecules. The packing of variable domains. J. Mol. Biol. 1985, 186, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Rabbitts, T.H. Interfering with RAS-effector protein interactions prevent RAS-dependent tumour initiation and causes stop-start control of cancer growth. Oncogene 2010, 29, 6064–6070. [Google Scholar] [CrossRef] [Green Version]
- Warren, A.J.; Bravo, J.; Williams, R.L.; Rabbitts, T.H. Structural basis for the heterodimeric interaction between the acute leukaemia-associated transcription factors AML1 and CBFbeta. EMBO J. 2000, 19, 3004–3015. [Google Scholar] [CrossRef] [Green Version]
- Sewell, H.; Tanaka, T.; El Omari, K.; Mancini, E.J.; Cruz, A.; Fernandez-Fuentes, N.; Chambers, J.; Rabbitts, T.H. Conformational flexibility of the oncogenic protein LMO2 primes the formation of the multi-protein transcription complex. Sci. Rep. 2014, 4, 3643. [Google Scholar] [CrossRef] [Green Version]
- Hammond, C.; Helenius, A. Quality control in the secretory pathway: Retention of a misfolded viral membrane glycoprotein involves cycling between the ER, intermediate compartment, and Golgi apparatus. J. Cell Biol. 1994, 126, 41–52. [Google Scholar] [CrossRef]
- Tanaka, T.; Williams, R.L.; Rabbitts, T.H. Tumour prevention by a single antibody domain targeting the interaction of signal transduction proteins with RAS. EMBO J. 2007, 26, 3250–3259. [Google Scholar] [CrossRef] [Green Version]
- Tse, E.; Rabbitts, T.H. Intracellular antibody-caspase-mediated cell killing: An approach for application in cancer therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 12266–12271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, J.S.; Brend, T.; Rabbitts, T.H. Cancer cell killing by target antigen engagement with engineered complementary intracellular antibody single domains fused to pro-caspase3. Sci. Rep. 2019, 9, 8553. [Google Scholar] [CrossRef] [Green Version]
- Tse, E.; Lobato, M.N.; Forster, A.; Tanaka, T.; Chung, G.T.; Rabbitts, T.H. Intracellular antibody capture technology: Application to selection of intracellular antibodies recognising the BCR-ABL oncogenic protein. J. Mol. Biol. 2002, 317, 85–94. [Google Scholar] [CrossRef]
- Zhou, P.; Bogacki, R.; McReynolds, L.; Howley, P.M. Harnessing the ubiquitination machinery to target the degradation of specific cellular proteins. Mol. Cell 2000, 6, 751–756. [Google Scholar] [CrossRef]
- Melchionna, T.; Cattaneo, A. A protein silencing switch by ligand-induced proteasome-targeting intrabodies. J. Mol. Biol. 2007, 374, 641–654. [Google Scholar] [CrossRef]
- Bekes, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Tanaka, T.; Sewell, H.; Waters, S.; Phillips, S.E.; Rabbitts, T.H. Single domain intracellular antibodies from diverse libraries: Emphasizing dual functions of LMO2 protein interactions using a single VH domain. J. Biol. Chem. 2011, 286, 3707–3716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bery, N.; Legg, S.; Debreczeni, J.; Breed, J.; Embrey, K.; Stubbs, C.; Kolasinska-Zwierz, P.; Barrett, N.; Marwood, R.; Watson, J.; et al. KRAS-specific inhibition using a DARPin binding to a site in the allosteric lobe. Nat. Commun. 2019, 10, 2607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bery, N.; Miller, A.; Rabbitts, T. A potent KRAS macromolecule degrader specifically targeting tumours with mutant KRAS. Nat. Commun. 2020, 11, 3233. [Google Scholar] [CrossRef]
- Teng, K.W.; Tsai, S.T.; Hattori, T.; Fedele, C.; Koide, A.; Yang, C.; Hou, X.; Zhang, Y.; Neel, B.G.; O’Bryan, J.P.; et al. Selective and noncovalent targeting of RAS mutants for inhibition and degradation. Nat. Commun. 2021, 12, 2656. [Google Scholar] [CrossRef]
