
Polyclonal Antibodies Derived from Transchromosomic Bovines Vaccinated with the Recombinant F1-V Vaccine Increase Bacterial Opsonization In Vitro and Protect Mice from Pneumonic Plague

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Mouse Monoclonal Antibodies
2.3. Production of Anti-rF1-V Human Polyclonal Antibodies SAB-183 from Transchromosomic (Tc) Bovines
2.4. Tc Bovine Immunization and Plasma Collection
2.5. cGMP Purification of SAB-183
2.6. Cell Culture
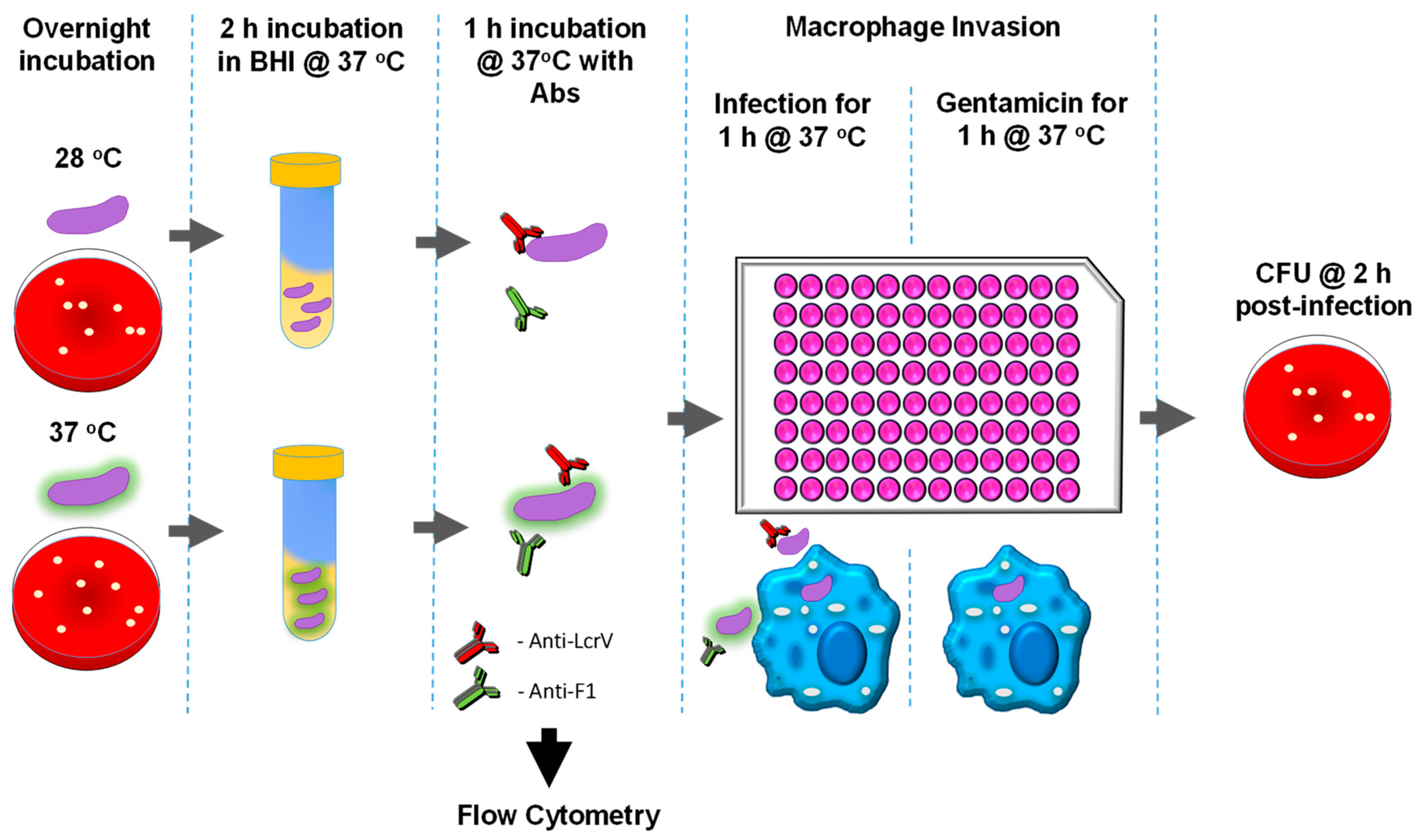
2.7. Quantification of Viable Intracellular Y. pestis (Gentamicin Protection Assay)
2.8. Exposure of Mice to Aerosolized Y. pestis
2.9. ELISA
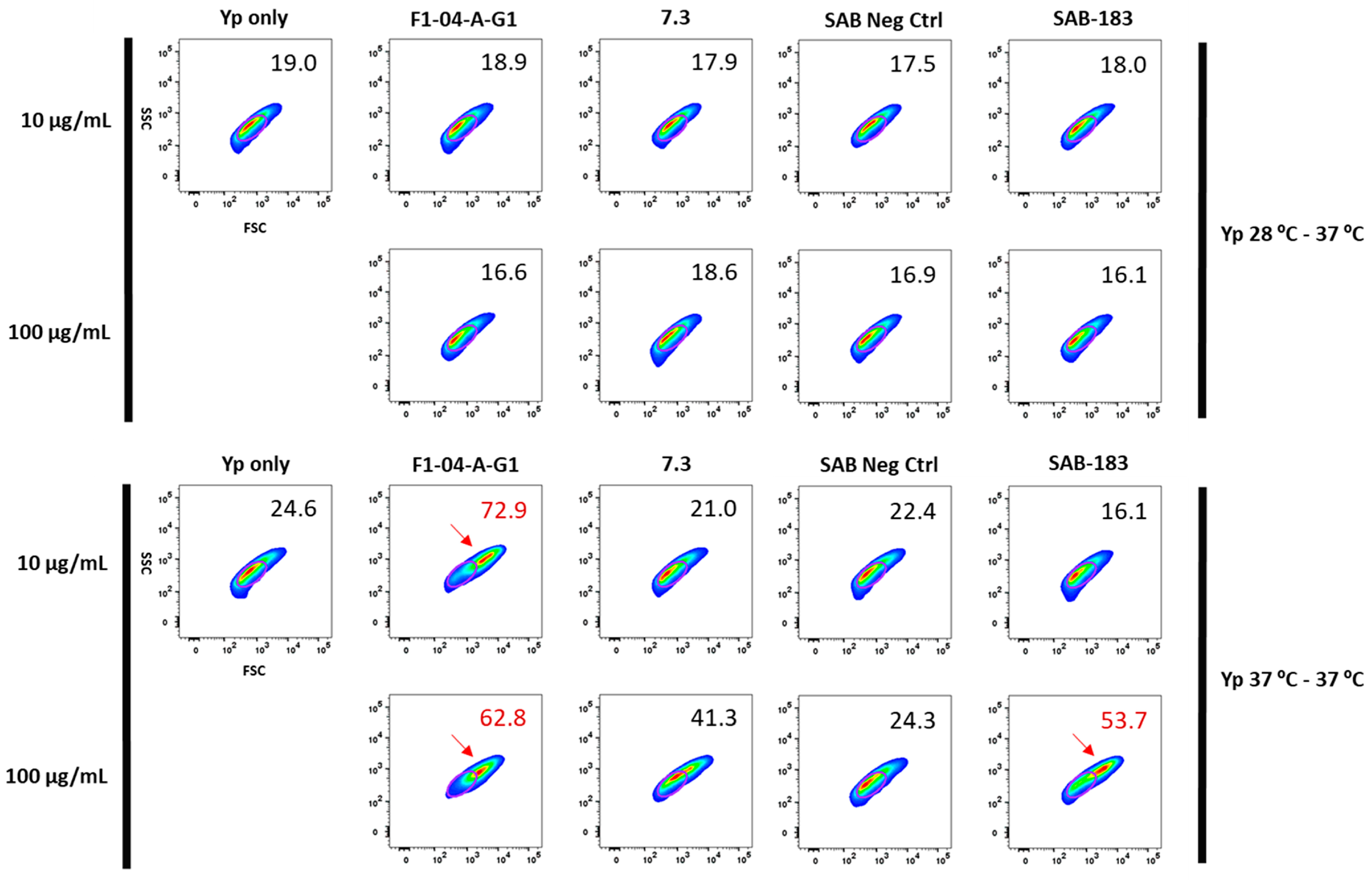
2.10. Flow Cytometry
2.11. Statistics
3. Results
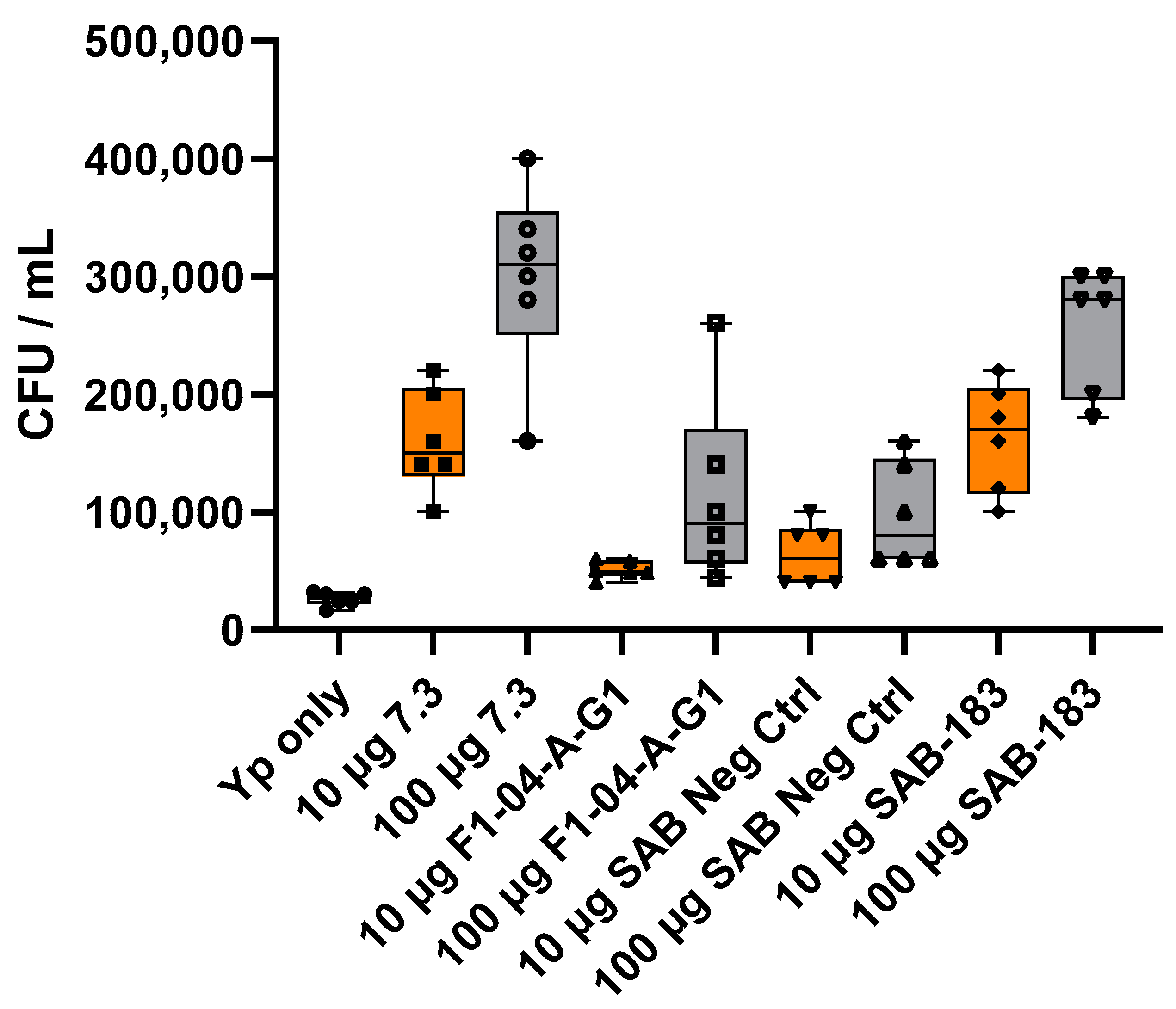
3.1. Mouse Anti-F1 and Anti-LcrV mAbs and Polyclonal Anti-rF1-V Antibodies Derived from Transchromosomic (Tc) Bovines Are Opsonic In Vitro
3.2. Transchromosomic (Tc) Bovines Immunized with rF1-V Elicit a Strong Human Antibody Response
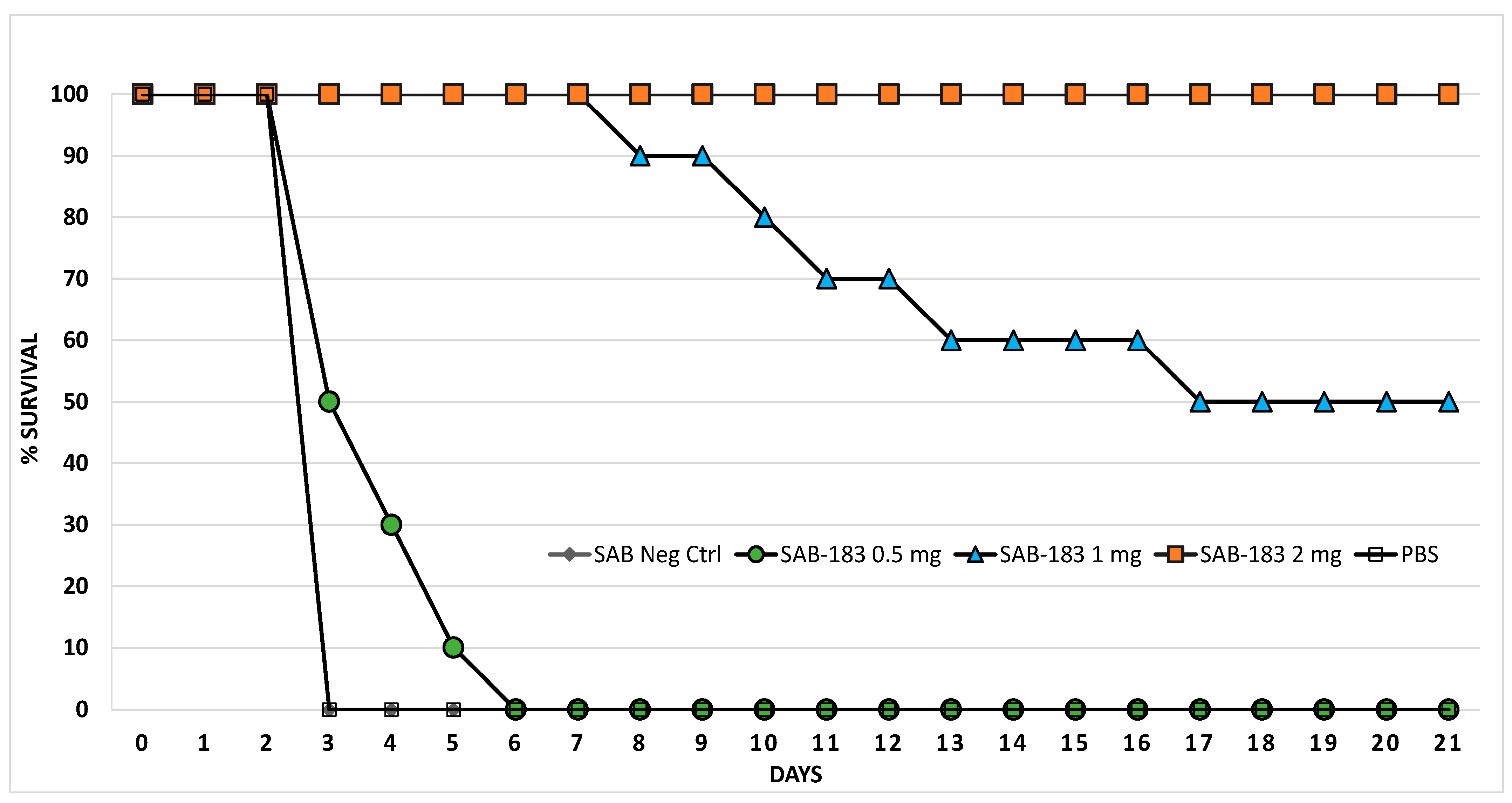
3.3. Human Anti-rF1-V Antibodies Derived from Transchromosomic (Tc) Bovines can Protect Mice after Exposure to Aerosolized Y. pestis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosario-Acevedo, R.; Biryukov, S.S.; Bozue, J.A.; Cote, C.K. Plague Prevention and Therapy: Perspectives on Current and Future Strategies. Biomedicines 2021, 9, 1421. [Google Scholar] [CrossRef] [PubMed]
- Demeure, C.; Dussurget, O.; Fiol, G.M.; Le Guern, A.S.; Savin, C.; Pizarro-Cerda, J. Yersinia pestis and plague: An updated view on evolution, virulence determinants, immune subversion, vaccination and diagnostics. Microbes Infect. 2019, 21, 202–212. [Google Scholar] [CrossRef]
- Titball, R.W.; Hill, J.; Lawton, D.G.; Brown, K.A. Yersinia pestis and plague. Biochem. Soc. Trans. 2003, 31, 104–107. [Google Scholar] [CrossRef]
- Pechous, R.D.; Sivaraman, V.; Stasulli, N.M.; Goldman, W.E. Pneumonic plague: The darker side of Yersinia pestis. Trends Microbiol. 2016, 24, 190–197. [Google Scholar] [CrossRef]
- Ligon, B.L. Plague: A review of its history and potential as a biological weapon. Semin. Pediatr. Infect. Dis. 2006, 17, 161–170. [Google Scholar] [CrossRef]
- Kugeler, K.J.; Mead, P.S.; Campbell, S.B.; Nelson, C.A. Antimicrobial Treatment Patterns and Illness Outcome Among United States Patients with Plague, 1942–2018. Clin. Infect. Dis. 2020, 70, S20–S26. [Google Scholar] [CrossRef] [PubMed]
- Godfred-Cato, S.; Cooley, K.M.; Fleck-Derderian, S.; Becksted, H.A.; Russell, Z.; Meaney-Delman, D.; Mead, P.S.; Nelson, C.A. Treatment of Human Plague: A Systematic Review of Published Aggregate Data on Antimicrobial Efficacy, 1939–2019. Clin. Infect. Dis. 2020, 70, S11–S19. [Google Scholar] [CrossRef]
- Lei, C.; Kumar, S. Yersinia pestis antibiotic resistance: A systematic review. Osong Public Health Res. Perspect. 2022, 13, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Andrianaivoarimanana, V.; Wagner, D.M.; Birdsell, D.N.; Nikolay, B.; Rakotoarimanana, F.; Randriantseheno, L.N.; Vogler, A.J.; Sahl, J.W.; Hall, C.M.; Somprasong, N.; et al. Transmission of antimicrobial resistant Yersinia pestis during a pneumonic plague outbreak. Clin. Infect. Dis. 2021, 74, 695–702. [Google Scholar] [CrossRef]
