Recent Trends in Active and Passive Immunotherapies of Alzheimer’s Disease


Abstract
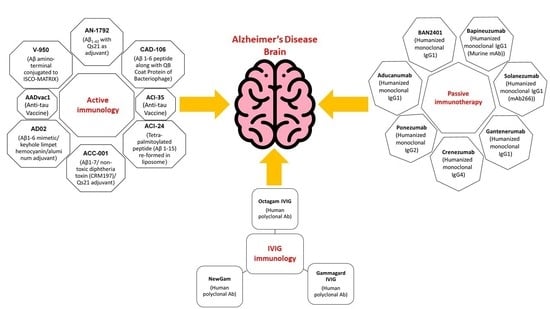
:
1. Introduction
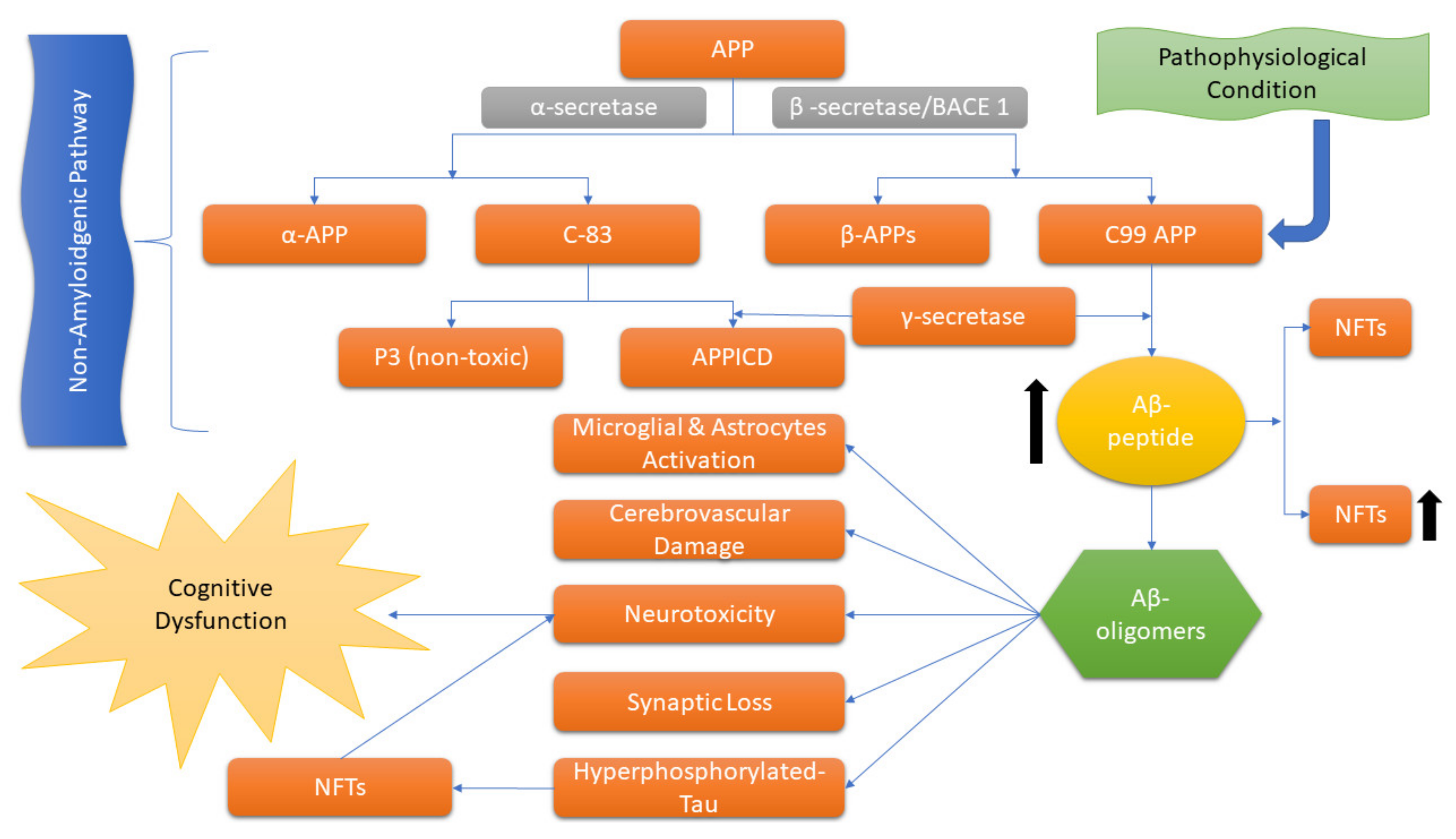
2. Etiopathophysiology of AD
3. Involvement of the Immune System and Inflammation in AD
4. Diagnoses
5. Current Treatments for AD
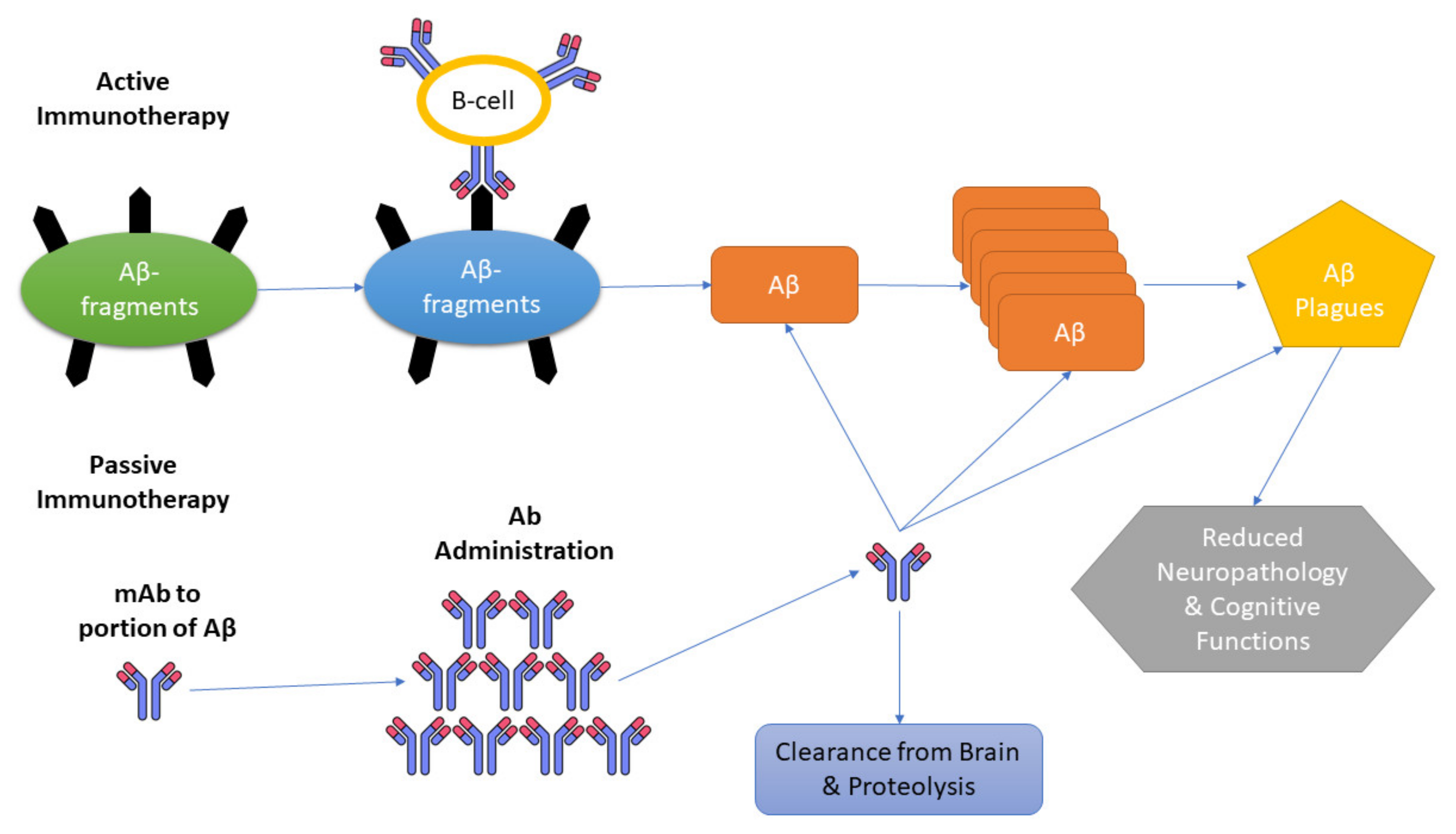
6. Immunotherapy and Its Types
7. Preclinical Immunotherapy Trials in Animal Models
8. Contributory Factors behind AD and Mechanisms of Their Clearance
8.1. Initiation of Microglia and Macrophages
8.2. Peripheral Sink Hypothesis
8.3. Aβ Oligomers Neutralization by Antibodies
9. Active Immunization (Vaccine) Clinical Trials
10. Active Immunotherapy Antibodies
10.1. Tau Immunotherapy
10.2. ACI-35
10.3. AADvac1

{kind=link}
{kind=link}
{kind=link}
| Aβ Active Immunotherapy Clinical Trials | ||||||||
|---|---|---|---|---|---|---|---|---|
| Drug | Sponsor | Vaccine Type | Target (Aβ/Tau) | Trial Phase and Status | Immunology | Positive Outcomes | Negative Outcomes | References |
| AN-1792 | ELAN (Dublin, Ireland) | Anti-Aβ vaccine (Aβ 1–42 with Qs21 as adjuvant). | Aβ N-terminus | II Halted, no improvement (NCT00021723) | Induction of anti-Aβ titers by B and T-cell activation. | ↓↓CSF tau and no change in CSF Aβ 42 level. | ~6% of cases developed Meningoencephalitis and cerebral microhemorrhage | [136] |
| CAD-106 | Novartis (Basel, Switzerland) | Anti-Aβ vaccine Aβ 1–6 peptide along with QB coat protein of bacteriophage. | Aβ N-terminus (AB1-6) | II Ongoing (NCT00956410, NCT01023685, NCT00795418, NCT01097096, NCT00733863, NCT00411580) | Induction of anti-Aβ titers without T-cell activation. | Safe and well-tolerated, ↑↑Total serum Aβ, ↓↓and free Aβ in plasma while CSF t-tau, p-tau, and Aβ-40 and 42 remain unchanged. | The occurrence of ARIA in a few cases. | [137,138,139] |
| ACI-24 | AC immune (Lausanne, Switzerland) | Tetra-palmitoylated peptide (Aβ 1–15) re-formed in liposome. | B sheet conformation of Aβ | I/II Ongoing (NCT02738450) | The non-inflammatory response of Th2 helper cells against Aβ. | ↓↓ insoluble Aβ40 and 42 and soluble Aβ42. | No significant adverse effects. | [117,137,138] |
| ACC-001 | Pfizer (New York, NY, USA)/Janssen (Titusville, NJ, USA) | Anti-Aβ vaccine Aβ 1–7/non-toxic diphtheria toxin (CRM197)/Qs21 adjuvant. | Aβ 1–7 | II (Completed) Additional Phase II is ongoing(NCT01284387, NCT00955409, NCT01227564, NCT00960531, NCT01238991, NCT00752232, NCT00959192, NCT00498602, NCT00479557) | Induces antibody’s response against Aβ. | Safe and well-tolerated, ↑↑plasma Aβ40, ↓↓CSF p-tau slightly, while other CSF biomarkers remain unchanged. | Local injection reactions and headaches; ARIA-E occurs in few cases. | [140] |
| AD02 | GlaxoSmithKline (Brentford, UK)/AFFiRiS (Vienna, Austria) | Aβ 1–6 mimetic/keyhole limpet hemocyanin/aluminum adjuvant. | Mimotope of Aβ N-terminal | II Ongoing (NCT01093664, NCT01117818, NCT02008513, NCT00633841, NCT00711321, NCT01357629, NCT01614132, NCT00003453, NCT00996008) | Stimulate the immune system to make antibodies against Aβ. | Safe; no detailed outcomes. | The non-endogenous nature of drugs avoids the development of tolerance. | [138] |
| V-950 | Merck and Co. (Kenilworth, NJ, USA) | Aβ amino-terminal conjugated to ISCO-MATRIX. | Aβ | I (Discontinued) (NCT00464334) | Production of anti-Aβ antibodies. | Results unpublished. | AE’s rate is high. Mostly Fatigue, nausea, anemia diarrhea, while in a few cases arrhythmia, dysphagia. | [138,141] |
| Tau Active Immunotherapy | ||||||||
| AADvac1 | Axon Neuroscience (Bratislava, Slovakia) | Anti-tau vaccine | Tau derived peptide (294–305 aa) | I (NCT02031198, NCT01850238, NCT02579252, NCT03174886) | Antibodies are directed against p-tau and promote tau clearance. | Safe; ↓↓tau aggregates. Improved cognition. | No significant adverse effects. | [135,138] |
| ACI-35 | AC immune (Lausanne, Switzerland) | Anti-tau vaccine | Tau derived peptide (294–305 a.a) | I (NCT04445831) | Stimulate immune system B and T-cell response. Antibodies are directed against p-tau and promote tau clearance. | ↓↓soluble and insoluble tau. | No significant adverse effects. | [138,142] |
11. Passive Immunotherapy
11.1. Antibody Bapineuzumab First-Generation Anti-Fibrillar Forms of Aβ
11.2. Antibody Solanezumab First-Generation against Soluble Monomeric Forms of Aβ
12. Intravenous Immunoglobulin (IVIG) Immunotherapy
13. Future Research and Limitations
14. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Srivastava, S.; Ahmad, R.; Khare, S.K. Alzheimer’s disease and its treatment by different approaches: A review. Eur. J. Med. Chem. 2021, 216, 113320. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, F.; Miraglia, F.; Piludu, F.; Granata, G.; Romanello, R.; Caulo, M.; Onofrj, V.; Bramanti, P.; Colosimo, C.; Rossini, P.M. “Small World” architecture in brain connectivity and hippocampal volume in Alzheimer’s disease: A study via graph theory from EEG data. Brain Imaging Behav. 2017, 11, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Association, A. 2017 Alzheimer’s disease facts and figures. Alzheimers Dement. 2017, 13, 325–373. [Google Scholar] [CrossRef]
- Association, A. 2021 Alzheimer’s disease facts and figures. Alzheimers Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef]
- Pérez-Palma, E.; Bustos, B.I.; Villamán, C.F.; Alarcón, M.A.; Avila, M.E.; Ugarte, G.D.; Reyes, A.E.; Opazo, C.; De Ferrari, G.V. Overrepresentation of glutamate signaling in Alzheimer’s disease: Network-based pathway enrichment using meta-analysis of genome-wide association studies. PLoS ONE 2014, 9, e95413. [Google Scholar] [CrossRef] [Green Version]
- Hatami, A.; Monjazeb, S.; Milton, S.; Glabe, C.G. Familial Alzheimer’s disease mutations within the amyloid precursor protein alter the aggregation and conformation of the amyloid-β peptide. J. Biol. Chem. 2017, 292, 3172–3185. [Google Scholar] [CrossRef] [Green Version]
- Portet, F.; Ousset, P.J.; Visser, P.J.; Frisoni, G.B.; Nobili, F.; Scheltens, P.; Vellas, B.; Touchon, J. Mild cognitive impairment (MCI) in medical practice: A critical review of the concept and new diagnostic procedure. Report of the MCI Working Group of the European Consortium on Alzheimer’s Disease. J. Neurol. Neurosurg. Psychiatry 2006, 77, 714–718. [Google Scholar] [CrossRef] [Green Version]