- Stumpp, M.T.; Dawson, K.M.; Binz, H.K. Beyond Antibodies: The DARPin((R)) Drug Platform. BioDrugs 2020, 34, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Sha, F.; Salzman, G.; Gupta, A.; Koide, S. Monobodies and other synthetic binding proteins for expanding protein science. Protein Sci. 2017, 26, 910–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Thomas, J.; Van Montfort, R.; Miller, A.; Rabbitts, T. Pan RAS-binding compounds selected from a chemical library by inhibiting interaction between RAS and a reduced affinity intracellular antibody. Sci. Rep. 2021, 11, 1712. [Google Scholar] [CrossRef]
- Bery, N.; Bataille, C.J.R.; Russell, A.; Hayes, A.; Raynaud, F.; Milhas, S.; Anand, S.; Tulmin, H.; Miller, A.; Rabbitts, T.H. A cell-based screening method using an intracellular antibody for discovering small molecules targeting the translocation protein LMO2. Sci. Adv. 2021, 7, eabg1950. [Google Scholar] [CrossRef]
- Tanaka, T.; Rabbitts, T.H. Interfering with protein-protein interactions: Potential for cancer therapy. Cell Cycle 2008, 7, 1569–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.M.; Choi, D.K.; Jung, K.; Bae, J.; Kim, J.S.; Park, S.W.; Song, K.H.; Kim, Y.S. Antibody targeting intracellular oncogenic Ras mutants exerts anti-tumour effects after systemic administration. Nat. Commun. 2017, 8, 15090. [Google Scholar] [CrossRef] [Green Version]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [CrossRef]
- Green, M.; Ishino, M.; Loewenstein, P.M. Mutational analysis of HIV-1 Tat minimal domain peptides: Identification of trans-dominant mutants that suppress HIV-LTR-driven gene expression. Cell 1989, 58, 215–223. [Google Scholar] [CrossRef]
- Patel, S.G.; Sayers, E.J.; He, L.; Narayan, R.; Williams, T.L.; Mills, E.M.; Allemann, R.K.; Luk, L.Y.P.; Jones, A.T.; Tsai, Y.H. Cell-penetrating peptide sequence and modification dependent uptake and subcellular distribution of green florescent protein in different cell lines. Sci. Rep. 2019, 9, 6298. [Google Scholar] [CrossRef] [Green Version]
- Tietz, O.; Cortezon-Tamarit, F.; Chalk, R.; Able, S.; Vallis, K.A. Tricyclic cell-penetrating peptides for efficient delivery of functional antibodies into cancer cells. Nat. Chem. 2022, 14, 284–293. [Google Scholar] [CrossRef]
- Rossotti, M.A.; Belanger, K.; Henry, K.A.; Tanha, J. Immunogenicity and humanization of single-domain antibodies. FEBS J. 2022, 289, 4304–4327. [Google Scholar] [CrossRef] [PubMed]
- Francis, A.I.; Ghany, S.; Gilkes, T.; Umakanthan, S. Review of COVID-19 vaccine subtypes, efficacy and geographical distributions. Postgrad Med. J. 2022, 98, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Boldicke, T. Therapeutic Potential of Intrabodies for Cancer Immunotherapy: Current Status and Future Directions. Antibodies 2022, 11, 49. [Google Scholar] [CrossRef] [PubMed]
- Town, J.; Pais, H.; Harrison, S.; Stead, L.F.; Bataille, C.; Bunjobpol, W.; Zhang, J.; Rabbitts, T.H. Exploring the surfaceome of Ewing sarcoma identifies a new and unique therapeutic target. Proc. Natl. Acad. Sci. USA 2016, 113, 3603–3608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pais, H.; Ruggero, K.; Zhang, J.; Al-Assar, O.; Bery, N.; Bhuller, R.; Weston, V.; Kearns, P.R.; Mecucci, C.; Miller, A.; et al. Surfaceome interrogation using an RNA-seq approach highlights leukemia initiating cell biomarkers in an LMO2 T cell transgenic model. Sci. Rep. 2019, 9, 5760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waite, J.C.; Wang, B.; Haber, L.; Hermann, A.; Ullman, E.; Ye, X.; Dudgeon, D.; Slim, R.; Ajithdoss, D.K.; Godin, S.J.; et al. Tumor-targeted CD28 bispecific antibodies enhance the antitumor efficacy of PD-1 immunotherapy. Sci. Transl. Med. 2020, 12, eaba2325. [Google Scholar] [CrossRef]
- Miller, A.; Carr, S.; Rabbitts, T.; Ali, H. Multimeric antibodies with increased valency surpassing functional affinity and potency thresholds using novel formats. MAbs 2020, 12, 1752529. [Google Scholar] [CrossRef] [Green Version]