- Rabaan, A.A.; Al-Ahmed, S.H.; Alsuliman, S.A.; Aldrazi, F.A.; Alfouzan, W.A.; Haque, S. The rise of pneumonic plague in Madagascar: Current plague outbreak breaks usual seasonal mould. J. Med. Microbiol. 2019, 68, 292–302. [Google Scholar] [CrossRef]
- Cabanel, N.; Bouchier, C.; Rajerison, M.; Carniel, E. Plasmid-mediated doxycycline resistance in a Yersinia pestis strain isolated from a rat. Int. J. Antimicrob. Agents 2018, 51, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Hinnebusch, B.J.; Rosso, M.L.; Schwan, T.G.; Carniel, E. High-frequency conjugative transfer of antibiotic resistance genes to Yersinia pestis in the flea midgut. Mol. Microbiol. 2002, 46, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Guiyoule, A.; Gerbaud, G.; Buchrieser, C.; Galimand, M.; Rahalison, L.; Chanteau, S.; Courvalin, P.; Carniel, E. Transferable plasmid-mediated resistance to streptomycin in a clinical isolate of Yersinia pestis. Emerg. Infect. Dis. 2001, 7, 43. [Google Scholar] [CrossRef]
- Williamson, E.D.; Oyston, P.C. Protecting against plague: Towards a next-generation vaccine. Clin. Exp. Immunol. 2013, 172, 1–8. [Google Scholar] [CrossRef]
- Little, S.; Webster, W.; Wilhelm, H.; Fisher, D.; Norris, S.; Powell, B.; Enama, J.; Adamovicz, J. Quantitative anti-F1 and anti-V IgG ELISAs as serological correlates of protection against plague in female Swiss Webster mice. Vaccine 2010, 28, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Galyov, E.E.; Smirnov, O.; Karlishev, A.V.; Volkovoy, K.I.; Denesyuk, A.I.; Nazimov, I.V.; Rubtsov, K.S.; Abramov, V.M.; Dalvadyanz, S.M.; Zav’yalov, V.P. Nucleotide sequence of the Yersinia pestis gene encoding F1 antigen and the primary structure of the protein. Putative T and B cell epitopes. FEBS Lett. 1990, 277, 230–232. [Google Scholar] [CrossRef]
- Han, Y.; Zhou, D.; Pang, X.; Song, Y.; Zhang, L.; Bao, J.; Tong, Z.; Wang, J.; Guo, Z.; Zhai, J.; et al. Microarray analysis of temperature-induced transcriptome of Yersinia pestis. Microbiol. Immunol. 2004, 48, 791–805. [Google Scholar] [CrossRef]
- Du, Y.; Rosqvist, R.; Forsberg, A. Role of fraction 1 antigen of Yersinia pestis in inhibition of phagocytosis. Infect. Immun. 2002, 70, 1453–1460. [Google Scholar] [CrossRef]
- Cavanaugh, D.C.; Randall, R. The role of multiplication of Pasteurella pestis in mononuclear phagocytes in the pathogenesis of flea-borne plague. J. Immunol. 1959, 83, 348–363. [Google Scholar] [CrossRef]
- Schotthoefer, A.M.; Bearden, S.W.; Holmes, J.L.; Vetter, S.M.; Montenieri, J.A.; Williams, S.K.; Graham, C.B.; Woods, M.E.; Eisen, R.J.; Gage, K.L. Effects of temperature on the transmission of Yersinia pestis by the flea, Xenopsylla cheopis, in the late phase period. Parasit Vectors 2011, 4, 191. [Google Scholar] [CrossRef]
- Liu, F.; Chen, H.; Galvan, E.M.; Lasaro, M.A.; Schifferli, D.M. Effects of Psa and F1 on the adhesive and invasive interactions of Yersinia pestis with human respiratory tract epithelial cells. Infect. Immun. 2006, 74, 5636–5644. [Google Scholar] [CrossRef] [PubMed]