- Sakai, K.; Boche, D.; Carare, R.; Johnston, D.; Holmes, C.; Love, S.; Nicoll, J.A.R. Aβ immunotherapy for Alzheimer’s disease: Effects on apoE and cerebral vasculopathy. Acta Neuropathol. 2014, 128, 777–789. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Gandy, S.; Heppner, F.L. Microglia as Dynamic and Essential Components of the Amyloid Hypothesis. Neuron 2013, 78, 575–577. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Perry, V.H.; Holmes, C. Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 2014, 10, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Angulo, E.; Casadó, V.; Mallol, J.; Canela, E.I.; Viñals, F.; Ferrer, I.; Lluis, C.; Franco, R. A1 Adenosine Receptors Accumulate in Neurodegenerative Structures in Alzheimer Disease and Mediate Both Amyloid Precursor Protein Processing and Tau Phosphorylation and Translocation. Brain Pathol. 2003, 13, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Prokop, S.; Miller, K.R.; Heppner, F.L. Microglia actions in Alzheimer’s disease. Acta Neuropathol. 2013, 126, 461–477. [Google Scholar] [CrossRef]
- Deture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction between Aβ and Tau in the Pathogenesis of Alzheimer’s Disease. Int. J. Biol. Sci. 2021, 2021, 2181–2192. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T.; et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D. If amyloid drives Alzheimer disease, why have anti-amyloid therapies not yet slowed cognitive decline? PLoS Biol. 2022, 20, e3001694. [Google Scholar] [CrossRef]
- Jorfi, M.; Maaser-Hecker, A.; Tanzi, R.E. The neuroimmune axis of Alzheimer’s disease. Genome Med. 2023, 15, 1–25. [Google Scholar] [CrossRef]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Willbold, D.; Strodel, B.; Schröder, G.F.; Hoyer, W.; Heise, H. Amyloid-type Protein Aggregation and Prion-like Properties of Amyloids. Chem. Rev. 2021, 121, 8285–8307. [Google Scholar] [CrossRef]
- Moreno-Gonzalez, I.; Edwards, G.; Morales, R.; Duran-Aniotz, C.; Escobedo, G.; Marquez, M.; Pumarola, M.; Soto, C. Aged Cattle Brain Displays Alzheimer’s Disease-Like Pathology and Promotes Brain Amyloidosis in a Transgenic Animal Model. Front. Aging Neurosci. 2022, 13, 963. [Google Scholar] [CrossRef] [PubMed]
- Jo, M.; Lee, S.; Jeon, Y.M.; Kim, S.; Kwon, Y.; Kim, H.J. The role of TDP-43 propagation in neurodegenerative diseases: Integrating insights from clinical and experimental studies. Exp. Mol. Med. 2020, 52, 1652–1662. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C. Prion-like mechanisms in Alzheimer disease. In Handbook of Clinical Neurology; NIH Public Access: Bethesda, MD, USA, 2018; Volume 153, pp. 303–319. [Google Scholar]
- Li, J.-W.; Zong, Y.; Cao, X.-P.; Tan, L.; Tan, L. Microglial priming in Alzheimer’s disease. Ann. Transl. Med. 2018, 6, 176. [Google Scholar] [CrossRef]
- Li, C.; Zhao, R.; Gao, K.; Wei, Z.; Yaoyao Yin, M.; Ting Lau, L.; Chui, D.; Cheung Hoi Yu, A. Astrocytes: Implications for Neuroinflammatory Pathogenesis of Alzheimers Disease. Curr. Alzheimer Res. 2011, 8, 67–80. [Google Scholar] [CrossRef] [Green Version]
- Vinters, H.V. Emerging concepts in alzheimer’s disease. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 291–319. [Google Scholar] [CrossRef]
- Aron, L.; Yankner, B.A. Neurodegenerative disorders: Neural synchronization in Alzheimer’s disease. Nature 2016, 540, 207–208. [Google Scholar] [CrossRef] [Green Version]
- Ku, M.C.; Wolf, S.A.; Respondek, D.; Matyash, V.; Pohlmann, A.; Waiczies, S.; Waiczies, H.; Niendorf, T.; Synowitz, M.; Glass, R.; et al. GDNF mediates glioblastoma-induced microglia attraction but not astrogliosis. Acta Neuropathol. 2013, 125, 609–620. [Google Scholar] [CrossRef]
- Liu, C.; Cui, G.; Zhu, M.; Kang, X.; Guo, H. Neuroinflammation in Alzheimer’s disease: Chemokines produced by astrocytes and chemokine receptors. Int. J. Clin. Exp. Pathol. 2014, 7, 8342–8355. [Google Scholar]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Thériault, P.; Elali, A.; Rivest, S. The dynamics of monocytes and microglia in Alzheimer’s disease. Alzheimers Res. Ther. 2015, 7, 41. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Pozo, A.; Muzikansky, A.; Gómez-Isla, T.; Growdon, J.H.; Betensky, R.A.; Frosch, M.P.; Hyman, B.T. Differential relationships of reactive astrocytes and microglia to fibrillar amyloid deposits in alzheimer disease. J. Neuropathol. Exp. Neurol. 2013, 72, 462–471. [Google Scholar] [CrossRef] [Green Version]
- Vandenbark, A.A.; Offner, H.; Matejuk, S.; Matejuk, A. Microglia and astrocyte involvement in neurodegeneration and brain cancer. J. Neuroinflamm. 2021, 18, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hickman, S.E.; Khoury, J. El Mechanisms of Mononuclear Phagocyte Recruitment in Alzheimers Disease. CNS Neurol. Disord. Drug Targets 2012, 9, 168–173. [Google Scholar] [CrossRef]
- Goldmann, T.; Wieghofer, P.; Jordão, M.J.C.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 2016, 17, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Olmos-Alonso, A.; Schetters, S.T.T.; Sri, S.; Askew, K.; Mancuso, R.; Vargas-Caballero, M.; Holscher, C.; Perry, V.H.; Gomez-Nicola, D. Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain 2016, 139, 891–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; O’Connor, T.; Vassar, R. The contribution of activated astrocytes to Aβ production: Implications for Alzheimer’s disease pathogenesis. J. Neuroinflamm. 2011, 8, 150. [Google Scholar] [CrossRef] [Green Version]
- Turola, E.; Furlan, R.; Bianco, F.; Matteoli, M.; Verderio, C. Microglial microvesicle secretion and intercellular signaling. Front. Physiol. 2012, 3, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetzl, E.J.; Mustapic, M.; Kapogiannis, D.; Eitan, E.; Lobach, I.V.; Goetzl, L.; Schwartz, J.B.; Miller, B.L. Cargo proteins of plasma astrocyte-derived exosomes in Alzheimer’s disease. FASEB J. 2016, 30, 3853–3859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.S.; Lee, H.J.; Lim, I.; Satoh, J.I.; Kim, S.U. Human astrocytes: Secretome profiles of cytokines and chemokines. PLoS ONE 2014, 9, e92325. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D. Astrocyte physiopathology: At the crossroads of intercellular networking, inflammation and cell death. Prog. Neurobiol. 2015, 130, 86–120. [Google Scholar] [CrossRef]
- Song, J.H.; Yu, J.T.; Tan, L. Brain-Derived Neurotrophic Factor in Alzheimer’s Disease: Risk, Mechanisms, and Therapy. Mol. Neurobiol. 2015, 52, 1477–1493. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Combs, C.K.; Colleen Karlo, J.; Kao, S.C.; Landreth, G.E. β-amyloid stimulation of microglia anti monocytes results in TNFα-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J. Neurosci. 2001, 21, 1179–1188. [Google Scholar] [CrossRef] [Green Version]
- Ho, G.J.; Drego, R.; Hakimian, E.; Masliah, E. Mechanisms of cell signaling and inflammation in Alzheimer’s disease. Curr. Drug Targets Inflamm. Allergy 2005, 4, 247–256. [Google Scholar] [CrossRef]
