

The Role of Antibody-Based Therapies in Neuro-Oncology
 , ,
, ,
Abstract
:1. Background
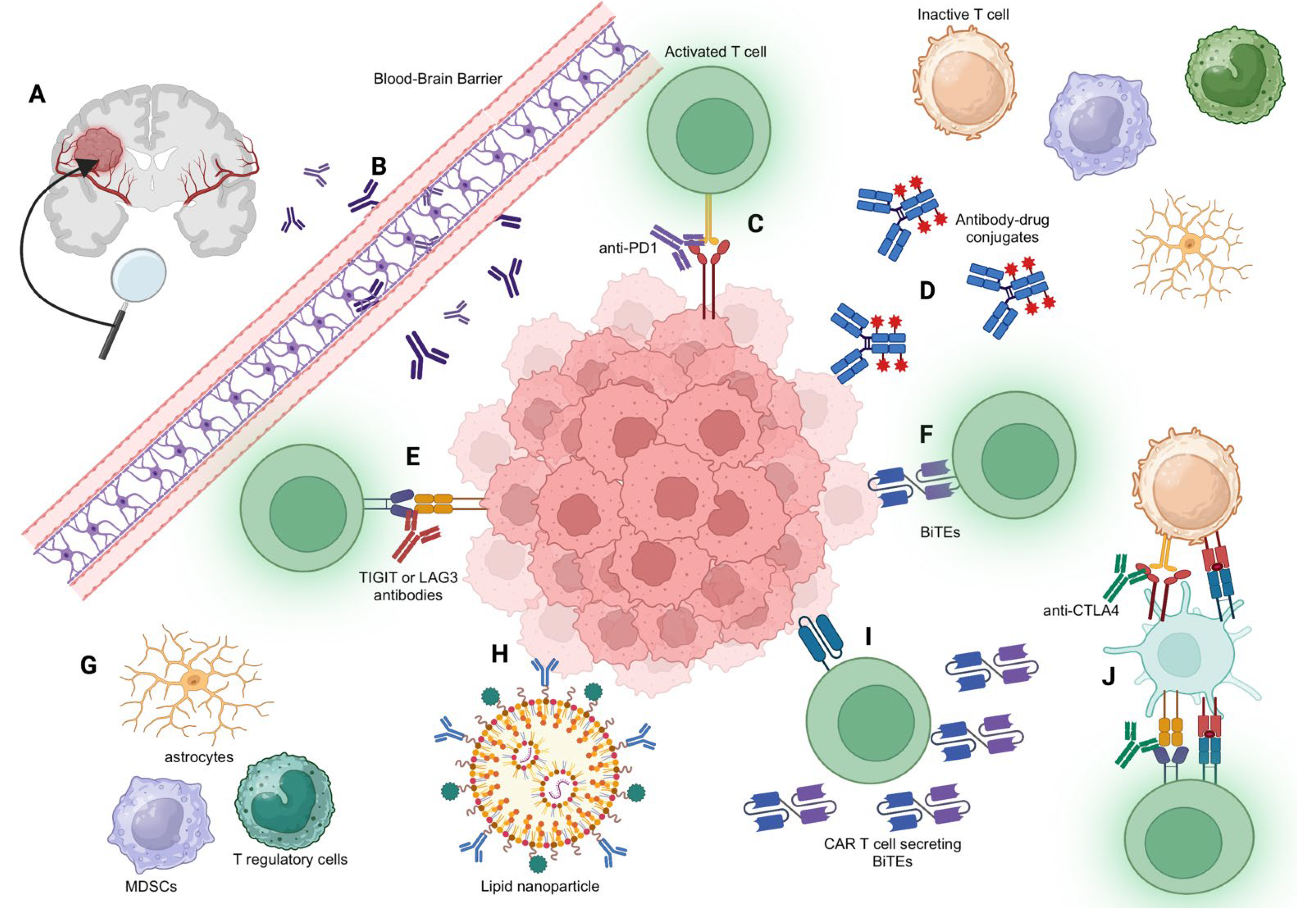
2. The Challenge of Crossing the BBB
3. Immune Checkpoint Inhibitors and Immunomodulatory Antibodies
3.1. Immune Checkpoint Inhibitors: Anti-PD-1 and Anti-CTLA-4
3.2. Combination Treatment Strategies
3.3. Clinical Trials for Anti-PD-1 and Anti-CTLA-4 Therapies
3.4. Additional Immune Checkpoints: TIGIT and LAG3
3.5. Modulating the Myeloid Compartment with Antibodies
3.6. Neurotoxicity Profile of Immune Checkpoint and Modulatory Antibodies
4. Tumor-Specific Targets for Monoclonal Antibody-Based Therapy
4.1. EGFRvIII
4.2. IL13Rα2
4.3. HER2
4.4. Disialoganglioside (GD2)
4.5. CD147
4.6. SMARCAL1 Mutations
4.7. Delta-Like Canonical Notch Ligand 3 (DLL3)
5. Advanced Antibody-Based Strategies
5.1. Antibody–Drug Conjugates (ADCs)
5.2. Bispecific T Cell Engagers (BiTEs)
5.3. Using Adoptive Cell Therapy to Enhance Antibody Delivery to the CNS
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cancer Facts & Figures 2023. Available online: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/2023-cancer-facts-figures.html (accessed on 24 July 2023).