- Andrews, G.P.; Strachan, S.T.; Benner, G.E.; Sample, A.K.; Anderson, G.W., Jr.; Adamovicz, J.J.; Welkos, S.L.; Pullen, J.K.; Friedlander, A.M. Protective efficacy of recombinant Yersinia outer proteins against bubonic plague caused by encapsulated and nonencapsulated Yersinia pestis. Infect. Immun. 1999, 67, 1533–1537. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, M.I.; Noel, B.L.; Rampersaud, R.; Mena, P.; Benach, J.L.; Bliska, J.B. Vaccination of mice with a Yop translocon complex elicits antibodies that are protective against infection with F1- Yersinia pestis. Infect. Immun. 2008, 76, 5181–5190. [Google Scholar] [CrossRef] [PubMed]
- Worsham, P.L.; Stein, M.P.; Welkos, S.L. Construction of defined F1 negative mutants of virulent Yersinia pestis. Contrib. Microbiol. Immunol. 1995, 13, 325–328. [Google Scholar]
- Welkos, S.L.; Davis, K.M.; Pitt, L.M.; Worsham, P.L.; Freidlander, A.M. Studies on the contribution of the F1 capsule-associated plasmid pFra to the virulence of Yersinia pestis. Contrib. Microbiol. Immunol. 1995, 13, 299–305. [Google Scholar]
- Biryukov, S.; Dankmeyer, J.L.; Shamsuddin, Z.; Velez, I.; Rill, N.O.; Rosario-Acevedo, R.; Klimko, C.P.; Shoe, J.L.; Hunter, M.; Ward, M.D.; et al. Impact of Toll-like receptor-specific agonists on the host immune response to the Yersinia pestis plague rF1V vaccine. Front. Immunol. 2021, 12, 726416. [Google Scholar] [CrossRef]
- Williamson, E.D.; Eley, S.M.; Griffin, K.F.; Green, M.; Russell, P.; Leary, S.E.; Oyston, P.C.; Easterbrook, T.; Reddin, K.M.; Robinson, A.; et al. A new improved sub-unit vaccine for plague: The basis of protection. FEMS Immunol. Med. Microbiol. 1995, 12, 223–230. [Google Scholar] [CrossRef]
- Anderson, G.W., Jr.; Leary, S.E.; Williamson, E.D.; Titball, R.W.; Welkos, S.L.; Worsham, P.L.; Friedlander, A.M. Recombinant V antigen protects mice against pneumonic and bubonic plague caused by F1-capsule-positive and -negative strains of Yersinia pestis. Infect. Immun. 1996, 64, 4580–4585. [Google Scholar] [CrossRef]
- Pettersson, J.; Holmstrom, A.; Hill, J.; Leary, S.; Frithz-Lindsten, E.; von Euler-Matell, A.; Carlsson, E.; Titball, R.; Forsberg, A.; Wolf-Watz, H. The V-antigen of Yersinia is surface exposed before target cell contact and involved in virulence protein translocation. Mol. Microbiol. 1999, 32, 961–976. [Google Scholar] [CrossRef]
- Mota, L.J. Type III secretion gets an LcrV tip. Trends Microbiol. 2006, 14, 197–200. [Google Scholar] [CrossRef]
- Weeks, S.; Hill, J.; Friedlander, A.; Welkos, S. Anti-V antigen antibody protects macrophages from Yersinia pestis -induced cell death and promotes phagocytosis. Microb. Pathog. 2002, 32, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Cowan, C.; Philipovskiy, A.V.; Wulff-Strobel, C.R.; Ye, Z.; Straley, S.C. Anti-LcrV antibody inhibits delivery of Yops by Yersinia pestis KIM5 by directly promoting phagocytosis. Infect. Immun. 2005, 73, 6127–6137. [Google Scholar] [CrossRef] [PubMed]