- Verri, M.; Pastoris, O.; Dossena, M.; Aquilani, R.; Guerriero, F.; Cuzzoni, G.; Venturini, L.; Ricevuti, G.; Bongiorno, A.I. Mitochondrial alterations, oxidative stress and neuroinflammation in Alzheimer’s disease. Int. J. Immunopathol. Pharmacol. 2012, 25, 345–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, S.; Sato, N.; Uchio-Yamada, K.; Sawada, K.; Kunieda, T.; Takeuchi, D.; Kurinami, H.; Shinohara, M.; Rakugi, H.; Morishita, R. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Aβ deposition in an Alzheimer mouse model with diabetes. Proc. Natl. Acad. Sci. USA 2010, 107, 7036–7041. [Google Scholar] [CrossRef] [Green Version]
- Holmes, C.; Cunningham, C.; Perry, V.H. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009, 74, 1157–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalermpalanupap, T.; Schroeder, J.P.; Rorabaugh, J.M.; Liles, L.C.; Lah, J.J.; Levey, A.I.; Weinshenker, D. Locus coeruleus ablation exacerbates cognitive deficits, neuropathology, and lethality in P301S tau transgenic mice. J. Neurosci. 2018, 38, 74–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiner, M.; Harvey, D.; Hayes, J.; Landau, S.; Aisen, P.; Petersen, R.; Tosun, D.; Veitch, D.; Jack, C.; Decarli, C.; et al. Effects of traumatic brain injury and posttraumatic stress disorder on development of Alzheimer’s disease in Vietnam Veterans using the Alzheimer’s Disease Neuroimaging Initiative: Preliminary report. Alzheimers Dement. Transl. Res. Clin. Interv. 2017, 3, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, E.M.; Chibnik, L.B.; Keenan, B.T.; Ottoboni, L.; Raj, T.; Tang, A.; Rosenkrantz, L.L.; Imboywa, S.; Lee, M.; Von Korff, A.; et al. CD33 Alzheimer’s disease locus: Altered monocyte function and amyloid biology. Nat. Neurosci. 2013, 16, 848–850. [Google Scholar] [CrossRef] [PubMed]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Singh Pahwa, J.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2013, 41, 1088–1093. [Google Scholar]
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.V.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.I.; Lah, J.J.; et al. Variant of TREM2 Associated with the Risk of Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 107–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thambisetty, M.; An, Y.; Nalls, M.; Sojkova, J.; Swaminathan, S.; Zhou, Y.; Singleton, A.B.; Wong, D.F.; Ferrucci, L.; Saykin, A.J.; et al. Effect of complement CR1 on brain amyloid burden during aging and its modification by APOE genotype. Biol. Psychiatry 2013, 73, 422–428. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, R.H.; Nagao, T.; Gouras, G.K. Plaque formation and the intraneuronal accumulation of β-amyloid in Alzheimer’s disease. Pathol. Int. 2017, 67, 185–193. [Google Scholar] [CrossRef]
- Rajan, K.B.; Wilson, R.S.; Weuve, J.; Barnes, L.L.; Evans, D.A. Cognitive impairment 18 years before clinical diagnosis of Alzheimer disease dementia. Neurology 2015, 85, 898–904. [Google Scholar] [CrossRef] [Green Version]
- Sperling, R.; Mormino, E.; Johnson, K. The evolution of preclinical Alzheimer’s disease: Implications for prevention trials. Neuron 2014, 84, 608–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mordechai, S.; Shufan, E.; Porat Katz, B.S.; Salman, A. Early diagnosis of Alzheimer’s disease using infrared spectroscopy of isolated blood samples followed by multivariate analyses. Analyst 2017, 142, 1276–1284. [Google Scholar] [CrossRef] [PubMed]
- Berti, V.; Polito, C.; Lombardi, G.; Ferrari, C.; Sorbi, S.; Pupi, A. Rethinking on the concept of biomarkers in preclinical Alzheimer’s disease. Neurol. Sci. 2016, 37, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Davatzikos, C.; Da, X.; Toledo, J.B.; Zee, J.; Wolk, D.A.; Xie, S.X.; Ou, Y.; Shacklett, A.; Parmpi, P.; Shaw, L.; et al. Integration and relative value of biomarkers for prediction of MCI to AD progression: Spatial patterns of brain atrophy, cognitive scores, APOE genotype and CSF biomarkers. NeuroImage Clin. 2014, 4, 164–173. [Google Scholar] [CrossRef] [Green Version]
- Henriksen, K.; O’Bryant, S.E.; Hampel, H.; Trojanowski, J.Q.; Montine, T.J.; Jeromin, A.; Blennow, K.; Lönneborg, A.; Wyss-Coray, T.; Soares, H.; et al. The future of blood-based biomarkers for Alzheimer’s disease. Alzheimers Dement. 2014, 10, 115–131. [Google Scholar] [CrossRef] [Green Version]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Frankfort, S.; Tulner, L.; van Campen, J.; Verbeek, M.; Jansen, R.; Beijnen, J. Amyloid Beta Protein and Tau in Cerebrospinal Fluid and Plasma as Biomarkers for Dementia: A Review of Recent Literature. Curr. Clin. Pharmacol. 2008, 3, 123–131. [Google Scholar] [CrossRef]
- Spies, P.E.; Verbeek, M.M.; Van Groen, T.; Claassen, J.A.H.R. Reviewing reasons for the decreased CSF Abeta42 concentration in Alzheimer disease. Front. Biosci. 2012, 17, 2024–2034. [Google Scholar] [CrossRef] [Green Version]
- Skillbäck, T.; Farahmand, B.Y.; Rosén, C.; Mattsson, N.; Nägga, K.; Kilander, L.; Religa, D.; Wimo, A.; Winblad, B.; Schott, J.M.; et al. Cerebrospinal fluid tau and amyloid-β1-42 in patients with dementia. Brain 2015, 138, 2716–2731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ringman, J.M.; Younkin, S.G.; Pratico, D.; Seltzer, W.; Cole, G.M.; Geschwind, D.H.; Rodriguez-Agudelo, Y.; Schaffer, B.; Fein, J.; Sokolow, S.; et al. Biochemical markers in persons with preclinical familial Alzheimer disease. Neurology 2008, 71, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Goel, T.; Tanveer, M.; Dwivedi, S.; Murugan, R. FAF-DRVFL: Fuzzy activation function based deep random vector functional links network for early diagnosis of Alzheimer disease. Appl. Soft Comput. 2021, 106, 107371. [Google Scholar] [CrossRef]
- Wenzler, S.; Knochel, C.; Balaban, C.; Kraft, D.; Kopf, J.; Alves, G.S.; Prvulovic, D.; Carvalho, A.F.; Oertel-Knochel, V. Integrated Biomarkers for Depression in Alzheimer’s Disease: A Critical Review. Curr. Alzheimer Res. 2017, 14, 441–452. [Google Scholar] [CrossRef]
- Durães, F.; Pinto, M.; Sousa, E. Old drugs as new treatments for neurodegenerative diseases. Pharmaceuticals 2018, 11, 44. [Google Scholar] [CrossRef] [Green Version]
- Allsop, D.; Taylor, M.; Fullwood, N.; Michael, M.; Aggidis, A.; Vincent, S.; Dale, M. A novel approach to the therapy of Alzheimer’s disease based on peptide nanoliposome inhibitors of Aβ and tau aggregation. J. Prev. Alzheimers Dis. 2017, 4, 385–386. [Google Scholar]
- U.S. Food and Drug Administration (FDA). FDA Grants Accelerated Approval for Alzheimer’s Drug|FDA. 2021. Available online: https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-drug (accessed on 14 June 2022).
