
Suppression of MUC1-Overexpressing Tumors by a Novel MUC1/CD3 Bispecific Antibody

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Isolation of PBMCs and T Cells
2.3. Antibody Design and Purification
2.4. HPLC-SEC and SDS-PAGE Analysis
2.5. Binding Assay
2.6. T Cell Activation Analysis
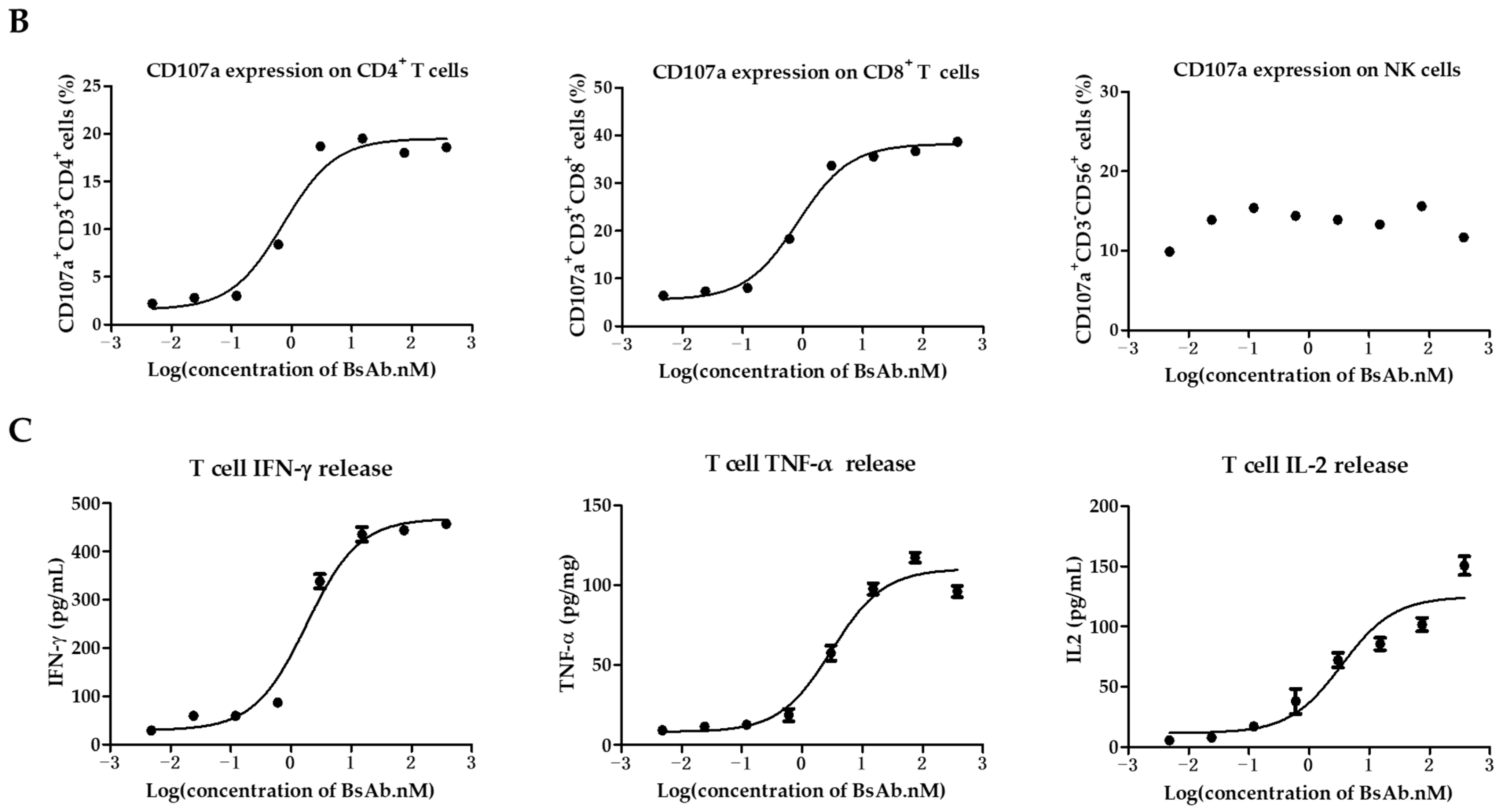
2.7. Cytokines Measurement
2.8. In Vitro Cytotoxicity Assay
2.9. In Vivo Efficacy Study
2.10. Statistical Analysis
3. Results
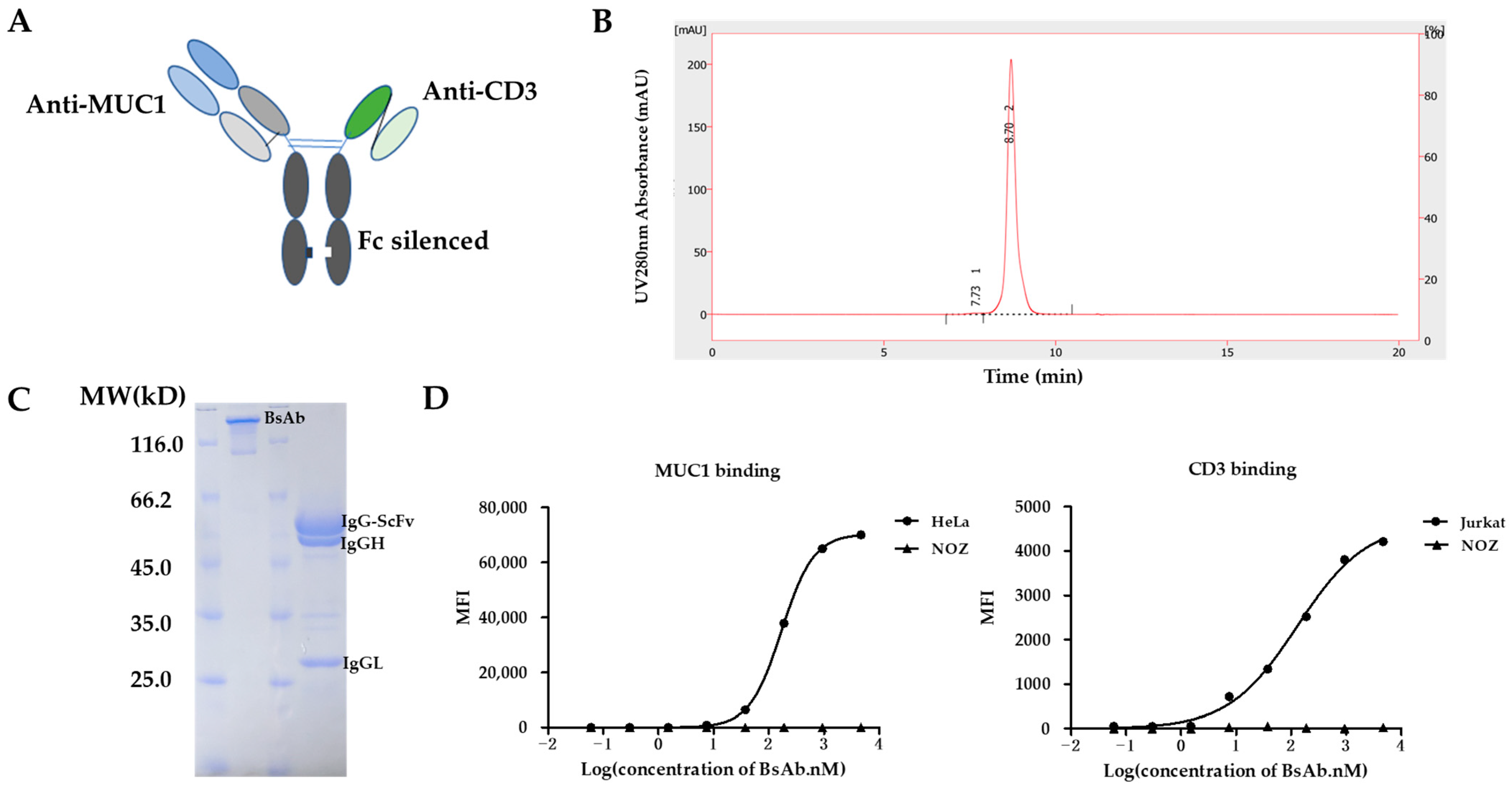
3.1. Design and Generation of MUC1/CD3 BsAb
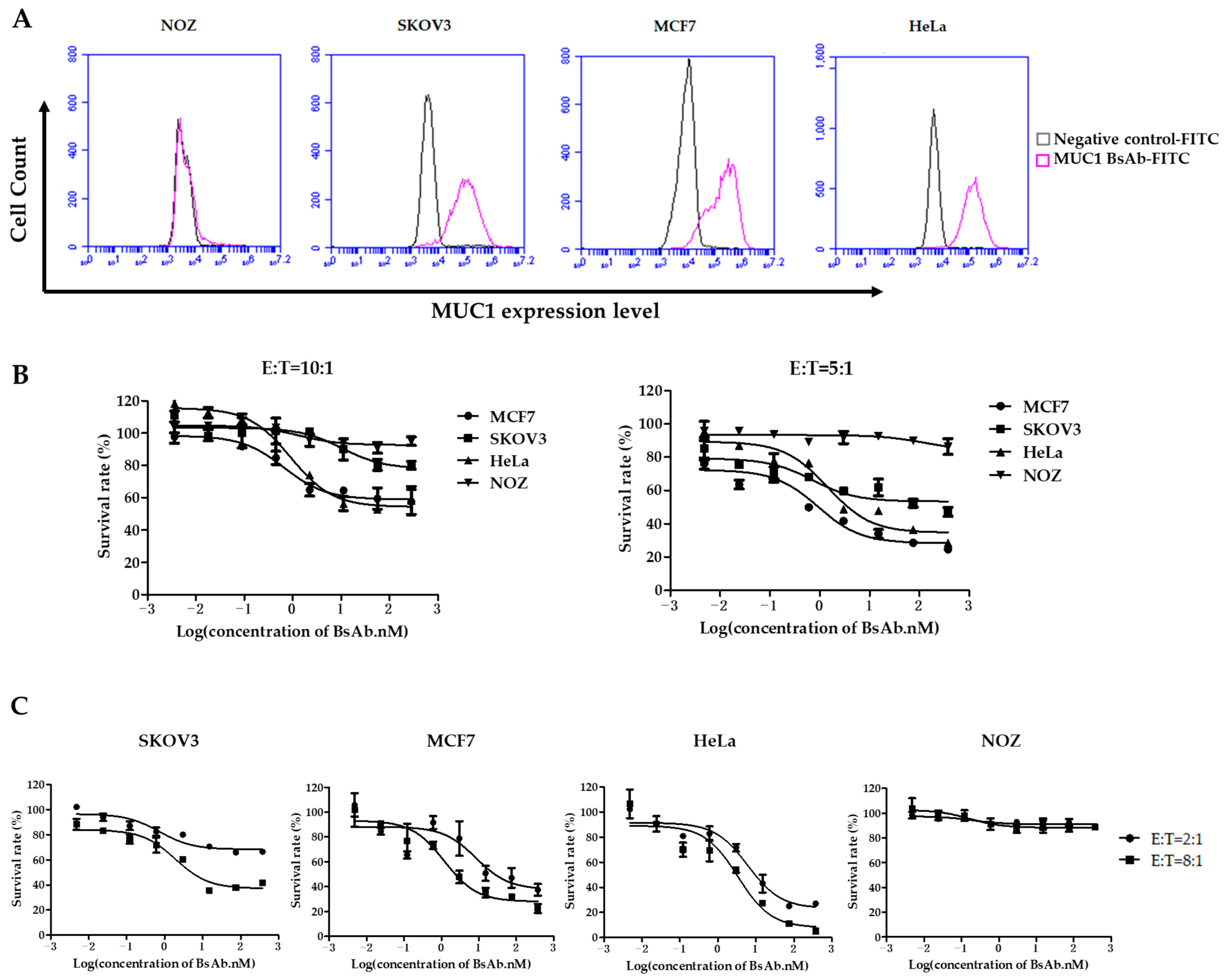
3.2. MUC1/CD3 BsAb Effectively Induces T Cell-Mediated Lysis of MUC1-Positive Tumor Cells In Vitro
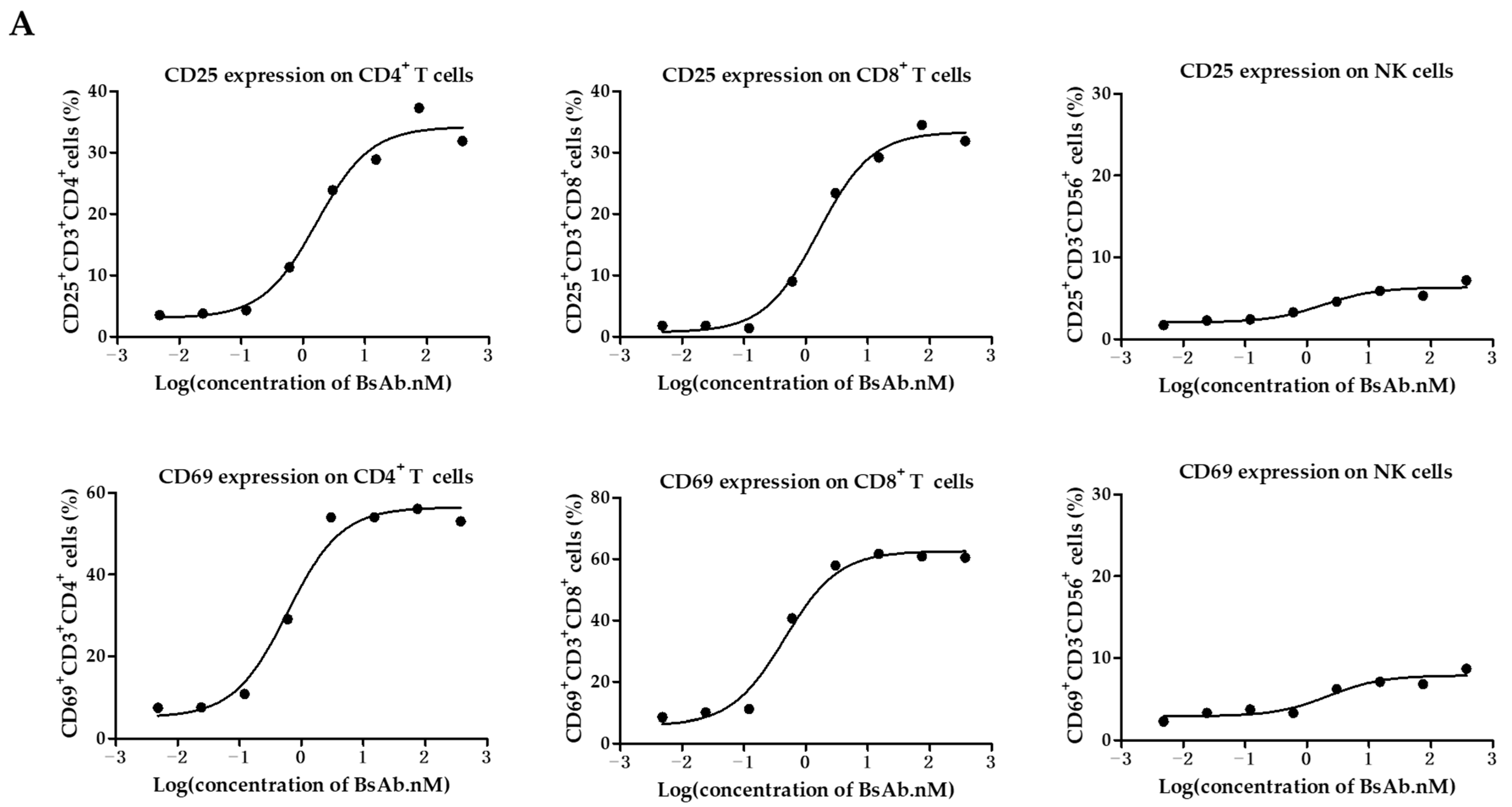
3.3. MUC1/CD3 BsAb Potently Activates T Cell In Vitro
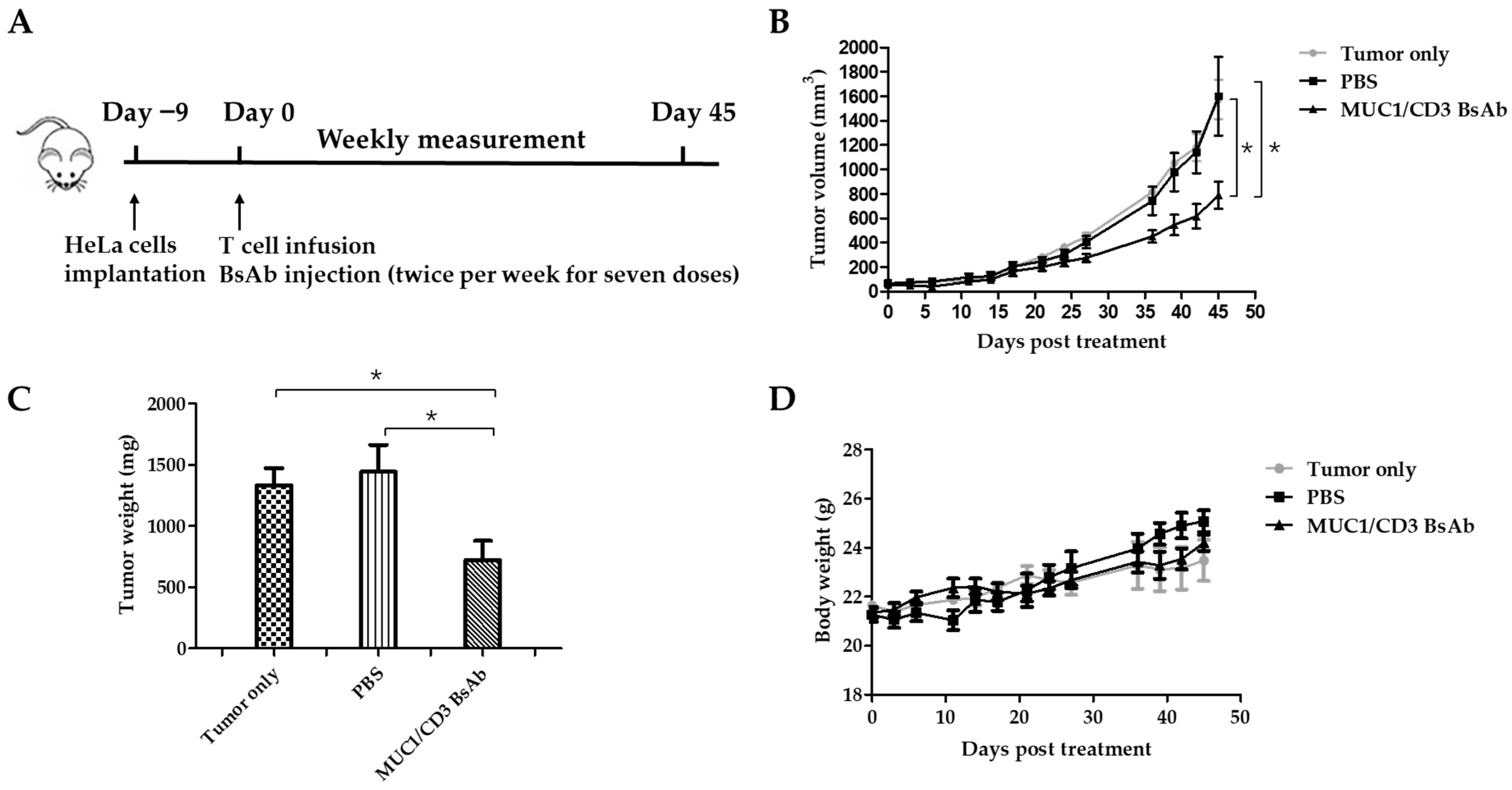
3.4. MUC1/CD3 BsAb Efficiently Inhibits MUC1-Positive Tumor Cell Growth In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dekker, J.; Rossen, J.W.; Büller, H.A.; Einerhand, A.W. The MUC family: An obituary. Trends Biochem. Sci. 2002, 27, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhang, Z.; Zhang, S.; Zhu, P.; Ko, J.K.; Yung, K.K. MUC1: Structure, Function, and Clinic Application in Epithelial Cancers. Int. J. Mol. Sci. 2021, 22, 6567. [Google Scholar] [CrossRef] [PubMed]
- Dhar, P.; McAuley, J. The Role of the Cell Surface Mucin MUC1 as a Barrier to Infection and Regulator of Inflammation. Front. Cell Infect. Microbiol. 2019, 9, 117. [Google Scholar] [CrossRef] [Green Version]
- Gendler, S.J. MUC1, the renaissance molecule. J. Mammary Gland. Biol. Neoplasia 2001, 6, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Bose, M.; Mukherjee, P. Microbe-MUC1 Crosstalk in Cancer-Associated Infections. Trends Mol. Med. 2020, 26, 324–336. [Google Scholar] [CrossRef]