- Eisele, N.A.; Anderson, D.M. Dual-function antibodies to Yersinia pestis LcrV required for pulmonary clearance of plague. Clin. Vaccine Immunol. 2009, 16, 1720–1727. [Google Scholar] [CrossRef]
- Pujol, C.; Bliska, J.B. Turning Yersinia pathogenesis outside in: Subversion of macrophage function by intracellular Yersiniae. Clin Immunol. 2005, 114, 216–226. [Google Scholar] [CrossRef]
- DiMezzo, T.L.; Ruthel, G.; Brueggemann, E.E.; Hines, H.B.; Ribot, W.J.; Chapman, C.E.; Powell, B.S.; Welkos, S.L. In vitro intracellular trafficking of virulence antigen during infection by Yersinia pestis. PLoS ONE 2009, 4, e6281. [Google Scholar] [CrossRef]
- Welkos, S.; Friedlander, A.; McDowell, D.; Weeks, J.; Tobery, S. V antigen of Yersinia pestis inhibits neutrophil chemotaxis. Microb. Pathog. 1998, 24, 185–196. [Google Scholar] [CrossRef]
- Hill, J.; Leary, S.E.; Griffin, K.F.; Williamson, E.D.; Titball, R.W. Regions of Yersinia pestis V antigen that contribute to protection against plague identified by passive and active immunization. Infect. Immun. 1997, 65, 4476–4482. [Google Scholar] [CrossRef]
- Anisimov, A.P.; Dentovskaya, S.V.; Panfertsev, E.A.; Svetoch, T.E.; Kopylov, P.K.; Segelke, B.W.; Zemla, A.; Telepnev, M.V.; Motin, V.L. Amino acid and structural variability of Yersinia pestis LcrV protein. Infect. Genet. Evol. 2010, 10, 137–145. [Google Scholar] [CrossRef]
- Roggenkamp, A.; Geiger, A.M.; Leitritz, L.; Kessler, A.; Heesemann, J. Passive immunity to infection with Yersinia spp. mediated by anti-recombinant V antigen is dependent on polymorphism of V antigen. Infect. Immun. 1997, 65, 446–451. [Google Scholar] [CrossRef]
- Daniel, C.; Dewitte, A.; Poiret, S.; Marceau, M.; Simonet, M.; Marceau, L.; Descombes, G.; Boutillier, D.; Bennaceur, N.; Bontemps-Gallo, S.; et al. Polymorphism in the Yersinia LcrV antigen enables immune escape from the protection conferred by an LcrV-secreting Lactococcus Lactis in a Pseudotuberculosis Mouse Model. Front Immunol. 2019, 2, 1830. [Google Scholar] [CrossRef]
- Williamson, E.D.; Packer, P.J.; Waters, E.L.; Simpson, A.J.; Dyer, D.; Hartings, J.; Twenhafel, N.; Pitt, M.L. Recombinant (F1+V) vaccine protects cynomolgus macaques against pneumonic plague. Vaccine 2011, 29, 4771–4777. [Google Scholar] [CrossRef]
- Goodin, J.L.; Powell, B.S.; Enama, J.T.; Raab, R.W.; McKown, R.L.; Coffman, G.L.; Andrews, G.P. Purification and characterization of a recombinant Yersinia pestis V-F1 “Reversed” fusion protein for use as a new subunit vaccine against plague. Protein Expr. Purif. 2011, 76, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Heath, D.G.; Anderson Jr, G.W.; Mauro, J.M.; Welkos, S.L.; Andrews, G.P.; Adamovicz, J.; Friedlander, A.M. Protection against experimental bubonic and pneumonic plague by a recombinant capsular F1-V antigen fusion protein vaccine. Vaccine 1998, 16, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Williamson, E.; Flick-Smith, H.; Waters, E.; Miller, J.; Hodgson, I.; Le Butt, C.; Hill, J. Immunogenicity of the rF1+ rV vaccine for plague with identification of potential immune correlates. Microb. Pathog. 2007, 42, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Williamson, E.; Stagg, A.; Eley, S.; Taylor, R.; Green, M.; Jones, S.; Titball, R. Kinetics of the immune response to the (F1+ V) vaccine in models of bubonic and pneumonic plague. Vaccine 2007, 25, 1142–1148. [Google Scholar] [CrossRef]