- Leach, A.; Miller, A.; Bentley, E.; Mattiuzzo, G.; Thomas, J.; McAndrew, C.; Van Montfort, R.; Rabbitts, T. Implementing a method for engineering multivalency to substantially enhance binding of clinical trial anti-SARS-CoV-2 antibodies to wildtype spike and variants of concern proteins. Sci. Rep. 2021, 11, 10475. [Google Scholar] [CrossRef]
- Wittrup, K.D.; Thurber, G.M.; Schmidt, M.M.; Rhoden, J.J. Practical theoretic guidance for the design of tumor-targeting agents. Methods Enzym. 2012, 503, 255–268. [Google Scholar] [CrossRef] [Green Version]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Quevedo, C.E.; Cruz-Migoni, A.; Bery, N.; Miller, A.; Tanaka, T.; Petch, D.; Bataille, C.J.R.; Lee, L.Y.W.; Fallon, P.S.; Tulmin, H.; et al. Small molecule inhibitors of RAS-effector protein interactions derived using an intracellular antibody fragment. Nat. Commun. 2018, 9, 3169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janin, J.; Chothia, C. The structure of protein-protein recognition sites. J. Biol. Chem. 1990, 265, 16027–16030. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Cruz-Migoni, A.; Canning, P.; Quevedo, C.E.; Bataille, C.J.R.; Bery, N.; Miller, A.; Russell, A.J.; Phillips, S.E.V.; Carr, S.B.; Rabbitts, T.H. Structure-based development of new RAS-effector inhibitors from a combination of active and inactive RAS-binding compounds. Proc. Natl. Acad. Sci. USA 2019, 116, 2545–2550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bery, N.; Cruz-Migoni, A.; Bataille, C.J.; Quevedo, C.E.; Tulmin, H.; Miller, A.; Russell, A.; Phillips, S.E.; Carr, S.B.; Rabbitts, T.H. BRET-based RAS biosensors that show a novel small molecule is an inhibitor of RAS-effector protein-protein interactions. Elife 2018, 7, 37122. [Google Scholar] [CrossRef]
- Boehm, T.; Foroni, L.; Kaneko, Y.; Perutz, M.F.; Rabbitts, T.H. The rhombotin family of cysteine-rich LIM-domain oncogenes: Distinct members are involved in T-cell translocations to human chromosomes 11p15 and 11p13. Proc. Natl. Acad. Sci. USA 1991, 88, 4367–4371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Royer-Pokora, B.; Loos, U.; Ludwig, W.D. TTG-2, a new gene encoding a cysteine-rich protein with the LIM motif, is overexpressed in acute T-cell leukaemia with the t(11;14)(p13;q11). Oncogene 1991, 6, 1887–1893. [Google Scholar]
- Chambers, J.; Rabbitts, T.H. LMO2 at 25 years: A paradigm of chromosomal translocation proteins. Open Biol. 2015, 5, 150062. [Google Scholar] [CrossRef] [Green Version]
- Wadman, I.A.; Osada, H.; Grutz, G.G.; Agulnick, A.D.; Westphal, H.; Forster, A.; Rabbitts, T.H. The LIM-only protein Lmo2 is a bridging molecule assembling an erythroid, DNA-binding complex which includes the TAL1, E47, GATA-1 and Ldb1/NLI proteins. EMBO J. 1997, 16, 3145–3157. [Google Scholar] [CrossRef]
- Xiao, T.; Frey, G.; Fu, Q.; Lavine, C.L.; Scott, D.A.; Seaman, M.S.; Chou, J.J.; Chen, B. HIV-1 fusion inhibitors targeting the membrane-proximal external region of Env spikes. Nat. Chem. Biol. 2020, 16, 529–537. [Google Scholar] [CrossRef]
- Vinogradov, A.A.; Yin, Y.; Suga, H. Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges. J. Am. Chem. Soc. 2019, 141, 4167–4181. [Google Scholar] [CrossRef] [PubMed]
- Lobato, M.N.; Rabbitts, T.H. Intracellular antibodies and challenges facing their use as therapeutic agents. Trends Mol. Med. 2003, 9, 390–396. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rabbitts, T.H. Intracellular Antibodies for Drug Discovery and as Drugs of the Future. Antibodies 2023, 12, 24. https://doi.org/10.3390/antib12010024
Rabbitts TH. Intracellular Antibodies for Drug Discovery and as Drugs of the Future. Antibodies. 2023; 12(1):24. https://doi.org/10.3390/antib12010024
Chicago/Turabian StyleRabbitts, T. H. 2023. "Intracellular Antibodies for Drug Discovery and as Drugs of the Future" Antibodies 12, no. 1: 24. https://doi.org/10.3390/antib12010024
APA StyleRabbitts, T. H. (2023). Intracellular Antibodies for Drug Discovery and as Drugs of the Future. Antibodies, 12(1), 24. https://doi.org/10.3390/antib12010024