- Wu, G.; Kim, D.; Kim, J.N.; Park, S.; Maharjan, S.; Koh, H.; Moon, K.; Lee, Y.; Kwon, H.J. A Mucin1 C-terminal Subunit-directed Monoclonal Antibody Targets Overexpressed Mucin1 in Breast Cancer. Theranostics 2018, 8, 78–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.Y.; Roh, S.J.; Kim, Y.N.; Kim, S.Z.; Park, H.S.; Jang, K.Y.; Chung, M.J.; Kang, M.J.; Lee, D.G.; Moon, W.S. Expression of MUC1, MUC2, MUC5AC and MUC6 in cholangiocarcinoma: Prognostic impact. Oncol. Rep. 2009, 22, 649–657. [Google Scholar]
- Jing, X.; Liang, H.; Hao, C.; Yang, X.; Cui, X. Overexpression of MUC1 predicts poor prognosis in patients with breast cancer. Oncol. Rep. 2019, 41, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; Boyce, R.W.; Abd El-Rehim, D.; Kurien, T.; Green, A.R.; Paish, E.C.; Robertson, J.F.; Ellis, I.O. Expression of mucins (MUC1, MUC2, MUC3, MUC4, MUC5AC and MUC6) and their prognostic significance in human breast cancer. Mod. Pathol. 2005, 18, 1295–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, S.; Mukherjee, P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol Med. 2014, 20, 332–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.H.; Choi, S.; Park, Y.; Jin, H.S. Mucin1 and Mucin16: Therapeutic Targets for Cancer Therapy. Pharmaceuticals 2021, 14, 1053. [Google Scholar] [CrossRef] [PubMed]
- Beckwith, D.M.; Cudic, M. Tumor-associated O-glycans of MUC1: Carriers of the glyco-code and targets for cancer vaccine design. Semin. Immunol. 2020, 47, 101389. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Bandyopadhyay, D. MUC1: A target molecule for cancer therapy. Cancer Biol. Ther. 2007, 6, 481–486. [Google Scholar] [CrossRef]