- Smith, K.; Garman, L.; Wrammert, J.; Zheng, N.-Y.; Capra, N.D.; Ahmed, R.; Wilson, P.C. Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat. Protoc. 2009, 4, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.U.; Morrison, S.L. Production and properties of chimeric antibody molecules. Methods Enzymol. 1989, 178, 459–476. [Google Scholar] [CrossRef] [PubMed]
- Riechmann, L.; Clark, M.; Waldmann, H.; Winter, G. Reshaping human antibodies for therapy. Nature 1988, 332, 323–327. [Google Scholar] [CrossRef]
- Himes, B.T.; Geiger, P.A.; Ayasoufi, K.; Bhargav, A.G.; Brown, D.A.; Parney, I.F. Immunosuppression in Glioblastoma: Current Understanding and Therapeutic Implications. Front. Oncol. 2021, 11, 770561. [Google Scholar] [CrossRef]
- Wu, D.; Chen, Q.; Chen, X.; Han, F.; Chen, Z.; Wang, Y. The blood–brain barrier: Structure, regulation, and drug delivery. Signal Transduct. Target. Ther. 2023, 8, 217. [Google Scholar] [CrossRef] [PubMed]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The blood-brain barrier and blood-tumour barrier in brain tumours and metastases. Nat. Rev. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Blood-Brain Barrier and Delivery of Protein and Gene Therapeutics to Brain. Front. Aging Neurosci. 2019, 11, 373. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-López, E.; Schuhmacher, A.J. Transportation of Single-Domain Antibodies through the Blood–Brain Barrier. Biomolecules 2021, 11, 1131. [Google Scholar] [CrossRef]
- Roopenian, D.C.; Akilesh, S. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715–725. [Google Scholar] [CrossRef]
- Tien, J.; Leonoudakis, D.; Petrova, R.; Trinh, V.; Taura, T.; Sengupta, D.; Jo, L.; Sho, A.; Yun, Y.; Doan, E.; et al. Modifying antibody-FcRn interactions to increase the transport of antibodies through the blood-brain barrier. mAbs 2023, 15, 2229098. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology 2018, 20, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Solar, P.; Hendrych, M.; Barak, M.; Valekova, H.; Hermanova, M.; Jancalek, R. Blood-Brain Barrier Alterations and Edema Formation in Different Brain Mass Lesions. Front. Cell Neurosci. 2022, 16, 922181. [Google Scholar] [CrossRef]
- Wolburg, H.; Noell, S.; Fallier-Becker, P.; Mack, A.F.; Wolburg-Buchholz, K. The disturbed blood-brain barrier in human glioblastoma. Mol. Asp. Med. 2012, 33, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Bard, F.; Cannon, C.; Barbour, R.; Burke, R.-L.; Games, D.; Grajeda, H.; Guido, T.; Hu, K.; Huang, J.; Johnson-Wood, K.; et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat. Med. 2000, 6, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Dalmau, J.; Rosenfeld, M.R. Paraneoplastic syndromes of the CNS. Lancet Neurol. 2008, 7, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Day, E.D.; Lassiter, S.; Woodhall, B.; Mahaley, J.L.; Mahaley, M.S. The localization of radioantibodies in human brain tumors: I. Preliminary exploration. Cancer Res. 1965, 25, 773–778. [Google Scholar] [PubMed]
- Zalutsky, M.R.; Moseley, R.P.; Coakham, H.B.; Coleman, R.E.; Bigner, D.D. Pharmacokinetics and tumor localization of 131I-labeled anti-tenascin monoclonal antibody 81C6 in patients with gliomas and other intracranial malignancies. Cancer Res. 1989, 49, 2807–2813. [Google Scholar] [PubMed]
- Faresjö, R.; Bonvicini, G.; Fang, X.T.; Aguilar, X.; Sehlin, D.; Syvänen, S. Brain pharmacokinetics of two BBB penetrating bispecific antibodies of different size. Fluids Barriers CNS 2021, 18, 26. [Google Scholar] [CrossRef]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Honda, T.; Egen, J.G.; Lämmermann, T.; Kastenmüller, W.; Torabi-Parizi, P.; Germain, R.N. Tuning of Antigen Sensitivity by T Cell Receptor-Dependent Negative Feedback Controls T Cell Effector Function in Inflamed Tissues. Immunity 2014, 40, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Sharma, A.; Subudhi, S.K.; Blando, J.; Scutti, J.; Vence, L.; Wargo, J.; Allison, J.P.; Ribas, A.; Sharma, P. Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3+ Regulatory T Cells (Tregs) in Human Cancers. Clin. Cancer Res. 2019, 25, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, V.A.; Dmello, C.; McGrail, D.J.; Brat, D.J.; Lee-Chang, C.; Heimberger, A.B.; Chand, D.; Stupp, R.; Sonabend, A.M. Immune checkpoint blockade in glioblastoma: From tumor heterogeneity to personalized treatment. J. Clin. Investig. 2023, 133. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Vandenberk, L.; Van Woensel, M.; Belmans, J.; Schaaf, M.; Boon, L.; De Vleeschouwer, S.; Agostinis, P. Preclinical efficacy of immune-checkpoint monotherapy does not recapitulate corresponding biomarkers-based clinical predictions in glioblastoma. OncoImmunology 2017, 6, e1295903. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kwon, M.; Kim, K.H.; Kim, T.-S.; Hong, S.-H.; Kim, C.G.; Kang, S.-G.; Moon, J.H.; Kim, E.H.; Park, S.-H.; et al. Immune Checkpoint Inhibitor-induced Reinvigoration of Tumor-infiltrating CD8+ T Cells is Determined by Their Differentiation Status in Glioblastoma. Clin. Cancer Res. 2019, 25, 2549–2559. [Google Scholar] [CrossRef] [PubMed]
- Mauldin, I.S.; Jo, J.; Wages, N.A.; Yogendran, L.V.; Mahmutovic, A.; Young, S.J.; Lopes, M.B.; Slingluff, C.L.; Erickson, L.D.; Fadul, C.E. Proliferating CD8+ T Cell Infiltrates Are Associated with Improved Survival in Glioblastoma. Cells 2021, 10, 3378. [Google Scholar] [CrossRef]