- Delrieu, J.; Piau, A.; Caillaud, C.; Voisin, T.; Vellas, B. Managing cognitive dysfunction through the continuum of alzheimers disease: Role of pharmacotherapy. CNS Drugs 2011, 25, 213–226. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Schulzer, M.; McGeer, E.G. Anti-inflammatory agents as possible protective factors for Alzheimer’s disease: Analysis of relevant epidemiological studies. In Neuroinflammatory Mechanisms in Alzheimer’s Disease Basic and Clinical Research; Birkhäuser: Basel, Switzerland, 2001; pp. 53–64. [Google Scholar]
- Galimberti, D.; Scarpini, E. Progress in Alzheimer’s disease. J. Neurol. 2012, 259, 201–211. [Google Scholar] [CrossRef]
- Fu, H.J.; Liu, B.; Frost, J.L.; Lemere, C.A. Amyloid-β Immunotherapy for Alzheimer’s Disease. CNS Neurol. Disord. Drug Targets 2010, 9, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Lemere, C.A.; Masliah, E. Can Alzheimer disease be prevented by amyloid-Β immunotherapy? Nat. Rev. Neurol. 2010, 6, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Kabir, M.T.; Uddin, M.S.; Mathew, B.; Das, P.K.; Perveen, A.; Ashraf, G.M. Emerging Promise of Immunotherapy for Alzheimer’s Disease: A New Hope for the Development of Alzheimer’s Vaccine. Curr. Top. Med. Chem. 2020, 20, 1214–1234. [Google Scholar] [CrossRef]
- Solomon, B.; Koppel, R.; Frankel, D.; Hanan-Aharon, E. Disaggregation of Alzheimer β-amyloid by site-directed mAb. Proc. Natl. Acad. Sci. USA 1997, 94, 4109–4112. [Google Scholar] [CrossRef] [Green Version]
- Schenk, D.; Barbour, R.; Dunn, W.; Gordon, G.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; Khan, K.; et al. Immunization with amyloid-[beta] attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 1999, 400, 173–177. [Google Scholar] [CrossRef]
- Asuni, A.A.; Boutajangout, A.; Scholtzova, H.; Knudsen, E.; Li, Y.S.; Quartermain, D.; Frangione, B.; Wisniewski, T.; Sigurdsson, E.M. Vaccination of Alzheimer’s model mice with Aβ derivative in alum adjuvant reduces Aβ burden without microhemorrhages. Eur. J. Neurosci. 2006, 24, 2530–2542. [Google Scholar] [CrossRef] [Green Version]
- Lemere, C.A. Immunotherapy for Alzheimer’s disease: Hoops and hurdles. Mol. Neurodegener. 2013, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Kotilinek, L.A.; Bacskai, B.; Westerman, M.; Kawarabayashi, T.; Younkin, L.; Hyman, B.T.; Younkin, S.; Ashe, K.H. Reversible memory loss in a mouse transgenic model of Alzheimer’s disease. J. Neurosci. 2002, 22, 6331–6335. [Google Scholar] [CrossRef]
- Rohn, T.T.; Vyas, V.; Hernandez-Estrada, T.; Nichol, K.E.; Christie, L.A.; Head, E. Lack of pathology in a triple transgenic mouse model of Alzheimer’s disease after overexpression of the anti-apoptotic protein Bcl-2. J. Neurosci. 2008, 28, 3051–3059. [Google Scholar] [CrossRef] [Green Version]
- Domert, J.; Rao, S.B.; Agholme, L.; Brorsson, A.C.; Marcusson, J.; Hallbeck, M.; Nath, S. Spreading of amyloid-β peptides via neuritic cell-to-cell transfer is dependent on insufficient cellular clearance. Neurobiol. Dis. 2014, 65, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Winslow, A.R.; Moussaud, S.; Zhu, L.; Post, K.L.; Dickson, D.W.; Berezovska, O.; McLean, P.J. Convergence of pathology in dementia with Lewy bodies and Alzheimer’s disease: A role for the novel interaction of alpha-synuclein and presenilin 1 in disease. Brain 2014, 137, 1958–1970. [Google Scholar] [CrossRef] [Green Version]
- Morales, R.; Callegari, K.; Soto, C. Prion-like features of misfolded Aβ and tau aggregates. Virus Res. 2015, 207, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Meraz-Ríos, M.A.; Lira-De León, K.I.; Campos-Peña, V.; De Anda-Hernández, M.A.; Mena-López, R. Tau oligomers and aggregation in Alzheimer’s disease. J. Neurochem. 2010, 112, 1353–1367. [Google Scholar] [CrossRef] [PubMed]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinton, L.K.; Blurton-Jones, M.; Myczek, K.; Trojanowski, J.Q.; LaFerla, F.M. Synergistic interactions between Aβ, tau, and α-synuclein: Acceleration of neuropathology and cognitive decline. J. Neurosci. 2010, 30, 7281–7289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, D. Immunotherapy for Alzheimer’s disease. Proc. J. Intern. Med. 2011, 269, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, D.; Sudduth, T.; Greenstein, A. P1–027: Intracranial administration of Gammagard IVIg lowers amyloid and modulates neuroinflammatory profiles along a different time-course than anti-beta-amyloid IgG: Implications for mechanism of action. Alzheimers Dement. 2013, 9, P162. [Google Scholar] [CrossRef]
- Wang, A.; Das, P.; Switzer, R.C.; Golde, T.E.; Jankowsky, J.L. Robust amyloid clearance in a mouse model of Alzheimer’s disease provides novel insights into the mechanism of amyloid-β immunotherapy. J. Neurosci. 2011, 31, 4124–4136. [Google Scholar] [CrossRef] [Green Version]
- Mandler, M.; Valera, E.; Rockenstein, E.; Mante, M.; Weninger, H.; Patrick, C.; Adame, A.; Schmidhuber, S.; Santic, R.; Schneeberger, A.; et al. Active immunization against alpha-synuclein ameliorates the degenerative pathology and prevents demyelination in a model of multiple system atrophy. Mol. Neurodegener. 2015, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- DeMattos, R.B.; Bales, K.R.; Cummins, D.J.; Paul, S.M.; Holtzman, D.M. Brain to plasma amyloid-β efflux: A measure of brain amyloid burden in a mouse model of Alzheimer’s disease. Science 2002, 295, 2264–2267. [Google Scholar] [CrossRef]
- MR, F.; JR, B. Immunotherapy for Alzheimer’s disease. Neurol. Clin. 2013, 31, 869–878. [Google Scholar]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef]
- Bao, F.; Wicklund, L.; Lacor, P.N.; Klein, W.L.; Nordberg, A.; Marutle, A. Different β-amyloid oligomer assemblies in Alzheimer brains correlate with age of disease onset and impaired cholinergic activity. Neurobiol. Aging 2012, 33, 825.e1–825.e13. [Google Scholar] [CrossRef] [PubMed]
- Alcantar, N.A.; Jimenez, J.; Morgan, D. P1-255: Direct observation of the kinetic mechanisms for Aß peptide aggregation: Towards elucidating Alzheimer plaque dissolution. Alzheimers Dement. 2010, 6, S247. [Google Scholar] [CrossRef]
- DeMattos, R.B.; Bales, K.R.; Cummins, D.J.; Dodart, J.C.; Paul, S.M.; Holtzman, D.M. Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 8850–8855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisniewski, T.; Goni, F. Immunotherapy for Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 499–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisniewski, T.; Frangione, B. Immunological and anti-chaperone therapeutic approaches for Alzheimer disease. Proc. Brain Pathol. 2005, 15, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.J.; Bullock, R.; Jones, R.W.; Wilkinson, D.; Paterson, K.R.; Jenkins, L.; Millais, S.B.; Donoghue, S. Evaluation of the safety and immunogenicity of synthetic Aβ42 (AN1792) in patients with AD. Neurology 2005, 64, 94–101. [Google Scholar] [CrossRef]