- Bouillez, A.; Rajabi, H.; Pitroda, S.; Jin, C.; Alam, M.; Kharbanda, A.; Tagde, A.; Wong, K.K.; Kufe, D. Inhibition of MUC1-C Suppresses MYC Expression and Attenuates Malignant Growth in KRAS Mutant Lung Adenocarcinomas. Cancer Res. 2016, 76, 1538–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besmer, D.M.; Curry, J.M.; Roy, L.D.; Tinder, T.L.; Sahraei, M.; Schettini, J.; Hwang, S.I.; Lee, Y.Y.; Gendler, S.J.; Mukherjee, P. Pancreatic ductal adenocarcinoma mice lacking mucin 1 have a profound defect in tumor growth and metastasis. Cancer Res. 2011, 71, 4432–4442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Wang, F.; Liu, G.; Yuan, H.; Chen, T.; Wang, J.; Xie, F.; Zhai, R.; Wang, F.; Guo, Y.; et al. Impact of Mucin1 knockdown on the phenotypic characteristics of the human hepatocellular carcinoma cell line SMMC-7721. Oncol. Rep. 2014, 31, 2811–2819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Li, D.; Ren, J.; Li, C.; Xiao, Z.X. MUC1 activates JNK1 and inhibits apoptosis under genotoxic stress. Biochem. Biophys. Res. Commun. 2013, 440, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Chaika, N.V.; Gebregiworgis, T.; Lewallen, M.E.; Purohit, V.; Radhakrishnan, P.; Liu, X.; Zhang, B.; Mehla, K.; Brown, R.B.; Caffrey, T.; et al. MUC1 mucin stabilizes and activates hypoxia-inducible factor 1 alpha to regulate metabolism in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 13787–13792. [Google Scholar] [CrossRef] [PubMed]
- Weiner, L.M.; Murray, J.C.; Shuptrine, C.W. Antibody-based immunotherapy of cancer. Cell 2012, 148, 1081–1084. [Google Scholar] [CrossRef] [Green Version]
- Danielczyk, A.; Stahn, R.; Faulstich, D.; Löffler, A.; Märten, A.; Karsten, U.; Goletz, S. PankoMab: A potent new generation anti-tumour MUC1 antibody. Cancer Immunol. Immunother. 2006, 55, 1337–1347. [Google Scholar] [CrossRef] [PubMed]