- Karimi, S.; Mansouri, S.; Mamatjan, Y.; Liu, J.; Nassiri, F.; Suppiah, S.; Singh, O.; Aldape, K.; Zadeh, G. Programmed death ligand-1 (PD-L1) expression in meningioma; prognostic significance and its association with hypoxia and NFKB2 expression. Sci. Rep. 2020, 10, 14115. [Google Scholar] [CrossRef]
- Ijad, N.; Dahal, A.; Kim, A.E.; Wakimoto, H.; Juratli, T.A.; Brastianos, P.K. Novel Systemic Approaches for the Management of Meningiomas: Immunotherapy and Targeted Therapies. Neurosurg. Clin. 2023, 34, 447–454. [Google Scholar] [CrossRef]
- Huang, J.; Liu, F.; Liu, Z.; Tang, H.; Wu, H.; Gong, Q.; Chen, J. Immune Checkpoint in Glioblastoma: Promising and Challenging. Front. Pharmacol. 2017, 8, 242. [Google Scholar] [CrossRef]
- Saha, D.; Martuza, R.L.; Rabkin, S.D. Oncolytic herpes simplex virus immunovirotherapy in combination with immune checkpoint blockade to treat glioblastoma. Immunotherapy 2018, 10, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Menon, H.; Ramapriyan, R.; Cushman, T.R.; Verma, V.; Kim, H.H.; Schoenhals, J.E.; Atalar, C.; Selek, U.; Chun, S.G.; Chang, J.Y.; et al. Role of Radiation Therapy in Modulation of the Tumor Stroma and Microenvironment. Front. Immunol. 2019, 10, 193. [Google Scholar] [CrossRef] [PubMed]
- Akkari, L.; Bowman, R.L.; Tessier, J.; Klemm, F.; Handgraaf, S.M.; de Groot, M.; Quail, D.F.; Tillard, L.; Gadiot, J.; Huse, J.T.; et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci. Transl. Med. 2020, 12, eaaw7843. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; See, A.P.; Phallen, J.; Jackson, C.M.; Belcaid, Z.; Ruzevick, J.; Durham, N.; Meyer, C.; Harris, T.J.; Albesiano, E.; et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Kim, T.M.; Frenel, J.; Simonelli, M.; Lopez, J.; Subramaniam, D.S.; Siu, L.L.; Wang, H.; Krishnan, S.; Stein, K.; et al. Treatment with pembrolizumab in programmed death ligand 1–positive recurrent glioblastoma: Results from the multicohort phase 1 KEYNOTE-028 trial. Cancer 2021, 127, 1620–1629. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs. Bevacizumab in Patients with Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003. [Google Scholar] [CrossRef] [PubMed]
- Duerinck, J.; Schwarze, J.K.; Awada, G.; Tijtgat, J.; Vaeyens, F.; Bertels, C.; Geens, W.; Klein, S.; Seynaeve, L.; Cras, L.; et al. Intracerebral administration of CTLA-4 and PD-1 immune checkpoint blocking monoclonal antibodies in patients with recurrent glioblastoma: A phase I clinical trial. J. Immunother. Cancer 2021, 9, e002296. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Nayak, L.; Molinaro, A.M.; Peters, K.; Clarke, J.L.; Jordan, J.T.; de Groot, J.; Nghiemphu, L.; Kaley, T.; Colman, H.; McCluskey, C.; et al. Randomized Phase II and Biomarker Study of Pembrolizumab plus Bevacizumab versus Pembrolizumab Alone for Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2021, 27, 1048–1057. [Google Scholar] [CrossRef]
- Sahebjam, S.; A Forsyth, P.; Tran, N.D.; A Arrington, J.; Macaulay, R.; Etame, A.B.; Walko, C.M.; Boyle, T.; Peguero, E.N.; Jaglal, M.; et al. Hypofractionated stereotactic re-irradiation with pembrolizumab and bevacizumab in patients with recurrent high-grade gliomas: Results from a phase I study. Neuro-Oncology 2021, 23, 677–686. [Google Scholar] [CrossRef]
- Omuro, A.; A Brandes, A.; Carpentier, A.F.; Idbaih, A.; A Reardon, D.; Cloughesy, T.; Sumrall, A.; Baehring, J.; Bent, M.v.D.; Bähr, O.; et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro-Oncology 2023, 25, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Weller, M.; Idbaih, A.; Steinbach, J.; Finocchiaro, G.; Raval, R.R.; Ansstas, G.; Baehring, J.; Taylor, J.W.; Honnorat, J.; et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro-Oncology 2022, 24, 1935–1949. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neuro-Oncology 2018, 20, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; López-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inogés, S.; De Andrea, C.; López-Diaz De Cerio, A.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Brastianos, P.K.; Kim, A.E.; Giobbie-Hurder, A.; Lee, E.Q.; Wang, N.; Eichler, A.F.; Chukwueke, U.; Forst, D.A.; Arrillaga-Romany, I.C.; Dietrich, J.; et al. Phase 2 study of pembrolizumab in patients with recurrent and residual high-grade meningiomas. Nat. Commun. 2022, 13, 1325. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.L.; Nayak, L.; Meredith, D.M.; Driver, J.; Du, Z.; Hoffman, S.; Li, Y.; Lee, E.Q.; Beroukim, R.; Rinne, M.; et al. Activity of PD-1 blockade with nivolumab among patients with recurrent atypical/anaplastic meningioma: Phase II trial results. Neuro-Oncology 2022, 24, 101–113. [Google Scholar] [CrossRef]
- Strickler, J.H.; Hanks, B.A.; Khasraw, M. Tumor Mutational Burden as a Predictor of Immunotherapy Response: Is More Always Better? Clin. Cancer Res. 2021, 27, 1236–1241. [Google Scholar] [CrossRef]
- Nassiri, F.; Patil, V.; Yefet, L.S.; Singh, O.; Liu, J.; Dang, R.M.A.; Yamaguchi, T.N.; Daras, M.; Cloughesy, T.F.; Colman, H.; et al. Oncolytic DNX-2401 virotherapy plus pembrolizumab in recurrent glioblastoma: A phase 1/2 trial. Nat. Med. 2023, 29, 1370–1378. [Google Scholar] [CrossRef]
- Ge, Z.; Peppelenbosch, M.P.; Sprengers, D.; Kwekkeboom, J. TIGIT, the Next Step Towards Successful Combination Immune Checkpoint Therapy in Cancer. Front. Immunol. 2021, 12, 699895. [Google Scholar] [CrossRef]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef]