- Pride, M.; Seubert, P.; Grundman, M.; Hagen, M.; Eldridge, J.; Black, R.S. Progress in the active immunotherapeutic approach to Alzheimer’s disease: Clinical investigations into AN1792-associated meningoencephalitis. Proc. Neurodegener. Dis. 2008, 5, 194–196. [Google Scholar] [CrossRef]
- Gilman, S.; Koller, M.; Black, R.S.; Jenkins, L.; Griffith, S.G.; Fox, N.C.; Eisner, L.; Kirby, L.; Boada Rovira, M.; Forette, F.; et al. Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005, 64, 1553–1562. [Google Scholar] [CrossRef]
- Boche, D.; Nicoll, J.A.R. SYMPOSIUM: Clearance of Aβ from the Brain in Alzheimer’Disease: The Role of the Immune System in Clearance of Aβ from the Brain. Brain Pathol. 2008, 18, 267–278. [Google Scholar] [CrossRef]
- Ferrer, I.; Rovira, M.B.; Guerra, M.L.S.; Rey, M.J.; Costa-Jussá, F. Neuropathology and Pathogenesis of Encephalitis following Amyloid β Immunization in Alzheimer’s Disease. Brain Pathol. 2004, 14, 11–20. [Google Scholar] [CrossRef]
- Masliah, E.; Hansen, L.; Adame, A.; Crews, L.; Bard, F.; Lee, C.; Seubert, P.; Games, D.; Kirby, L.; Schenk, D. Aβ vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology 2005, 64, 129–131. [Google Scholar] [CrossRef]
- Nicoll, J.A.R.; Barton, E.; Boche, D.; Neal, J.W.; Ferrer, I.; Thompson, P.; Vlachouli, C.; Wilkinson, D.; Bayer, A.; Games, D.; et al. Aβ species removal after Aβ42 immunization. J. Neuropathol. Exp. Neurol. 2006, 65, 1040–1048. [Google Scholar] [CrossRef] [Green Version]
- Bombois, S.; Maurage, C.A.; Gompel, M.; Deramecourt, V.; Mackowiak-Cordoliani, M.A.; Black, R.S.; Lavielle, R.; Delacourte, A.; Pasquier, F. Absence of β-amyloid deposits after immunization in Alzheimer disease with Lewy body dementia. Arch. Neurol. 2007, 64, 583–587. [Google Scholar] [CrossRef] [Green Version]
- Godyń, J.; Jończyk, J.; Panek, D.; Malawska, B. Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol. Reports 2016, 68, 127–138. [Google Scholar] [CrossRef] [PubMed]
- St-Amour, I.; Cicchetti, F.; Calon, F. Immunotherapies in Alzheimer’s disease: Too much, too little, too late or off-target? Acta Neuropathol. 2016, 131, 481–504. [Google Scholar] [CrossRef] [PubMed]
- Wiessner, C.; Wiederhold, K.H.; Tissot, A.C.; Frey, P.; Danner, S.; Jacobson, L.H.; Jennings, G.T.; Lüönd, R.; Ortmann, R.; Reichwald, J.; et al. The second-generation active Aβ immunotherapy CAD106 reduces amyloid accumulation in APP transgenic mice while minimizing potential side effects. J. Neurosci. 2011, 31, 9323–9331. [Google Scholar] [CrossRef] [PubMed]
- Winblad, B.; Andreasen, N.; Minthon, L.; Floesser, A.; Imbert, G.; Dumortier, T.; Maguire, R.P.; Blennow, K.; Lundmark, J.; Staufenbiel, M.; et al. Safety, tolerability, and antibody response of active Aβ immunotherapy with CAD106 in patients with Alzheimer’s disease: Randomised, double-blind, placebo-controlled, first-in-human study. Lancet Neurol. 2012, 11, 597–604. [Google Scholar] [CrossRef]
- Wisniewski, T. Active immunotherapy for Alzheimer’s disease. Lancet Neurol. 2012, 11, 571–572. [Google Scholar] [CrossRef] [Green Version]
- Ryan, J.M.; Grundman, M. Anti-amyloid-β immunotherapy in alzheimer’s disease: Acc-001 clinical trials are ongoing. J. Alzheimers Dis. 2009, 17, 243. [Google Scholar] [CrossRef]
- Schneeberger, A.; Mandler, M.; Otava, O.; Zauner, W.; Mattner, F.; Schmidt, W. Development of AFFITOPE vaccines for Alzheimer’s Disease (AD)—From concept to clinical testing. J. Nutr. Health Aging 2009, 13, 264–267. [Google Scholar] [CrossRef]
- Rosenmann, H. Immunotherapy for Targeting Tau Pathology in Alzheimer’s Disease and Tauopathies. Curr. Alzheimer Res. 2013, 10, 217–228. [Google Scholar] [CrossRef]
- Galimberti, D.; Scarpini, E. Disease-modifying treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2011, 4, 203–216. [Google Scholar] [CrossRef] [Green Version]
- Collin, L.; Bohrmann, B.; Göpfert, U.; Oroszlan-Szovik, K.; Ozmen, L.; Grüninger, F. Neuronal uptake of tau/pS422 antibody and reduced progression of tau pathology in a mouse model of Alzheimer’s disease. Brain 2014, 137, 2834–2846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, S.; Wegmann, S.; Cho, H.; Devos, S.L.; Commins, C.; Roe, A.D.; Nicholls, S.B.; Carlson, G.A.; Pitstick, R.; Nobuhara, C.K.; et al. Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat. Commun. 2015, 6, 8490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobuhara, C.K.; DeVos, S.L.; Commins, C.; Wegmann, S.; Moore, B.D.; Roe, A.D.; Costantino, I.; Frosch, M.P.; Pitstick, R.; Carlson, G.A.; et al. Tau Antibody Targeting Pathological Species Blocks Neuronal Uptake and Interneuron Propagation of Tau in Vitro. Am. J. Pathol. 2017, 187, 1399–1412. [Google Scholar] [CrossRef] [Green Version]
- Yanamandra, K.; Kfoury, N.; Jiang, H.; Mahan, T.E.; Ma, S.; Maloney, S.E.; Wozniak, D.F.; Diamond, M.I.; Holtzman, D.M. Anti-tau antibodies that block tau aggregate seeding invitro markedly decrease pathology and improve cognition in vivo. Neuron 2013, 80, 402–414. [Google Scholar] [CrossRef] [Green Version]
- Boutajangout, A.; Quartermain, D.; Sigurdsson, E.M. Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. J. Neurosci. 2010, 30, 16559–16566. [Google Scholar] [CrossRef] [Green Version]
- Sahara, N.; Murayama, M.; Higuchi, M.; Suhara, T.; Takashima, A. Biochemical distribution of tau protein in synaptosomal fraction of transgenic mice expressing human p301l tau. Front. Neurol. 2014, 5, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asuni, A.A.; Boutajangout, A.; Quartermain, D.; Sigurdsson, E.M. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J. Neurosci. 2007, 27, 9115–9129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boimel, M.; Grigoriadis, N.; Lourbopoulos, A.; Haber, E.; Abramsky, O.; Rosenmann, H. Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Exp. Neurol. 2010, 224, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E.; Petrucelli, L.; Lewis, J. Targeting Aβ and tau in Alzheimer’s disease, an early interim report. Exp. Neurol. 2010, 223, 252–266. [Google Scholar] [CrossRef] [Green Version]