- Williamson, E.; Flick-Smith, H.; Lebutt, C.; Rowland, C.; Jones, S.; Waters, E.; Gwyther, R.; Miller, J.; Packer, P.; Irving, M. Human immune response to a plague vaccine comprising recombinant F1 and V antigens. Infect. Immun. 2005, 73, 3598–3608. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.; Copse, C.; Leary, S.; Stagg, A.J.; Williamson, E.D.; Titball, R.W. Synergistic protection of mice against plague with monoclonal antibodies specific for the F1 and V antigens of Yersinia pestis. Infect. Immun. 2003, 71, 2234–2238. [Google Scholar] [CrossRef]
- Xiao, X.; Zhu, Z.; Dankmeyer, J.L.; Wormald, M.M.; Fast, R.L.; Worsham, P.L.; Cote, C.K.; Amemiya, K.; Dimitrov, D.S. Human anti-plague monoclonal antibodies protect mice from Yersinia pestis in a bubonic plague model. PLoS ONE 2010, 5, e13047. [Google Scholar] [CrossRef]
- Anderson, G.W., Jr.; Worsham, P.L.; Bolt, C.R.; Andrews, G.P.; Welkos, S.L.; Friedlander, A.M.; Burans, J.P. Protection of mice from fatal bubonic and pneumonic plague by passive immunization with monoclonal antibodies against the F1 protein of Yersinia pestis. Am. J. Trop. Med. Hyg. 1997, 56, 471–473. [Google Scholar] [CrossRef]
- Zauberman, A.; Cohen, S.; Levy, Y.; Halperin, G.; Lazar, S.; Velan, B.; Shafferman, A.; Flashner, Y.; Mamroud, E. Neutralization of Yersinia pestis-mediated macrophage cytotoxicity by anti-LcrV antibodies and its correlation with protective immunity in a mouse model of bubonic plague. Vaccine 2008, 26, 1616–1625. [Google Scholar] [CrossRef]
- Fellows, P.; Adamovicz, J.; Hartings, J.; Sherwood, R.; Mega, W.; Brasel, T.; Barr, E.; Holland, L.; Lin, W.; Rom, A.; et al. Protection in mice passively immunized with serum from cynomolgus macaques and humans vaccinated with recombinant plague vaccine (rF1V). Vaccine 2010, 28, 7748–7756. [Google Scholar] [CrossRef]
- Friedlander, A.M.; Welkos, S.L.; Worsham, P.L.; Andrews, G.P.; Heath, D.G.; Anderson Jr, G.W.; Pitt, M.L.; Estep, J.; Davis, K. Relationship between virulence and immunity as revealed in recent studies of the Fl capsule of Yersinia pestis. Clin. Infect. Dis. 1995, 21, S178–S181. [Google Scholar] [CrossRef] [PubMed]
- Graham, V.; Hatch, G.; Bewley, K.; Steeds, K.; Lansley, A.; Bate, S.; Funnell, S. Efficacy of primate humoral passive transfer in a murine model of pneumonic plague is mouse strain-dependent. J. Immunol. Res. 2014, 2014, 807564. [Google Scholar] [CrossRef]
- Motin, V.L.; Nakajima, R.; Smirnov, G.B.; Brubaker, R.R. Passive immunity to yersiniae mediated by anti-recombinant V antigen and protein AV antigen fusion peptide. Infect. Immun. 1994, 62, 4192–4201. [Google Scholar] [CrossRef]
- Walker, R.V. Studies on the immune response of guinea pigs to the envelope substance of Pasteurella pestis. I. Immunogenicity and persistence of large doses of fraction I in guinea pigs observed with fluorescent antibody. J. Immunol. 1962, 88, 153–163. [Google Scholar] [CrossRef]