- Hung, A.L.; Maxwell, R.; Theodros, D.; Belcaid, Z.; Mathios, D.; Luksik, A.S.; Kim, E.; Wu, A.; Xia, Y.; Garzon-Muvdi, T.; et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. OncoImmunology 2018, 7, e1466769. [Google Scholar] [CrossRef] [PubMed]
- Harris-Bookman, S.; Mathios, D.; Martin, A.M.; Xia, Y.; Kim, E.; Xu, H.; Belcaid, Z.; Polanczyk, M.; Barberi, T.; Theodros, D.; et al. Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma. Int. J. Cancer 2018, 143, 3201–3208. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Prins, R.M.; Heimberger, A.B. The immune landscape of common CNS malignancies: Implications for immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 729–744. [Google Scholar] [CrossRef] [PubMed]
- Birge, R.B.; Boeltz, S.; Kumar, S.; Carlson, J.; Wanderley, J.; Calianese, D.; Barcinski, M.; A Brekken, R.; Huang, X.; Hutchins, J.T.; et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016, 23, 962–978. [Google Scholar] [CrossRef] [PubMed]
- Budhu, S.; Giese, R.; Gupta, A.; Fitzgerald, K.; Zappasodi, R.; Schad, S.; Hirschhorn, D.; Campesato, L.F.; De Henau, O.; Gigoux, M.; et al. Targeting Phosphatidylserine Enhances the Anti-tumor Response to Tumor-Directed Radiation Therapy in a Preclinical Model of Melanoma. Cell Rep. 2021, 34, 108620. [Google Scholar] [CrossRef]
- He, J.; Yin, Y.; Luster, T.A.; Watkins, L.; Thorpe, P.E. Antiphosphatidylserine antibody combined with irradiation damages tumor blood vessels and induces tumor immunity in a rat model of glioblastoma. Clin. Cancer Res. 2009, 15, 6871–6880. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Huang, X.; Lynn, K.D.; Thorpe, P.E. Phosphatidylserine-targeting antibody induces M1 macrophage polarization and promotes myeloid-derived suppressor cell differentiation. Cancer Immunol. Res. 2013, 1, 256–268. [Google Scholar] [CrossRef]
- Ly, K.I.; Richardson, L.G.; Liu, M.; Muzikansky, A.; Cardona, J.; Lou, K.; Beers, A.L.; Chang, K.; Brown, J.M.; Ma, X.; et al. Bavituximab decreases immunosuppressive myeloid-derived suppressor cells in newly diagnosed glioblastoma patients. Clin Cancer Res. 2023, 29, 3017–3025. [Google Scholar] [CrossRef]
- Zhang, M.; Hutter, G.; Kahn, S.A.; Azad, T.D.; Gholamin, S.; Xu, C.Y.; Liu, J.; Achrol, A.S.; Richard, C.; Sommerkamp, P.; et al. Anti-CD47 Treatment Stimulates Phagocytosis of Glioblastoma by M1 and M2 Polarized Macrophages and Promotes M1 Polarized Macrophages In Vivo. PLoS ONE 2016, 11, e0153550. [Google Scholar] [CrossRef]
- Gholamin, S.; Mitra, S.S.; Feroze, A.H.; Liu, J.; Kahn, S.A.; Zhang, M.; Esparza, R.; Richard, C.; Ramaswamy, V.; Remke, M.; et al. Disrupting the CD47-SIRPα anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 2017, 9, eaaf2968. [Google Scholar] [CrossRef]
- Wang, X.; Guo, G.; Guan, H.; Yu, Y.; Lu, J.; Yu, J. Challenges and potential of PD-1/PD-L1 checkpoint blockade immunotherapy for glioblastoma. J. Exp. Clin. Cancer Res. 2019, 38, 87. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, Q.; Xiao, W.; Zhao, Y.; Pi, J.; Xu, H.; Zhao, H.; Xu, J.; Evans, C.E.; Jin, H. Advances in Anti-Tumor Treatments Targeting the CD47/SIRPα Axis. Front. Immunol. 2020, 11, 18. [Google Scholar] [CrossRef] [PubMed]
- Oldenborg, P.A. Role of CD47 in erythroid cells and in autoimmunity. Leuk. Lymphoma 2004, 45, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Mirabile, A.; Brioschi, E.; Ducceschi, M.; Piva, S.; Lazzari, C.; Bulotta, A.; Viganò, M.G.; Petrella, G.; Gianni, L.; Gregorc, V. PD-1 Inhibitors-Related Neurological Toxicities in Patients with Non-Small-Cell Lung Cancer: A Literature Review. Cancers 2019, 11, 296. [Google Scholar] [CrossRef]
- Mangan, B.L.; McAlister, R.K.; Balko, J.M.; Johnson, D.B.; Moslehi, J.J.; Gibson, A.; Phillips, E.J. Evolving insights into the mechanisms of toxicity associated with immune checkpoint inhibitor therapy. Br. J. Clin. Pharmacol. 2020, 86, 1778–1789. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Manouchehri, A.; Haugh, A.M.; Quach, H.T.; Balko, J.M.; Lebrun-Vignes, B.; Mammen, A.; Moslehi, J.J.; Salem, J.-E. Neurologic toxicity associated with immune checkpoint inhibitors: A pharmacovigilance study. J. Immunother. Cancer 2019, 7, 134. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.; Johnson, D.B.; Davis, E.J. Toxicities associated with PD-1/PD-L1 blockade. Cancer J. 2018, 24, 36–40. [Google Scholar] [CrossRef]
- Cuzzubbo, S.; Javeri, F.; Tissier, M.; Roumi, A.; Barlog, C.; Doridam, J.; Lebbe, C.; Belin, C.; Ursu, R.; Carpentier, A. Neurological adverse events associated with immune checkpoint inhibitors: Review of the literature. Eur. J. Cancer 2017, 73, 1–8. [Google Scholar] [CrossRef]
- Kolb, N.A.; Trevino, C.R.; Waheed, W.; Sobhani, F.; Landry, K.K.; Thomas, A.A.; Hehir, M. Neuromuscular complications of immune checkpoint inhibitor therapy. Muscle Nerve 2018, 58, 10–22. [Google Scholar] [CrossRef]
- Touat, M.; Talmasov, D.; Ricard, D.; Psimaras, D. Neurological toxicities associated with immune-checkpoint inhibitors. Curr. Opin. Neurol. 2017, 30, 659–668. [Google Scholar] [CrossRef]
- Hottinger, A.F. Neurologic complications of immune checkpoint inhibitors. Curr. Opin. Neurol. 2016, 29, 806–812. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE). Version 5.0. Published Online November 27, 2017. Available online: https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcae_v5_quick_reference_5×7.pdf (accessed on 6 November 2023).