- Theunis, C.; Crespo-Biel, N.; Gafner, V.; Pihlgren, M.; López-Deber, M.P.; Reis, P.; Hickman, D.T.; Adolfsson, O.; Chuard, N.; Ndao, D.M.; et al. Efficacy and safety of a liposome-based vaccine against protein Tau, assessed in Tau.P301L mice that model tauopathy. PLoS ONE 2013, 8, e0072301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, K.; Sabbagh, M. Early investigational drugs targeting tau protein for the treatment of Alzheimers disease. Expert Opin. Investig. Drugs 2015, 24, 1355–1360. [Google Scholar] [CrossRef] [Green Version]
- Patton, R.L.; Kalback, W.M.; Esh, C.L.; Kokjohn, T.A.; Van Vickle, G.D.; Luehrs, D.C.; Kuo, Y.M.; Lopez, J.; Brune, D.; Ferrer, I.; et al. Amyloid-β peptide remnants in AN-1792-immunized Alzheimer’s disease patients: A biochemical analysis. Am. J. Pathol. 2006, 169, 1048–1063. [Google Scholar] [CrossRef] [Green Version]
- Blennow, K.; Hampel, H.; Zetterberg, H. Biomarkers in amyloid-β immunotherapy trials in Alzheimer’s disease. Neuropsychopharmacology 2014, 39, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Winblad, B.; Graf, A.; Riviere, M.E.; Andreasen, N.; Ryan, J.M. Active immunotherapy options for Alzheimer’s disease. Alzheimers Res. Ther. 2014, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Farlow, M.R.; Andreasen, N.; Riviere, M.E.; Vostiar, I.; Vitaliti, A.; Sovago, J.; Caputo, A.; Winblad, B.; Graf, A. Long-term treatment with active Aβ immunotherapy with CAD106 in mild Alzheimer’s disease. Alzheimers Res. Ther. 2015, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Pasquier, F.; Sadowsky, C.; Holstein, A.; Leterme, G.L.P.; Peng, Y.; Jackson, N.; Fox, N.C.; Ketter, N.; Liu, E.; Ryan, J.M. Two phase 2 multiple ascending-dose studies of vanutide cridificar (ACC-001) and QS-21 adjuvant in mild-to-moderate Alzheimer’s disease. J. Alzheimers Dis. 2016, 51, 1131–1143. [Google Scholar] [CrossRef]
- Mo, J.J.; Li, J.Y.; Yang, Z.; Liu, Z.; Feng, J.S. Efficacy and safety of anti-amyloid-β immunotherapy for Alzheimer’s disease: A systematic review and network meta-analysis. Ann. Clin. Transl. Neurol. 2017, 4, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Pihlgren, M.; Vukicevic, M.; Gafner, V.; Piorkowska, K.; Chuard, N.; Giriens, V.; Valdes, P.; Nazeeruddin, S.; Colin, P.; Pfeifer, A.; et al. O2-13-03: Efficacy of ACI-35, A Liposomal Anti-Phospho Tau Vaccine in Two Different Mouse Models of Alzheimer’s Disease. Alzheimers Dement. 2016, 12, P260–P261. [Google Scholar] [CrossRef]
- Cattepoel, S.; Hanenberg, M.; Kulic, L.; Nitsch, R.M. Chronic intranasal treatment with an anti-Aβ30-42 scFv antibody ameliorates amyloid pathology in a transgenic mouse model of Alzheimer’s disease. PLoS ONE 2011, 6, e18296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrushina, I.; Ghochikyan, A.; Mkrtichyan, M.; Mamikonyan, G.; Movsesyan, N.; Ajdari, R.; Vasilevko, V.; Karapetyan, A.; Lees, A.; Agadjanyan, M.G.; et al. Mannan-Abeta28 conjugate prevents Abeta-plaque deposition, but increases microhemorrhages in the brains of vaccinated Tg2576 (APPsw) mice. J. Neuroinflamm. 2008, 5, 42. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Lebson, L.; Lee, D.C.; Nash, K.; Grimm, J.; Rosenthal, A.; Selenica, M.L.B.; Morgan, D.; Gordon, M.N. Chronological age impacts immunotherapy and monocyte uptake independent of amyloid load. J. Neuroimmune Pharmacol. 2012, 7, 202–214. [Google Scholar] [CrossRef]
- Moreth, J.; Mavoungou, C.; Schindowski, K. Passive anti-amyloid immunotherapy in Alzheimer’s disease: What are the most promising targets? Immun. Ageing 2013, 10, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperling, R.; Salloway, S.; Brooks, D.J.; Tampieri, D.; Barakos, J.; Fox, N.C.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; et al. Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: A retrospective analysis. Lancet Neurol. 2012, 11, 241–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S.; et al. Phase 3 Trials of Solanezumab for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Two Phase 3 Trials of Bapineuzumab in Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinovici, G.D.; Jagust, W.J. Amyloid imaging in aging and dementia: Testing the amyloid hypothesis in vivo. Behav. Neurol. 2009, 21, 117–128. [Google Scholar] [CrossRef]
- Blennow, K.; Zetterberg, H.; Wei, J.; Liu, E.; Black, R.; Grundman, M. Immunotherapy with bapineuzumab lowers CSF tau protein levels in patients with Alzheimer’s disease. Alzheimers Dement. 2010, 6, S134–S135. [Google Scholar] [CrossRef]
- Laskowitz, D.T.; Kolls, B.J. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate alzheimer disease. Neurology 2010, 74, 2026. [Google Scholar] [CrossRef] [Green Version]
- Salloway, S.; Sperling, R.; Gilman, S.; Fox, N.C.; Blennow, K.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Doody, R.; van Dyck, C.H.; et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology 2009, 73, 2061–2070. [Google Scholar] [CrossRef]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [Green Version]
- Farlow, M.; Arnold, S.E.; Van Dyck, C.H.; Aisen, P.S.; Snider, B.J.; Porsteinsson, A.P.; Friedrich, S.; Dean, R.A.; Gonzales, C.; Sethuraman, G.; et al. Safety and biomarker effects of solanezumab in patients with Alzheimer’s disease. Alzheimers Dement. 2012, 8, 261–271. [Google Scholar] [CrossRef]
- Siemers, E.; Holdridge, K.C.; Sundell, K.L.; Liu-Seifert, H. Function and clinical meaningfulness of treatments for mild Alzheimer’s disease. Alzheimers Dement. Diagnosis Assess. Dis. Monit. 2016, 2, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Abbott, A.; Dolgin, E. Failed Alzheimer’s trial does not kill leading theory of disease. Nature 2016, 540, 15–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacks, C.A.; Avorn, J.; Kesselheim, A.S. The Failure of Solanezumab—How the FDA Saved Taxpayers Billions. N. Engl. J. Med. 2017, 376, 1706–1708. [Google Scholar] [CrossRef]
- Ritter, A.; Cummings, J. Fluid biomarkers in clinical trials of Alzheimer’s disease therapeutics. Front. Neurol. 2015, 6, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penninkilampi, R.; Brothers, H.M.; Eslick, G.D. Safety and Efficacy of Anti-Amyloid-β Immunotherapy in Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Neuroimmune Pharmacol. 2017, 12, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Ivanoiu, A.; Pariente, J.; Booth, K.; Lobello, K.; Luscan, G.; Hua, L.; Lucas, P.; Styren, S.; Yang, L.; Li, D.; et al. Long-term safety and tolerability of bapineuzumab in patients with Alzheimer’s disease in two phase 3 extension studies. Alzheimers Res. Ther. 2016, 8, 24. [Google Scholar] [CrossRef] [Green Version]
- Pfizer Study Evaluating the Safety and Efficacy of Bapineuzumab in Alzheimer Disease Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT00667810 (accessed on 15 October 2021).