- Dian, D.; Janni, W.; Kuhn, C.; Mayr, D.; Karsten, U.; Mylonas, I.; Friese, K.; Jeschke, U. Evaluation of a novel anti-mucin 1 (MUC1) antibody (PankoMab) as a potential diagnostic tool in human ductal breast cancer; comparison with two established antibodies. Onkologie 2009, 32, 238–244. [Google Scholar] [CrossRef]
- Fiedler, W.; DeDosso, S.; Cresta, S.; Weidmann, J.; Tessari, A.; Salzberg, M.; Dietrich, B.; Baumeister, H.; Goletz, S.; Gianni, L.; et al. A phase I study of PankoMab-GEX, a humanised glyco-optimised monoclonal antibody to a novel tumour-specific MUC1 glycopeptide epitope in patients with advanced carcinomas. Eur. J. Cancer 2016, 63, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledermann, J.; Sehouli, J.; Zurawski, B.; Raspagliesi, F.; De Giorgi, U.; Banerjee, S.; Arija, J.A.; Marin, M.R.; Lisyanskaya, A.; Póka, R.L.; et al. A double-blind, placebo-controlled, randomized, phase 2 study to evaluate the efficacy and safety of switch maintenance therapy with the anti-TA-MUC1 antibody PankoMab-GEX after chemotherapy in patients with recurrent epithelial ovarian carcinoma. Ann. Oncol. 2017, 28, v626. [Google Scholar] [CrossRef] [Green Version]
- Ochsenreither, S.; Fiedler, W.M.; Conte, G.D.; Macchini, M.; Matos, I.; Habel, B.; Ahrens-Fath, I.; Raspagliesi, F.; Lorusso, D.; Keilholz, U.; et al. Safety and preliminary activity results of the GATTO study, a phase Ib study combining the anti-TA-MUC1 antibody gatipotuzumab with the anti-EGFR tomuzotuximab in patients with refractory solid tumors. ESMO Open 2022, 7, 100447. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, N.K.; Yariz, K.O.; Bondarenko, I.; Manikhas, A.; Semiglazov, V.; Alyasova, A.; Komisarenko, V.; Shparyk, Y.; Murray, J.L.; Jones, D.; et al. Randomized phase II trial of letrozole plus anti-MUC1 antibody AS1402 in hormone receptor-positive locally advanced or metastatic breast cancer. Clin. Cancer Res. 2011, 17, 6822–6830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulec, S.; Pennington, K.; Bruetman, D.; Garl, S.; Horne, H.; Gold, D.; Wegener, W.; Goldenberg, D. A phase-I study of 90Y-hPAM4 (humanized anti-MUC1 monoclonal antibody) in patients with unresectable and metastatic pancreatic cancer. J. Nucl. Med. 2007, 48, 393P. [Google Scholar]
- Kramer, E.L.; Liebes, L.; Wasserheit, C.; Noz, M.E.; Blank, E.W.; Zabalegui, A.; Melamed, J.; Furmanski, P.; Peterson, J.A.; Ceriani, R.L. Initial clinical evaluation of radiolabeled MX-DTPA humanized BrE-3 antibody in patients with advanced breast cancer. Clin. Cancer Res. 1998, 4, 1679–1688. [Google Scholar]
- Chan, S.Y.; Gordon, A.N.; Coleman, R.E.; Hall, J.B.; Berger, M.S.; Sherman, M.L.; Eten, C.B.; Finkler, N.J. A phase 2 study of the cytotoxic immunoconjugate CMB-401 (hCTM01-calicheamicin) in patients with platinum-sensitive recurrent epithelial ovarian carcinoma. Cancer Immunol. Immunother. 2003, 52, 243–248. [Google Scholar] [CrossRef]
- Fischer, N.; Léger, O. Bispecific antibodies: Molecules that enable novel therapeutic strategies. Pathobiology 2007, 74, 3–14. [Google Scholar] [CrossRef]
- Nie, S.; Wang, Z.; Moscoso-Castro, M.; D’Souza, P.; Lei, C.; Xu, J.; Gu, J. Biology drives the discovery of bispecific antibodies as innovative therapeutics. Antib. Ther. 2020, 3, 18–62. [Google Scholar] [CrossRef] [Green Version]