- Hill, J.; Eyles, J.E.; Elvin, S.J.; Healey, G.D.; Lukaszewski, R.A.; Titball, R.W. Administration of antibody to the lung protects mice against pneumonic plague. Infect. Immun. 2006, 74, 3068–3070. [Google Scholar] [CrossRef]
- Gardner, C.L.; Sun, C.; Luke, T.; Raviprakash, K.; Wu, H.; Jiao, J.A.; Sullivan, E.; Reed, D.S.; Ryman, K.D.; Klimstra, W.B. Antibody Preparations from Human Transchromosomic Cows Exhibit Prophylactic and Therapeutic Efficacy against Venezuelan Equine Encephalitis Virus. J. Virol. 2017, 91, e00226-17. [Google Scholar] [CrossRef]
- Gilliland, T.; Liu, Y.; Li, R.; Dunn, M.; Cottle, E.; Terada, Y.; Ryckman, Z.; Alcorn, M.; Vasilatos, S.; Lundy, J.; et al. Protection of human ACE2 transgenic Syrian hamsters from SARS CoV-2 variants by human polyclonal IgG from hyper-immunized transchromosomic bovines. bioRxiv 2021. [Google Scholar] [CrossRef]
- Liu, Z.; Wu, H.; Egland, K.A.; Gilliland, T.C.; Dunn, M.D.; Luke, T.C.; Sullivan, E.J.; Klimstra, W.B.; Bausch, C.L.; Whelan, S.P.J. Human immunoglobulin from transchromosomic bovines hyperimmunized with SARS-CoV-2 spike antigen efficiently neutralizes viral variants. Hum. Vaccin. Immunother. 2022, 18, 1940652. [Google Scholar] [CrossRef]
- Luke, T.; Bennett, R.S.; Gerhardt, D.M.; Burdette, T.; Postnikova, E.; Mazur, S.; Honko, A.N.; Oberlander, N.; Byrum, R.; Ragland, D.; et al. Fully Human Immunoglobulin G From Transchromosomic Bovines Treats Nonhuman Primates Infected With Ebola Virus Makona Isolate. J. Infect. Dis. 2018, 218, S636–S648. [Google Scholar] [CrossRef]
- Luke, T.; Wu, H.; Zhao, J.; Channappanavar, R.; Coleman, C.M.; Jiao, J.A.; Matsushita, H.; Liu, Y.; Postnikova, E.N.; Ork, B.L.; et al. Human polyclonal immunoglobulin G from transchromosomic bovines inhibits MERS-CoV in vivo. Sci. Transl. Med. 2016, 8, 326ra321. [Google Scholar] [CrossRef]
- Perley, C.C.; Brocato, R.L.; Wu, H.; Bausch, C.; Karmali, P.P.; Vega, J.B.; Cohen, M.V.; Somerville, B.; Kwilas, S.A.; Principe, L.M.; et al. Anti-HFRS Human IgG Produced in Transchromosomic Bovines Has Potent Hantavirus Neutralizing Activity and Is Protective in Animal Models. Front. Microbiol. 2020, 11, 832. [Google Scholar] [CrossRef]
- Welkos, S.; Pitt, M.; Martinez, M.; Friedlander, A.; Vogel, P.; Tammariello, R. Determination of the virulence of the pigmentation-deficient and pigmentation-/plasminogen activator-deficient strains of Yersinia pestis in non-human primate and mouse models of pneumonic plague. Vaccine 2002, 20, 2206–2214. [Google Scholar] [CrossRef]
- Doll, J.M.; Zeitz, P.S.; Ettestad, P.; Bucholtz, A.L.; Davis, T.; Gage, K. Cat-transmitted fatal pneumonic plague in a person who traveled from Colorado to Arizona. Am. J. Trop. Med. Hyg. 1994, 51, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, H.; Sano, A.; Wu, H.; Jiao, J.A.; Kasinathan, P.; Sullivan, E.J.; Wang, Z.; Kuroiwa, Y. Triple immunoglobulin gene knockout transchromosomic cattle: Bovine lambda cluster deletion and its effect on fully human polyclonal antibody production. PLoS ONE 2014, 9, e90383. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, H.; Sano, A.; Wu, H.; Wang, Z.; Jiao, J.A.; Kasinathan, P.; Sullivan, E.J.; Kuroiwa, Y. Species-Specific Chromosome Engineering Greatly Improves Fully Human Polyclonal Antibody Production Profile in Cattle. PLoS ONE 2015, 10, e0130699. [Google Scholar] [CrossRef] [PubMed]
