Combined Anti-Cancer Strategies Based on Anti-Checkpoint Inhibitor Antibodies



Abstract
:1. Introduction
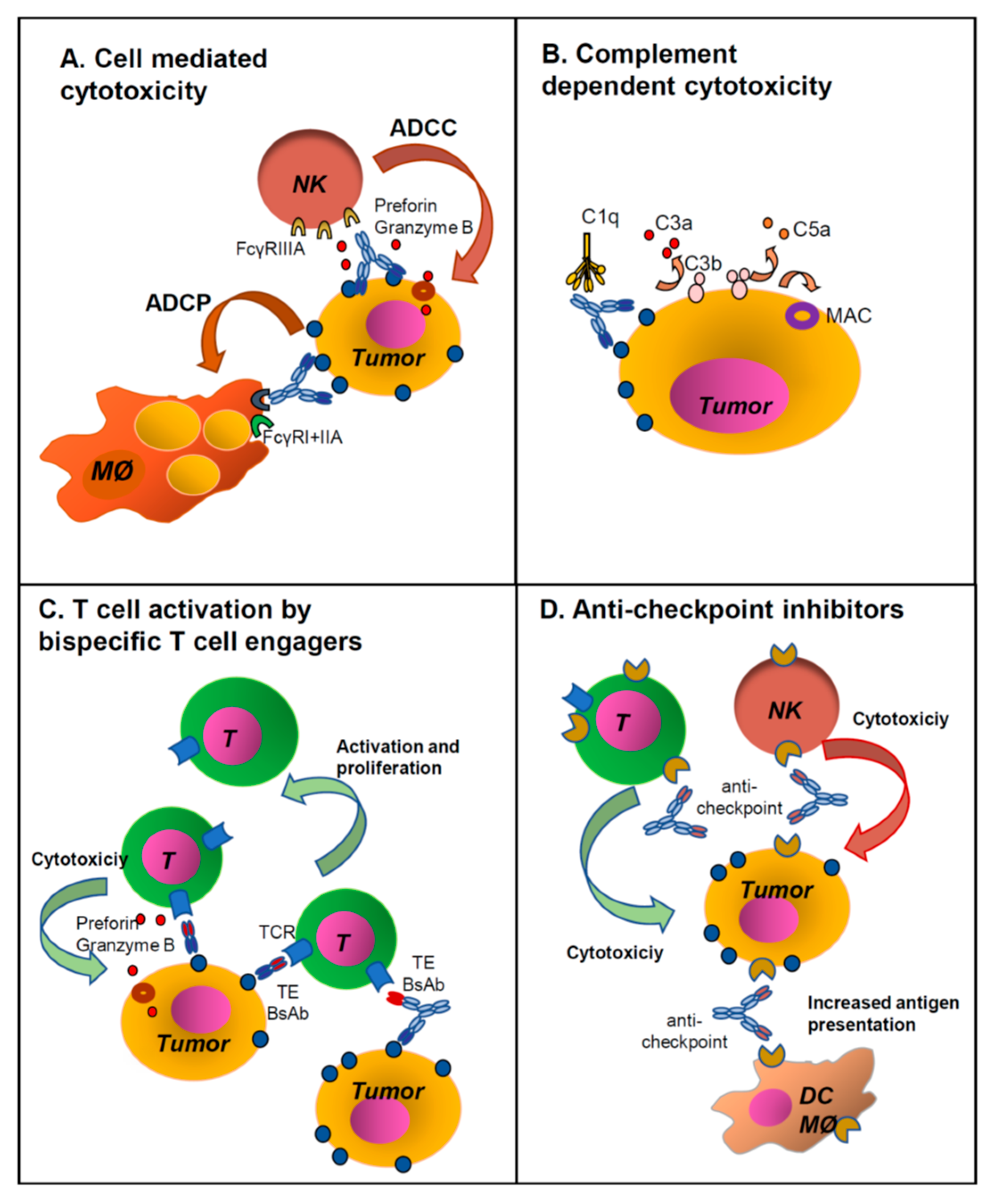
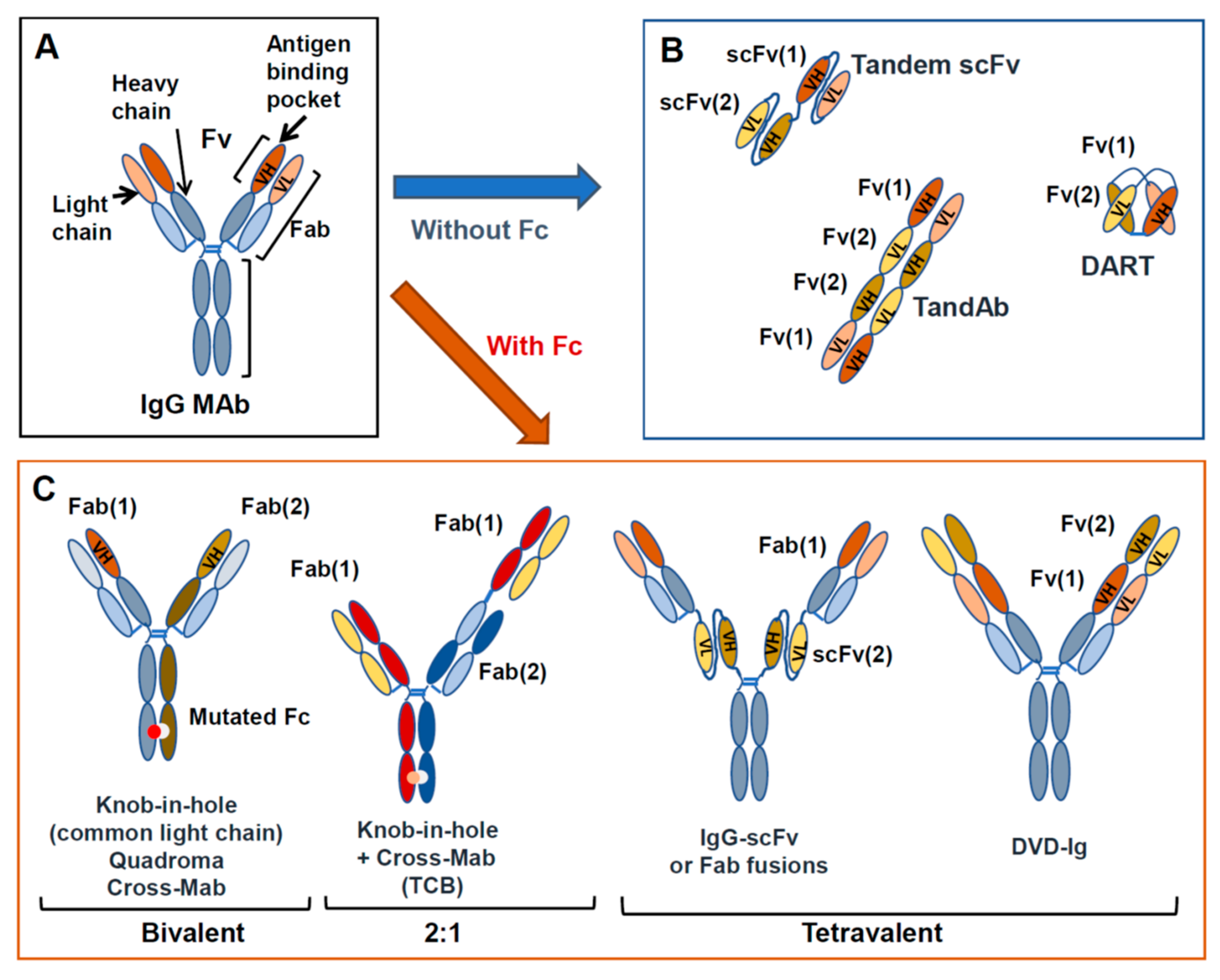
2. Unconjugated MAb Structure and Function
3. Other Antibody Formats
4. Immune Checkpoint Inhibitors
5. Brief Overview of Clinical Data with Anti-Checkpoint Inhibitors as Monotherapy
5.1. CTLA-4
5.2. PD-1
5.3. PD-L1
6. The Possible Role of Antibody Isotypes in the Efficacy of ICI Antibodies
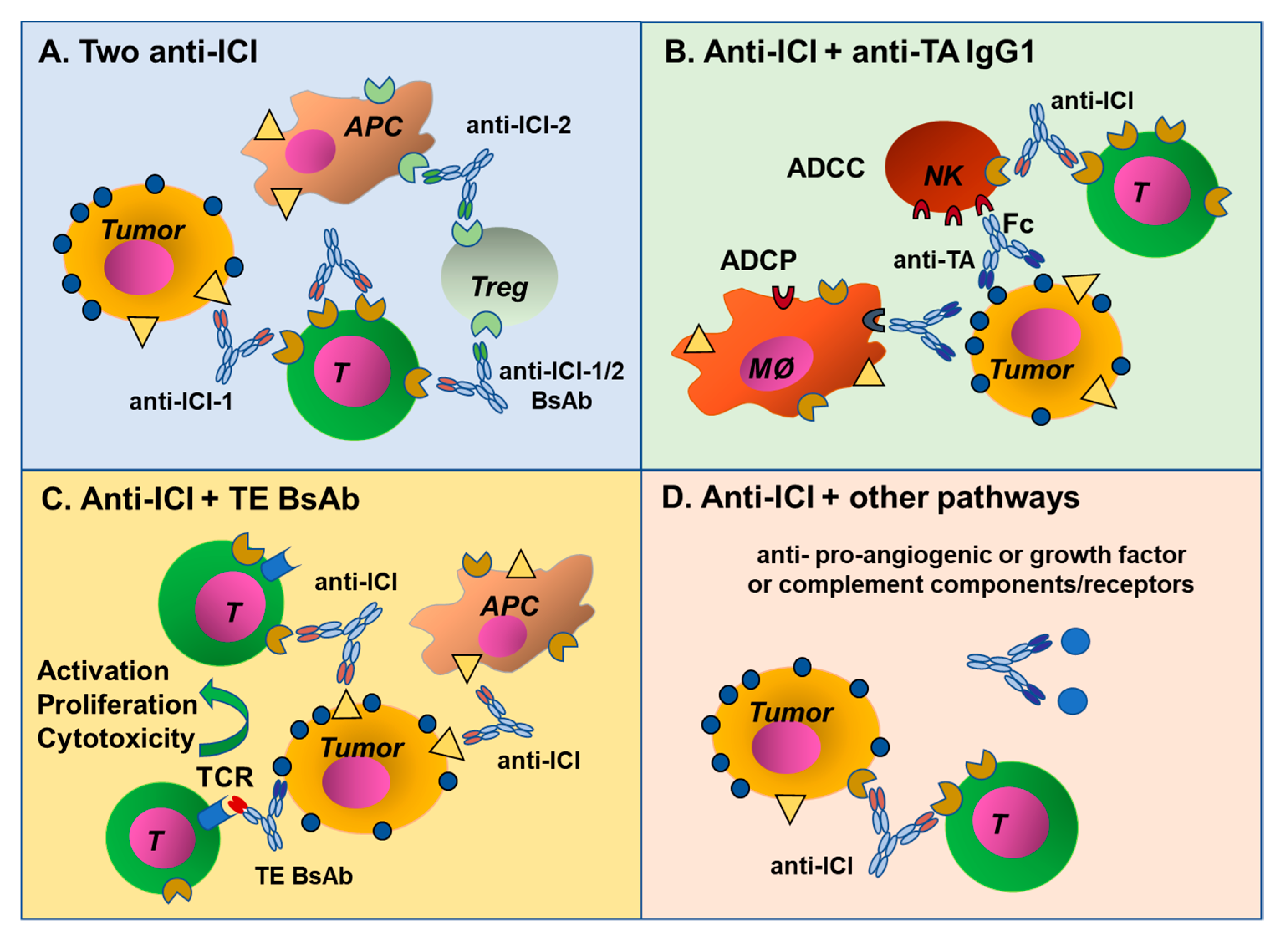
7. Combination of ICI Antibodies and Other MAbs or BsAbs
7.1. Rationale for Combining Anti-CTLA-4 with Anti-PD-1/PD-L1
7.2. Clinical Results Obtained with Anti-CTLA-4 and Anti-PD-1/PD-L1 Combinations
7.3. Combinations Using Novel ICI Antibodies
7.4. Combining ICI and Immune Stimulating Antibodies
7.5. Combination of an ICI Antibody with a Standard Anti-Tumor MAb
7.6. Combination of an ICI Antibody with a T Cell Engager Antibody