- Brahmer, J.R.; Lacchetti, C.; Schneider, B.J.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; Ernstoff, M.S.; Gardner, J.M.; Ginex, P.; et al. Management of Immune-Related Adverse Events in Patients Treated With Immune Checkpoint Inhibitor Therapy: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2018, 36, 1714–1768. [Google Scholar] [CrossRef]
- Schneider, B.J.; Naidoo, J.; Santomasso, B.D.; Lacchetti, C.; Adkins, S.; Anadkat, M.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; et al. Management of Immune-Related Adverse Events in Patients Treated With Immune Checkpoint Inhibitor Therapy: ASCO Guideline Update. J. Clin. Oncol. 2021, 39, 4073–4126. [Google Scholar] [CrossRef]
- Thompson, J.A.; Schneider, B.J.; Brahmer, J.; Achufusi, A.; Armand, P.; Berkenstock, M.K.; Bhatia, S.; Budde, L.E.; Chokshi, S.; Davies, M.; et al. Management of Immunotherapy-Related Toxicities, Version 1.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2022, 20, 387–405. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Coward, J.; McCaffrey, E.; Coucher, J.; Kalokerinos, P.; O’Byrne, K. Pembrolizumab-Induced Encephalopathy: A Review of Neurological Toxicities with Immune Checkpoint Inhibitors. J. Thorac. Oncol. 2017, 12, 1626–1635. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, J.; Dietrich, J.; Straathof, K.; Roddie, C.; Scott, B.J.; Davidson, T.B.; Prolo, L.M.; Batchelor, T.T.; Campen, C.J.; Davis, K.L.; et al. Tumor inflammation-associated neurotoxicity. Nat. Med. 2023, 29, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Sheth, V.; Gauthier, J. Taming the Beast: CRS and ICANS after CAR T-cell therapy for ALL. Bone Marrow Transplant. 2021, 56, 552–566. [Google Scholar] [CrossRef]
- Santomasso, B.; Bachier, C.; Westin, J.; Rezvani, K.; Shpall, E.J. The Other Side of CAR T-Cell Therapy: Cytokine Release Syndrome, Neurologic Toxicity, and Financial Burden. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Orentas, R.J.; Lee, D.W.; Mackall, C. Immunotherapy targets in pediatric cancer. Front. Oncol. 2012, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.D.; Tambaro, F.P.; Kohorst, M.; Chi, L.; Mahadeo, K.M.; Tewari, P.; Petropoulos, D.; Slopis, J.M.; Sadighi, Z.; Khazal, S. Immune Effector Cell Associated Neurotoxicity (ICANS) in Pediatric and Young Adult Patients Following Chimeric Antigen Receptor (CAR) T-Cell Therapy: Can We Optimize Early Diagnosis? Front. Oncol. 2021, 11, 634445. [Google Scholar] [CrossRef] [PubMed]
- Rees, J.H. Table 27.1, [Immune Effector Cell Encephalopathy (ICE) Score]. 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK584157/table/ch27.Tab1/ (accessed on 26 July 2023).
- Sugawa, N.; Ekstrand, A.J.; James, C.D.; Collins, V.P. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc. Natl. Acad. Sci. USA 1990, 87, 8602–8606. [Google Scholar] [CrossRef] [PubMed]
- Ekstrand, A.J.; Sugawa, N.; James, C.D.; Collins, V.P. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc. Natl. Acad. Sci. USA 1992, 89, 4309–4313. [Google Scholar] [CrossRef] [PubMed]
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal Growth Factor Receptor in Glioma: Signal Transduction, Neuropathology, Imaging, and Radioresistance. Neoplasia 2010, 12, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Rutkowska, A.; Stoczyńska-Fidelus, E.; Janik, K.; Włodarczyk, A.; Rieske, P. EGFRvIII: An Oncogene with Ambiguous Role. J. Oncol. 2019, 2019, 1092587. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; Archer, G.E.; Mitchell, D.A.; Heimberger, A.B.; McLendon, R.E.; Bigner, D.D.; Sampson, J.H. EGFRvIII-Targeted Vaccination Therapy of Malignant Glioma. Brain Pathol. 2009, 19, 713–723. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Li, G.; Wong, A.J. EGF receptor variant III as a target antigen for tumor immunotherapy. Expert. Rev. Vaccines 2008, 7, 977–985. [Google Scholar] [CrossRef]
- Kim, G.B.; Aragon-Sanabria, V.; Randolph, L.; Jiang, H.; Reynolds, J.A.; Webb, B.S.; Madhankumar, A.; Lian, X.; Connor, J.R.; Yang, J.; et al. High-affinity mutant Interleukin-13 targeted CAR T cells enhance delivery of clickable biodegradable fluorescent nanoparticles to glioblastoma. Bioact. Mater. 2020, 5, 624–635. [Google Scholar] [CrossRef]
- Zeng, J.; Zhang, J.; Yang, Y.-Z.; Wang, F.; Jiang, H.; Chen, H.-D.; Wu, H.-Y.; Sai, K.; Hu, W.-M. IL13RA2 is overexpressed in malignant gliomas and related to clinical outcome of patients. Am. J. Transl. Res. 2020, 12, 4702–4714. [Google Scholar] [PubMed]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang, X.; Chang, W.-C.; Weng, L.; Chang, B.; Sarkissian, A.; Brito, A.; Sanchez, J.F.; et al. Optimization of IL13Rα2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Mol. Ther. 2018, 26, 31–44. [Google Scholar] [CrossRef] [PubMed]
- IL13RA2 Protein Expression Summary—The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000123496-IL13RA2 (accessed on 26 July 2023).