- Seimetz, D.; Lindhofer, H.; Bokemeyer, C. Development and approval of the trifunctional antibody catumaxomab (anti-EpCAM× anti-CD3) as a targeted cancer immunotherapy. Cancer Treat. Rev. 2010, 36, 458–467. [Google Scholar] [CrossRef]
- Oak, E.; Bartlett, N.L. Blinatumomab for the treatment of B-cell lymphoma. Expert Opin. Investig. Drugs 2015, 24, 715–724. [Google Scholar] [CrossRef]
- Kang, C. Mosunetuzumab: First Approval. Drugs 2022, 82, 1229–1234. [Google Scholar] [CrossRef]
- Kang, C. Teclistamab: First Approval. Drugs 2022, 82, 1613–1619. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Tebentafusp: First approval. Drugs 2022, 82, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Chari, A.; Minnema, M.C.; Berdeja, J.G.; Oriol, A.; van de Donk, N.W.C.J.; Rodríguez-Otero, P.; Askari, E.; Mateos, M.V.; Costa, L.J.; Caers, J.; et al. Talquetamab, a T-Cell-Redirecting GPRC5D Bispecific Antibody for Multiple Myeloma. N. Engl. J. Med. 2022, 387, 2232–2244. [Google Scholar] [CrossRef] [PubMed]
- Taki, S.; Kamada, H.; Inoue, M.; Nagano, K.; Mukai, Y.; Higashisaka, K.; Yoshioka, Y.; Tsutsumi, Y.; Tsunoda, S.I. A novel bispecific antibody against human CD3 and ephrin receptor A10 for breast cancer therapy. PLoS ONE 2015, 10, e0144712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Pan, Z.; Han, L.; Zhou, Y.; Zong, H.; Wang, L.; Sun, R.; Jiang, H.; Xie, Y.; Yuan, Y.; et al. A novel bispecific antibody targeting CD3 and Lewis Y with potent therapeutic efficacy against gastric cancer. Biomedicines 2021, 9, 1059. [Google Scholar] [CrossRef] [PubMed]