- Pfizer A Long-Term Safety And Tolerability Study of Bapineuzumab in Alzheimer Disease Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT00996918 (accessed on 15 October 2021).
- Roher, A.E.; Maarouf, C.L.; Kokjohn, T.A.; Belden, C.; Serrano, G.; Sabbagh, M.S.; Beach, T.G. Chemical and neuropathological analyses of an alzheimer’s disease patient treated with solanezumab. Am. J. Neurodegener. Dis. 2016, 5, 158–170. [Google Scholar]
- ClinicalTrials.gov Identifier: NCT01900665. Progress of Mild Alzheimer’s Disease in Participants on Solanezumab Versus Placebo (EXPEDITION 3). Available online: https://clinicaltrials.gov/ct2/show/NCT01900665 (accessed on 15 October 2021).
- U.S. National Institutes of Health Clinical Trial of Solanezumab for Older Individuals Who May be at Risk for Memory Loss—Full Text View. Available online: https://clinicaltrials.gov/ct2/show/NCT02008357 (accessed on 14 June 2022).
- Blennow, K.; Nikolcheva, T.; Lasser, R.A.; Dukart, J.; Rabe, C.; Volz, D.; Scheltens, P. O1-10-01: Gantenerumab Treatment Reduces Biomarkers of Neuronal and Synaptic Degeneration in Alzheimer’s Disease. Alzheimers Dement. 2016, 12, P198. [Google Scholar] [CrossRef]
- Cummings, J.; Cho, W.; Ward, M.; Friesenhahn, M.; Brunstein, F.; Honigberg, L.; Clayton, D.; Mortensen, D.; Ho, C.; Paul, R. O4-11-06: A Randomized, Double-Blind, Placebo-Controlled Phase 2 Study To Evaluate The Efficacy And Safety Of Crenezumab In Patients With Mild To Moderate Alzheimer’s Disease. Alzheimers Dement. 2014, 10, P275. [Google Scholar] [CrossRef]
- Lannfelt, M.I. Immunotherapy and Biomarkers in Neurodegenerative Disorders; Methods in Pharmacology and Toxicology; Ingelsson, M., Lannfelt, L., Eds.; Springer: New York, NY, USA, 2016; Part F1. [Google Scholar]
- Genentech, I. A Study of Crenezumab Versus Placebo in Preclinical Presenilin1 (PSEN1) E280A Mutation Carriers to Evaluate Efficacy and Safety in the Treatment of Autosomal-Dominant Alzheimer’s (Clinical Trial Identifier NCT0199884). Available online: https://clinicaltrials.gov/ct2/show/NCT01998841 (accessed on 15 October 2021).
- NCT03114657 A Study of Crenezumab Versus Placebo to Evaluate the Efficacy and Safety in Participants With Prodromal to Mild Alzheimer’s Disease (AD). 2017. Available online: https://ClinicalTrials.gov/show/NCT03114657 (accessed on 14 June 2022).
- Pfizer A Multiple Dose Study of PF-04360365 In Patients With Mild to Moderate Alzheimer’s Disease. Available online: https://clinicaltrials.gov/ct2/show/NCT00945672 (accessed on 16 October 2021).
- Biogen Inc. 221AD302 Phase 3 Study of Aducanumab (BIIB037) in Early Alzheimer’s Disease (EMERGE). Available online: https://clinicaltrials.gov/ct2/show/NCT02484547 (accessed on 14 June 2022).
- Tucker, S.; Möller, C.; Tegerstedt, K.; Lord, A.; Laudon, H.; Sjödahl, J.; Söderberg, L.; Spens, E.; Sahlin, C.; Waara, E.R.; et al. The murine Version of BAN2401 (mAb158) selectively reduces amyloid-β protofibrils in brain and cerebrospinal fluid of tg-ArcSwe Mice. J. Alzheimer’s Dis. 2015, 43, 575–588. [Google Scholar] [CrossRef]
- Dickson, S.P.; Hendrix, S.B.; Ellison, N. O3-10-04: A statistical translation of the public ban2401 study results from a bayesian to a traditional framework. Alzheimers Dement. 2019, 15, P909–P910. [Google Scholar] [CrossRef]
- Cummings, J.; Vegas, L. Second Look at BAN2401 Data Still Positive, Despite Snafu|ALZFORUM. 2018. Available online: https://www.alzforum.org/news/conference-coverage/second-look-ban2401-data-still-positive-despite-snafu (accessed on 14 June 2022).