- Dye, J.M.; Wu, H.; Hooper, J.W.; Khurana, S.; Kuehne, A.I.; Coyle, E.M.; Ortiz, R.A.; Fuentes, S.; Herbert, A.S.; Golding, H.; et al. Production of Potent Fully Human Polyclonal Antibodies against Ebola Zaire Virus in Transchromosomal Cattle. Sci. Rep. 2016, 6, 24897. [Google Scholar] [CrossRef]
- Rosqvist, R.; Bolin, I.; Wolf-Watz, H. Inhibition of phagocytosis in Yersinia pseudotuberculosis: A virulence plasmid-encoded ability involving the Yop2b protein. Infect. Immun. 1988, 56, 2139–2143. [Google Scholar] [CrossRef]
- Wiley, D.J.; Rosqvist, R.; Schesser, K. Induction of the Yersinia type 3 secretion system as an all-or-none phenomenon. J. Mol. Biol. 2007, 373, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Bozue, J.; Cote, C.K.; Chance, T.; Kugelman, J.; Kern, S.J.; Kijek, T.K.; Jenkins, A.; Mou, S.; Moody, K.; Fritz, D.; et al. A Yersinia pestis tat mutant is attenuated in bubonic and small-aerosol pneumonic challenge models of infection but not as attenuated by intranasal challenge. PLoS ONE 2014, 9, e104524. [Google Scholar] [CrossRef]
- Bozue, J.; Cote, C.K.; Webster, W.; Bassett, A.; Tobery, S.; Little, S.; Swietnicki, W. A Yersinia pestis YscN ATPase mutant functions as a live attenuated vaccine against bubonic plague in mice. FEMS Microbiol. Lett. 2012, 332, 113–121. [Google Scholar] [CrossRef]
- Heine, H.S.; Louie, A.; Sorgel, F.; Bassett, J.; Miller, L.; Sullivan, L.J.; Kinzig-Schippers, M.; Drusano, G.L. Comparison of 2 antibiotics that inhibit protein synthesis for the treatment of infection with Yersinia pestis delivered by aerosol in a mouse model of pneumonic plague. J. Infect. Dis. 2007, 196, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Maeda, D.; Batista, M.T.; Pereira, L.R.; de Jesus Cintra, M.; Amorim, J.H.; Mathias-Santos, C.; Pereira, S.A.; Boscardin, S.B.; Silva, S.D.R.; Faquim-Mauro, E.L.; et al. Adjuvant-Mediated Epitope Specificity and Enhanced Neutralizing Activity of Antibodies Targeting Dengue Virus Envelope Protein. Front. Immunol. 2017, 8, 1175. [Google Scholar] [CrossRef] [PubMed]
- Amemiya, K.; Dankmeyer, J.L.; Keasey, S.L.; Trevino, S.R.; Wormald, M.M.; Halasohoris, S.A.; Ribot, W.J.; Fetterer, D.P.; Cote, C.K.; Worsham, P.L.; et al. Binding Sites of Anti-Lcr V Monoclonal Antibodies Are More Critical than the Avidities and Affinities for Passive Protection against Yersinia pestis Infection in a Bubonic Plague Model. Antibodies 2020, 9, 37. [Google Scholar] [CrossRef]
- Anisimov, A.P.; Panfertsev, E.A.; Svetoch, T.E.; Dentovskaya, S.V. Variability of the protein sequences of lcrV between epidemic and atypical rhamnose-positive strains of Yersinia pestis. Adv. Exp. Med. Biol. 2007, 603, 23–27. [Google Scholar] [CrossRef]