- Mineo, J.-F.; Bordron, A.; Baroncini, M.; Maurage, C.-A.; Ramirez, C.; Siminski, R.-M.; Berthou, C.; Hieu, P.D. Low HER2-expressing glioblastomas are more often secondary to anaplastic transformation of low-grade glioma. J. Neurooncol. 2007, 85, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, C.; Schiff, R. HER 2: Biology, Detection, and Clinical Implications. Arch. Pathol. Lab. Med. 2011, 135, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced with a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Nazha, B.; Inal, C.; Owonikoko, T.K. Disialoganglioside GD2 Expression in Solid Tumors and Role as a Target for Cancer Therapy. Front. Oncol. 2020, 10, 1000. [Google Scholar] [CrossRef] [PubMed]
- Del Bufalo, F.; De Angelis, B.; Caruana, I.; Del Baldo, G.; De Ioris, M.A.; Serra, A.; Mastronuzzi, A.; Cefalo, M.G.; Pagliara, D.; Amicucci, M.; et al. GD2-CART01 for Relapsed or Refractory High-Risk Neuroblastoma. N. Engl. J. Med. 2023, 388, 1284–1295. [Google Scholar] [CrossRef]
- Majzner, R.G.; Ramakrishna, S.; Yeom, K.W.; Patel, S.; Chinnasamy, H.; Schultz, L.M.; Richards, R.M.; Jiang, L.; Barsan, V.; Mancusi, R.; et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 2022, 603, 934–941. [Google Scholar] [CrossRef]
- Yang, M.; Yuan, Y.; Zhang, H.; Yan, M.; Wang, S.; Feng, F.; Ji, P.; Li, Y.; Li, B.; Gao, G.; et al. Prognostic significance of CD147 in patients with glioblastoma. J. Neurooncol. 2013, 115, 19–26. [Google Scholar] [CrossRef]
- Brosnan-Cashman, J.A.; Davis, C.M.; Diplas, B.H.; Meeker, A.K.; Rodriguez, F.J.; Heaphy, C.M. SMARCAL1 loss and alternative lengthening of telomeres (ALT) are enriched in giant cell glioblastoma. Mod. Pathol. 2021, 34, 1810–1819. [Google Scholar] [CrossRef]
- Spino, M.; Kurz, S.C.; Chiriboga, L.; Serrano, J.; Zeck, B.; Sen, N.; Patel, S.; Shen, G.; Vasudevaraja, V.; Tsirigos, A.; et al. Cell Surface Notch Ligand DLL3 is a Therapeutic Target in Isocitrate Dehydrogenase-mutant Glioma. Clin. Cancer Res. 2019, 25, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Noor, H.; Whittaker, S.; McDonald, K.L. DLL3 expression and methylation are associated with lower-grade glioma immune microenvironment and prognosis. Genomics 2022, 114, 110289. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, A.S.; Hong, D.S.; Hann, C.L.; Farago, A.F.; Beltran, H.; Waqar, S.N.; Hendifar, A.E.; Anthony, L.B.; Taylor, M.H.; Bryce, A.H.; et al. A phase I/II study of rovalpituzumab tesirine in delta-like 3—Expressing advanced solid tumors. NPJ Precis. Oncol. 2021, 5, 74. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Champiat, S.; Lai, W.V.; Izumi, H.; Govindan, R.; Boyer, M.; Hummel, H.-D.; Borghaei, H.; Johnson, M.L.; Steeghs, N.; et al. Tarlatamab, a First-in-Class DLL3-Targeted Bispecific T-Cell Engager, in Recurrent Small-Cell Lung Cancer: An Open-Label, Phase I Study. J. Clin. Oncol. 2023, 41, 2893–2903. [Google Scholar] [CrossRef] [PubMed]
- Von Achenbach, C.; Silginer, M.; Blot, V.; Weiss, W.A.; Weller, M. Depatuxizumab Mafodotin (ABT-414)-induced Glioblastoma Cell Death Requires EGFR Overexpression, but not EGFRY1068 Phosphorylation. Mol. Cancer Ther. 2020, 19, 1328–1339. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, M.; Curry, R.; Reardon, D.A.; Rasmussen, E.; Upreti, V.V.; Damore, M.A.; Henary, H.A.; Hill, J.S.; Cloughesy, T. Safety, tolerability, and pharmacokinetics of anti-EGFRvIII antibody-drug conjugate AMG 595 in patients with recurrent malignant glioma expressing EGFRvIII. Cancer Chemother. Pharmacol. 2019, 84, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Mut, M.; Sherman, J.H.; Shaffrey, M.E.; Schiff, D. Cintredekin besudotox in treatment of malignant glioma. Expert. Opin. Biol. Ther. 2008, 8, 805–812. [Google Scholar] [CrossRef]
- Weber, F.W.; Floeth, F.; Asher, A.; Bucholz, R.; Berge, M.; Pradoss, M.; Chang, S.; Bruces, J.; Hall, W.; Raino, N.G.; et al. Local convection enhanced delivery of IL4-Pseudomonas exotoxin (NBI-3001) for treatment of patients with recurrent malignant glioma. In Therapies for Glioma Present Status and Future Developments; Springer: Vienna, Austria, 2003; Volume 88, pp. 93–103. [Google Scholar] [CrossRef]