- Bacac, M.; Fauti, T.; Sam, J.; Colombetti, S.; Weinzierl, T.; Ouaret, D.; Bodmer, W.; Lehmann, S.; Hofer, T.; Hosse, R.J.; et al. A Novel Carcinoembryonic Antigen T-Cell Bispecific Antibody (CEA TCB) for the Treatment of Solid Tumors. Clin. Cancer Res. 2016, 22, 3286–3297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iizuka, A.; Nonomura, C.; Ashizawa, T.; Kondou, R.; Ohshima, K.; Sugino, T.; Mitsuya, K.; Hayashi, N.; Nakasu, Y.; Maruyama, K.; et al. A T-cell-engaging B7-H4/CD3-bispecific Fab-scFv Antibody Targets Human Breast Cancer. Clin. Cancer Res. 2019, 25, 2925–2934. [Google Scholar] [CrossRef] [Green Version]
- Junttila, T.T.; Li, J.; Johnston, J.; Hristopoulos, M.; Clark, R.; Ellerman, D.; Wang, B.E.; Li, Y.; Mathieu, M.; Li, G.; et al. Antitumor Efficacy of a Bispecific Antibody That Targets HER2 and Activates T Cells. Cancer Res. 2014, 74, 5561–5571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Zhang, L.; Zeng, Z.; Yan, T.; Cheng, J.; Miao, X.; Lu, Y. Complete Response to PD-1 Inhibitor in Primary Hepatocellular Carcinoma Patients Post-Progression on Bi-Specific Antibody Conjugated CIK Cell Treatment: A Report of Two Cases. Onco. Targets Ther. 2021, 14, 5447–5453. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, S.; Wu, Y.; Wu, D.; Wang, J.; Li, F. The combination therapy with EpCAM/CD3 BsAb and MUC-1/CD3 BsAb elicited antitumor immunity by T-cell adoptive immunotherapy in lung cancer. Int. J. Med. Sci. 2021, 18, 3380–3388. [Google Scholar] [CrossRef]
- Pang, X.; Huang, Z.; Zhong, T.; Zhang, P.; Wang, Z.M.; Xia, M.; Li, B. Cadonilimab, a tetravalent PD-1/CTLA-4 bispecific antibody with trans-binding and enhanced target binding avidity. MAbs 2023, 15, 2180794. [Google Scholar] [CrossRef]