- Lannfelt, L.; Relkin, N.R.; Siemers, E.R. Amyloid-ß-directed immunotherapy for Alzheimer’s disease. J. Intern. Med. 2014, 275, 284–295. [Google Scholar] [CrossRef] [Green Version]
- Griesenauer, R.H.; Kinch, M.S. An overview of FDA-approved vaccines & their innovators. Expert Rev. Vaccines 2017, 16, 1253–1266. [Google Scholar] [CrossRef] [PubMed]
- Dodel, R.; Bacher, M.; Przybylski, M.; Stefanescu, R.; Manea, M. Method of Treatment of Neurodementing Diseases Using Isolated, Monoclonal, Human, Anti-B-Amyloid Antibody. U.S. Patent No. 8,491,903, 23 July 2013. [Google Scholar]
- Dodel, R.C.; Du, Y.; Depboylu, C.; Hampel, H.; Frölich, L.; Haag, A.; Hemmeter, U.; Paulsen, S.; Teipel, S.J.; Brettschneider, S.; et al. Intravenous immunoglobulins containing antibodies against β-amyloid for the treatment of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1472–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Relkin, N. Clinical trials of Intravenous Immunoglobulin for Alzheimer’s disease. J. Clin. Immunol. 2014, 34, S74–S79. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, D.A. Intravenous immunoglobulin and Alzheimer’s disease: What now? J. Neuroinflamm. 2013, 10, 853. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Huo, L.; Shi, X.; Bai, R.; Zhang, Z.; Zhao, Y.; Chang, Y.; Chen, C. Endoplasmic reticulum stress induced by zinc oxide nanoparticles is an earlier biomarker for nanotoxicological evaluation. ACS Nano 2014, 8, 2562–2574. [Google Scholar] [CrossRef]
- Hughes, R.A.C.; Dalakas, M.C.; Cornblath, D.R.; Latov, N.; Weksler, M.E.; Relkin, N. Clinical applications of intravenous immunoglobulins in neurology. Clin. Exp. Immunol. 2009, 158, 34–42. [Google Scholar] [CrossRef]
- Loeffler, D.A. Erratum: Should development of Alzheimer’s disease-specific intravenous immunoglobulin be considered? J. Neuroinflamm. 2015, 12, 68. [Google Scholar] [CrossRef] [Green Version]
- Dodel, R.; Rominger, A.; Bartenstein, P.; Barkhof, F.; Blennow, K.; Förster, S.; Winter, Y.; Bach, J.-P.; Popp, J.; Alferink, J.; et al. Intravenous immunoglobulin for treatment of mild-to-moderate Alzheimer’s disease: A phase 2, randomised, double-blind, placebo-controlled, dose-finding. Lancet Neurol. 2013, 12, 233–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kile, S.; Au, W.; Parise, C.; Rose, K.; Donnel, T.; Hankins, A.; Chan, M.; Ghassemi, A. IVIG treatment of mild cognitive impairment due to Alzheimer’s disease: A randomised double-blinded exploratory study of the effect on brain atrophy, cognition and conversion to dementia. J. Neurol. Neurosurg. Psychiatry 2017, 88, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Flllit, H.; Hess, G.; Hill, J.; Bonnet, P.; Toso, C. IV immunoglobulin is associated with a reduced risk of Alzheimer disease and related disorders. Neurology 2009, 73, 180–185. [Google Scholar] [CrossRef] [PubMed]


| Aβ Passive Immunotherapy Clinical Trials | ||||||||
|---|---|---|---|---|---|---|---|---|
| Drug | Sponsor | Vaccine Type | Target (Aβ/Tau) | Trial Phase and Status | Immunology | Positive Outcomes | Negative Outcomes | References |
| Bapineuzumab | Janssen/Pfizer | Humanized monoclonal IgG1 (murine mAb) | N-terminal (Aβ1-5) | Two Phase III studies completed. Terminated, No improvements (NCT00606476, NCT01254773, NCT00996918, NCT00676143, NCT00998764, NCT00667810, NCT00575055, NCT00574132, NCT00916617, NCT00112073) | Fc-mediated activation of microglial phagocytosis and cytokine production. Bind to Aβ monomers, oligomers, and fibrils. | PII: ↓↓CSF t-tau and p-tau while Aβ-40 and 42 remain unchanged. PIII: ↓↓CSF p-tau. In carriers Aβ-42 while in non-carriers Aβ-42, t-tau, and p-tau remain unchanged. | Vasogenic cerebral edema. | [149,159,160,161,162,163] |
| Solanezumab | Eli Lilly (Indianapolis, IN, USA) | Humanized monoclonal IgG1 (mAb266) | Middle domain (Aβ 16-24) (Aβ monomers) | Two Phase III studies were completed; other phases III tests are ongoing (NCT02760602, NCT01900665, NCT01127633, NCT01148498, NCT02008357, NCT00905372, NCT00749216, NCT00904683, NCT00329082, NCT04623242) | Sequestration of soluble monomers of Aβ thus removes synaptotoxic fragments of Aβ. | PII: ↑↑CSF and Serum Aβ40 and 42 while CSF p-tau and t-tau remain unchanged. | Effects on cognition failed to reach clinical outcomes. | [117,160,164,165,166] |
| Gantenerumab | Hoffmam- La Rochi (Basel, Switzerland) | Humanized monoclonal IgG1 | N terminal and Mid Domain (Aβ 3-12; 18-27) | III Ongoing (NCT02051608, NCT02133937, NCT03236844, NCT03443973, NCT04592341, NCT04339413, NCT02882009, NCT02711423) | Microglia uptake and degradation. Preferentially interacts with fibrillar Aβ, microglial recruitment, and activation. | ↓↓Aβ fibrillation. | Vasogenic edema, discontinued after a futility analysis. | [160,167] Scarlet RoAD (NCT01224106; WN25203) |
| Crenezumab | Genentech (South San Francisco, CA, USA) | Humanized monoclonal IgG4 | Mid-domain (oligomers and fibrils) | II/III Ongoing (NCT03491150, NCT03114657, NCT02670083, NCT02427243, NCT01998841, NCT01723826, NCT02353598) | IgG4- Aβ interactions. Selectively targets Aβ oligomers and fibrils. | ↑↑Total plasma Aβ level, well-tolerated in mild to moderated AD cases. | Elevated vascular risk (B.P, CVD). | [168,169,170,171] |
| Ponezumab | Pfizer | Humanized monoclonal IgG2 | Aβ 1-40 (C- terminal amino acids) (Plasmatic monomer) | II (Halted) (NCT01125631, NCT00733642, NCT01821118, NCT00455000, NCT01005862, NCT00607308, NCT00722046, NCT00945672) | Peripheral sink. | Safe and well-tolerated. ↑↑plasma Aβ level, ↑↑CSF total Aβ level and free Aβ-42. | Failed to reach primary cognitive endpoints. | [137,172] |
| Aducanumab | Biogen Idec (Baar, Switzerland) | Humanized monoclonal IgG1 | N terminal and Mid Domain (Aβ oligomers and fibrils) | III Ongoing (NCT04241068, NCT02782975, NCT03639987, NCT01677572, NCT01397539, NCT02484547, NCT02477800) | Microglial recruitment and activation. | Brain penetration occurs and ↓↓Aβ stabilization in MMSE and CDR-sb. | Increased ARIA chances, usually in APOE e-carriers. | [173] |
| BAN2401 | BioArtic (Stockholm, Sweden)/Eisai (Nutley, NJ, USA) | Humanized monoclonal IgG1 | Protofibrils (≥100 kDa) | II (b) (NCT01230853, NCT02094729, NCT01767311) | Selectively targets soluble Aβ protofibrils. | ↓↓CSF-soluble Aβ. Shows a favorable safety profile. protofibrils/oligomers. | No significant adverse effects. | [174,175,176] |
| IVIG Immunotherapy Clinical Trials | ||||||||
|---|---|---|---|---|---|---|---|---|
| Drug | Sponsor | Vaccine Type | Target (Aβ/Tau) | Trial Phase and Status | Immunology | Positive Outcomes | Negative Outcomes | References |
| Octagam IVIG | Octapharma (Charlotte, NC, USA) | Human polyclonal Ab. | Multiple sites on conformational Aβ epitopes | II (Completed, No improvement) (NCT02303093, NCT00504075, NCT02637700, NCT00750867, NCT01859754, NCT01854827, NCT00722475) | Increased Aβ clearance by microglia-mediated phagocytosis. | ↓↓Aβ plaques and plasma Aβ-42 level. ↑↑ cognitive functions. | Ischemic stroke and microbleeds. | [182,184] |
| Gammagard IVIG | Baxter Healthcare (Deerfield, IL, USA) | Human polyclonal Ab | Multiple sites on conformational Aβ epitopes | III (Abandoned) (NCT04153422, NCT00504075, NCT02637700, NCT00750867, NCT01854827, NCT00722475, NCT02042027) | Increased microglial activation and promote Aβ clearance by phagocytosis. | Safe. ↑↑CSF total Aβ, ↑↑plasma Aβ 42 and 40. ↓↓Aβ fibril and oligomer levels. | No improvement. | [97,182] (NCT00818662) |
| NewGam | Sutter Health (Sacramento, CA, USA) | Human polyclonal Ab | Multiple sites on conformational Aβ epitopes | III Ongoing (NCT02638207, NCT01349790, NCT01012323, NCT01313507, NCT01225276) | Increased Aβ fibril clearance by microglia-mediated phagocytosis. Prevent the formation of soluble Aβ oligomers. | ↓↓Aβ fibril and oligomer levels. | - | [185] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshamrani, M. Recent Trends in Active and Passive Immunotherapies of Alzheimer’s Disease. Antibodies 2023, 12, 41. https://doi.org/10.3390/antib12020041
Alshamrani M. Recent Trends in Active and Passive Immunotherapies of Alzheimer’s Disease. Antibodies. 2023; 12(2):41. https://doi.org/10.3390/antib12020041
Chicago/Turabian StyleAlshamrani, Meshal. 2023. "Recent Trends in Active and Passive Immunotherapies of Alzheimer’s Disease" Antibodies 12, no. 2: 41. https://doi.org/10.3390/antib12020041
APA StyleAlshamrani, M. (2023). Recent Trends in Active and Passive Immunotherapies of Alzheimer’s Disease. Antibodies, 12(2), 41. https://doi.org/10.3390/antib12020041