- Sampson, J.H.; Reardon, D.A.; Friedman, A.H.; Friedman, H.S.; Coleman, R.E.; McLendon, R.E.; Pastan, I.; Bigner, D.D. Sustained radiographic and clinical response in patient with bifrontal recurrent glioblastoma multiforme with intracerebral infusion of the recombinant targeted toxin TP-38: Case study. Neuro-Oncology 2005, 7, 90–96. [Google Scholar] [CrossRef]
- Weaver, M.; Laske, D.W. Transferrin receptor ligand-targeted toxin conjugate (Tf-CRM107) for therapy of malignant gliomas. J. Neurooncol. 2003, 65, 3–13. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Juweid, M.; Dunn, R.M.; Sharkey, R.M. Cancer imaging with radiolabeled antibodies: New advances with technetium-99m-labeled monoclonal antibody Fab’ fragments, especially CEA-Scan and prospects for therapy. J. Nucl. Med. Technol. 1997, 25, 18–23. [Google Scholar]
- Lee, Y.; Bullard, D.E.; Humphrey, P.A.; Colapinto, E.V.; Friedman, H.S.; Zalutsky, M.R.; Coleman, R.E.; Bigner, D.D. Treatment of Intracranial Human Glioma Xenografts with 131I-labeled Anti-Tenascin Monoclonal Antibody 81C61. Cancer Res. 1988, 48, 2904–2910. [Google Scholar] [PubMed]
- Leshem, Y.; Pastan, I. Pseudomonas Exotoxin Immunotoxins and Anti-Tumor Immunity: From Observations at the Patient’s Bedside to Evaluation in Preclinical Models. Toxins 2019, 11, 20. [Google Scholar] [CrossRef]
- Mair, M.J.; Bartsch, R.; Le Rhun, E.; Berghoff, A.S.; Brastianos, P.K.; Cortes, J.; Gan, H.K.; Lin, N.U.; Lassman, A.B.; Wen, P.Y.; et al. Understanding the activity of antibody–drug conjugates in primary and secondary brain tumours. Nat. Rev. Clin. Oncol. 2023, 20, 372–389. [Google Scholar] [CrossRef] [PubMed]
- Parakh, S.; Nicolazzo, J.; Scott, A.M.; Gan, H.K. Antibody Drug Conjugates in Glioblastoma—Is There a Future for Them? Front. Oncol. 2021, 11, 718590. [Google Scholar] [CrossRef] [PubMed]
- Lassman, A.B.; Pugh, S.L.; Wang, T.J.C.; Aldape, K.; Gan, H.K.; Preusser, M.; A Vogelbaum, M.; Sulman, E.P.; Won, M.; Zhang, P.; et al. Depatuxizumab mafodotin in EGFR-amplified newly diagnosed glioblastoma: A phase III randomized clinical trial. Neuro-Oncol. 2022, 25, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Bargou, R.; Leo, E.; Zugmaier, G.; Klinger, M.; Goebeler, M.; Knop, S.; Noppeney, R.; Viardot, A.; Hess, G.; Schuler, M.; et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008, 321, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; Kuan, C.-T.; Cai, M.; Archer, G.E.; Mitchell, D.A.; Gedeon, P.C.; Sanchez-Perez, L.; Pastan, I.; Bigner, D.D.; Sampson, J.H. Systemic administration of a bispecific antibody targeting EGFRvIII successfully treats intracerebral glioma. Proc. Natl. Acad. Sci. USA 2013, 110, 270–275. [Google Scholar] [CrossRef]
- Choi, B.D.; Gedeon, P.C.; Ii, J.E.H.; Archer, G.E.; Reap, E.A.; Sanchez-Perez, L.; Mitchell, D.A.; Bigner, D.D.; Sampson, J.H. Human regulatory T cells kill tumor cells through granzyme-dependent cytotoxicity upon retargeting with a bispecific antibody. Cancer Immunol. Res. 2013, 1, 163. [Google Scholar] [CrossRef]
- Choi, B.D.; Pastan, I.; Bigner, D.D.; Sampson, J.H. A novel bispecific antibody recruits T cells to eradicate tumors in the “immunologically privileged” central nervous system. OncoImmunology 2013, 2, e23639. [Google Scholar] [CrossRef]
- Gedeon, P.C.; Suryadevara, C.M.; Choi, B.D.; Sampson, J.H. The effect of adoptive transfer of ex vivo activated T cells on the efficacy and tumor penetrance of intravenously-administered CD3-engaging bispecific antibody. J. Clin. Oncol. 2019, 37, 30. [Google Scholar] [CrossRef]
- Chacko, A.M.; Li, C.; Pryma, D.A.; Brem, S.; Coukos, G.; Muzykantov, V. Targeted delivery of antibody-based therapeutic and imaging agents to CNS tumors: Crossing the blood-brain barrier divide. Expert. Opin. Drug Deliv. 2013, 10, 907–926. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Zhang, N.; An, Z. Engineering antibody and protein therapeutics to cross the blood-brain barrier. Antib. Ther. 2022, 5, 311–331. [Google Scholar] [CrossRef] [PubMed]