- Pourjafar, M.; Samadi, P.; Saidijam, M. MUC1 antibody-based therapeutics: The promise of cancer immunotherapy. Immunotherapy 2020, 12, 1269–1286. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portell, C.A.; Wenzell, C.M.; Advani, A.S. Clinical and pharmacologic aspects of blinatumomab in the treatment of B-cell acute lymphoblastic leukemia. Clin. Pharmacol. 2013, 5 (Suppl. S1), 5–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvaris, R.; Ong, J.; Gregory, G.P. Bispecific Antibodies: A Review of Development, Clinical Efficacy and Toxicity in B-Cell Lymphomas. J. Pers. Med. 2021, 11, 355. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Dees, S.; Grewal, I.S. Overcoming the challenges associated with CD3+ T-cell redirection in cancer. Br. J. Cancer 2021, 124, 1037–1048. [Google Scholar] [CrossRef]
- Li, H.; Er Saw, P.; Song, E. Challenges and strategies for next-generation bispecific antibody-based antitumor therapeutics. Cell Mol. Immunol. 2020, 17, 451–461. [Google Scholar] [CrossRef]
- Golay, J.; Andrea, A.E. Combined Anti-Cancer Strategies Based on Anti-Checkpoint Inhibitor Antibodies. Antibodies 2020, 9, 17. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, J.; Lai, S.; Yu, H.; Ma, L. Suppression of MUC1-Overexpressing Tumors by a Novel MUC1/CD3 Bispecific Antibody. Antibodies 2023, 12, 47. https://doi.org/10.3390/antib12030047
Fang J, Lai S, Yu H, Ma L. Suppression of MUC1-Overexpressing Tumors by a Novel MUC1/CD3 Bispecific Antibody. Antibodies. 2023; 12(3):47. https://doi.org/10.3390/antib12030047
Chicago/Turabian StyleFang, Jun, Shifa Lai, Haoyang Yu, and Lan Ma. 2023. "Suppression of MUC1-Overexpressing Tumors by a Novel MUC1/CD3 Bispecific Antibody" Antibodies 12, no. 3: 47. https://doi.org/10.3390/antib12030047
APA StyleFang, J., Lai, S., Yu, H., & Ma, L. (2023). Suppression of MUC1-Overexpressing Tumors by a Novel MUC1/CD3 Bispecific Antibody. Antibodies, 12(3), 47. https://doi.org/10.3390/antib12030047