- Baker, S.; Kellam, P.; Krishna, A.; Reece, S. Protecting intubated patients from the threat of antimicrobial resistant infections with monoclonal antibodies. Lancet Microbe 2020, 1, e191–e192. [Google Scholar] [CrossRef]
- Fan, G.; Li, J. Engineering Antibodies for the Treatment of Infectious Diseases. Adv. Exp. Med. Biol. 2017, 1053, 207–220. [Google Scholar] [CrossRef]
- Khan, A.A.; Manzoor, K.N.; Sultan, A.; Saeed, M.; Rafique, M.; Noushad, S.; Talib, A.; Rentschler, S.; Deigner, H.P. Pulling the Brakes on Fast and Furious Multiple Drug-Resistant (MDR) Bacteria. Int. J. Mol. Sci. 2021, 22, 859. [Google Scholar] [CrossRef] [PubMed]
- Burki, T. Plague in Madagascar. Lancet Infect. Dis. 2017, 17, 1241. [Google Scholar] [CrossRef]
- Kmietowicz, Z. Pneumonic plague outbreak hits cities in Madagascar. BMJ 2017, 359, j4595. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SAB-183 Neg. Ctrl. | SAB-183 a | |||
|---|---|---|---|---|
| Antigen | Antibody Titer b | |||
| Median (Q1, Q3) | GEO Mean (GSE) | Median (Q1, Q3) | GEO Mean (GSE) | |
| F1 | 5.0 (5.0, 5.0) | 5.0 (1.0) | 16,612.6 (16,612.6, 16,612.6) | 16,612.6 (1.0) |
| LcrV | 10.0 (10.0, 20.0) | 12.6 (1.3) | 83,063.0 (83,063.0, 83,063.0) | 83,063.0 (1.0) |
| TS CO92 | 320.0 (320.0, 320.0) | 320.0 (1.0) | 4153.2 (4153.2, 4153.2) | 4153.2 (1.0) |
| TS C12 | 160.0 (160.0, 320.0) | 201.6 (1.3) | 1038.3 (1308.3, 2076.6) | 1308.2 (1.3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biryukov, S.S.; Wu, H.; Dankmeyer, J.L.; Rill, N.O.; Klimko, C.P.; Egland, K.A.; Shoe, J.L.; Hunter, M.; Fetterer, D.P.; Qiu, J.; et al. Polyclonal Antibodies Derived from Transchromosomic Bovines Vaccinated with the Recombinant F1-V Vaccine Increase Bacterial Opsonization In Vitro and Protect Mice from Pneumonic Plague. Antibodies 2023, 12, 33. https://doi.org/10.3390/antib12020033
Biryukov SS, Wu H, Dankmeyer JL, Rill NO, Klimko CP, Egland KA, Shoe JL, Hunter M, Fetterer DP, Qiu J, et al. Polyclonal Antibodies Derived from Transchromosomic Bovines Vaccinated with the Recombinant F1-V Vaccine Increase Bacterial Opsonization In Vitro and Protect Mice from Pneumonic Plague. Antibodies. 2023; 12(2):33. https://doi.org/10.3390/antib12020033
Chicago/Turabian StyleBiryukov, Sergei S., Hua Wu, Jennifer L. Dankmeyer, Nathaniel O. Rill, Christopher P. Klimko, Kristi A. Egland, Jennifer L. Shoe, Melissa Hunter, David P. Fetterer, Ju Qiu, and et al. 2023. "Polyclonal Antibodies Derived from Transchromosomic Bovines Vaccinated with the Recombinant F1-V Vaccine Increase Bacterial Opsonization In Vitro and Protect Mice from Pneumonic Plague" Antibodies 12, no. 2: 33. https://doi.org/10.3390/antib12020033
APA StyleBiryukov, S. S., Wu, H., Dankmeyer, J. L., Rill, N. O., Klimko, C. P., Egland, K. A., Shoe, J. L., Hunter, M., Fetterer, D. P., Qiu, J., Davies, M. L., Bausch, C. L., Sullivan, E. J., Luke, T., & Cote, C. K. (2023). Polyclonal Antibodies Derived from Transchromosomic Bovines Vaccinated with the Recombinant F1-V Vaccine Increase Bacterial Opsonization In Vitro and Protect Mice from Pneumonic Plague. Antibodies, 12(2), 33. https://doi.org/10.3390/antib12020033