- Karmur, B.S.; Philteos, J.; Abbasian, A.; Zacharia, B.E.; Lipsman, N.; Levin, V.; Grossman, S.; Mansouri, A. Blood-Brain Barrier Disruption in Neuro-Oncology: Strategies, Failures, and Challenges to Overcome. Front. Oncol. 2020, 10, 563840. [Google Scholar] [CrossRef] [PubMed]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Gardner, T.J.; Bourne, C.M.; Dacek, M.M.; Kurtz, K.; Malviya, M.; Peraro, L.; Silberman, P.C.; Vogt, K.C.; Unti, M.J.; Brentjens, R.; et al. Targeted Cellular Micropharmacies: Cells Engineered for Localized Drug Delivery. Cancers 2020, 12, 2175. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.N.; Lin, H.K.; Hossian, A.K.M.N.; Rafiq, S. Using Adoptive Cellular Therapy for Localized Protein Secretion. Cancer J. 2021, 27, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; Yu, X.; Castano, A.P.; Bouffard, A.A.; Schmidts, A.; Larson, R.C.; Bailey, S.R.; Boroughs, A.C.; Frigault, M.J.; Leick, M.B.; et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019, 37, 1049–1058. [Google Scholar] [CrossRef]


{kind=link}
| Author | Year | Phase | Tumor Type | Immunotherapy Agent | Combination Treatment | Number of Patients | Progression-Free Survival | Overall Survival |
|---|---|---|---|---|---|---|---|---|
| Brastianos PK, et al. [46] | 2022 | Phase II | recurrent high-grade meningioma | Pembrolizumab | None | 25 | 7.6 months (90% CI, 3.4–12.9) | 20.2 months (90% CI, 14.8–25.8) |
| Nassiri F, et al. [49] | 2023 | Phase I/II | recurrent glioblastoma | Pembrolizumab | Oncolytic Virotherapy | 49 | Not reported | 12.5 months (10.7–13.5) |
| Reardon DA, et al. [36] | 2021 | Phase I | recurrent glioblastoma | Pembrolizumab | None | 111 | 2.8 months (95% Cl, 1.8–8.1) | 13.1 months (95% CI 8.0–26.6) |
| Reardon DA, et al. [37] | 2020 | Phase III | recurrent glioblastoma | Nivolumab | Ipilimumab | 369 | 1.5 months (95% CI, 1.5–1.6) | 9.8 months (95% CI, 8.2–11.8) |
| Duerinck J, et al. [38] | 2021 | Phase I | recurrent glioblastoma | Ipilimumab | Nivolumab | 27 | 2.9 months (95% CI, 2.5–3) | 9.5 months (95% CI: 6.8–12.3) |
| Cloughesy TF, et al. [39] | 2019 | Phase II | recurrent glioblastoma | Pembrolizumab | None | 35 | 3.3 months | 13.7 months |
| Nayak L, et al. [40] | 2021 | Phase II | recurrent glioblastoma | Pembrolizumab | Bevacizumab | 80 | 4.1 months (95% CI, 2.8–8.6) | 8.8 months (95% CI, 7.7–14.2) |
| Sahebjam S, et al. [41] | 2020 | Phase I | recurrent high-grade glioma | Pembrolizumab | Hypofractionated Stereotactic Irradiation | 32 | 7.9 months (95% CI, 6.3–12.5) | 13.34 months (95% CI, 9.5–18.5) |
| Bi WL, et al. [47] | 2022 | Phase II | recurrent high-grade meningioma | Nivolumab | None | 25 | 5.6 months (95% CI, 3.2–7.4) | 30.9 months (95% CI, 17.6, NA) |
| Omuro A, et al. [42] | 2023 | Phase III | newly diagnosed glioblastoma | Nivolumab | Radiation Therapy | 560 | 6.0 months (95% CI, 5.7–6.2) | 13.4 (95% CI, 1.1–1.6) |
| Lim M, et al. [43] | 2022 | Phase III | newly diagnosed glioblastoma | Nivolumab | Temozolomide Plus Radiation Therapy | 716 | 10.6 months (95% CI, 8.9–11.8) | 28.9 months (95% CI, 24.4–31.6) |
| Omuro A, et al. [44] | 2018 | Phase I | recurrent glioblastoma | Nivolumab | Ipilimumab | 40 | 1.5 months (95% CI, 0.5–2.8) | 9.2 months (95% CI, 3.9–12.7) |
| Schalper KA, et al. [45] | 2019 | Phase II | recurrent glioblastoma | Nivolumab | None | 30 | 4.1 months (95% CI, 2.8–5.5) | 7.3 months (95% CI, 5.4–7.9) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramapriyan, R.; Sun, J.; Curry, A.; Richardson, L.G.; Ramesh, T.; Gaffey, M.A.; Gedeon, P.C.; Gerstner, E.R.; Curry, W.T.; Choi, B.D. The Role of Antibody-Based Therapies in Neuro-Oncology. Antibodies 2023, 12, 74. https://doi.org/10.3390/antib12040074
Ramapriyan R, Sun J, Curry A, Richardson LG, Ramesh T, Gaffey MA, Gedeon PC, Gerstner ER, Curry WT, Choi BD. The Role of Antibody-Based Therapies in Neuro-Oncology. Antibodies. 2023; 12(4):74. https://doi.org/10.3390/antib12040074
Chicago/Turabian StyleRamapriyan, Rishab, Jing Sun, Annabel Curry, Leland G. Richardson, Tarun Ramesh, Matthew A. Gaffey, Patrick C. Gedeon, Elizabeth R. Gerstner, William T. Curry, and Bryan D. Choi. 2023. "The Role of Antibody-Based Therapies in Neuro-Oncology" Antibodies 12, no. 4: 74. https://doi.org/10.3390/antib12040074
APA StyleRamapriyan, R., Sun, J., Curry, A., Richardson, L. G., Ramesh, T., Gaffey, M. A., Gedeon, P. C., Gerstner, E. R., Curry, W. T., & Choi, B. D. (2023). The Role of Antibody-Based Therapies in Neuro-Oncology. Antibodies, 12(4), 74. https://doi.org/10.3390/antib12040074