7.7. Combination of Antibodies Targeting ICI and ReceptorActivator of Nuclear Factor kB Ligand (RANKL) Antibodies
7.8. Combination of ICI and Anti-Angiogenic Antibodies
7.9. Combination with Antibodies Targeting the Complement System
7.10. Combining Different Specificities Using Bispecific or Two Monospecific Antibodies?
8. Conclusions and Future Prospects
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Golay, J. Direct targeting of cancer cells with antibodies: What can we learn from the successes and failure of unconjugated antibodies for lymphoid neoplasias? J. Autoimmun. 2017, 85, 6–19. [Google Scholar] [CrossRef] [PubMed]
- Weiner, G.J. Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 2015, 15, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, R.P.; Lindorfer, M.A. Cytotoxic mechanisms of immunotherapy: Harnessing complement in the action of anti-tumor monoclonal antibodies. Semin. Immunol. 2016, 28, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Sedykh, S.E.; Prinz, V.V.; Buneva, V.N.; Nevinsky, G.A. Bispecific antibodies: Design, therapy, perspectives. Drug Des. Devel. Ther. 2018, 12, 195–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansell, S.M.; Vonderheide, R.H. Cellular composition of the tumor microenvironment. Am. Soc. Clin. Oncol. Educ. Book 2013, 33, e91–e97. [Google Scholar] [CrossRef]
- Guerrouahen, B.S.; Maccalli, C.; Cugno, C.; Rutella, S.; Akporiaye, E.T. Reverting Immune Suppression to Enhance Cancer Immunotherapy. Front. Oncol. 2020, 9. [Google Scholar] [CrossRef] [Green Version]
- Sanmamed, M.F.; Pastor, F.; Rodriguez, A.; Perez-Gracia, J.L.; Rodriguez-Ruiz, M.E.; Jure-Kunkel, M.; Melero, I. Agonists of Co-stimulation in Cancer Immunotherapy Directed Against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin. Oncol. 2015, 42, 640–655. [Google Scholar] [CrossRef]
- Waldmann, T.A. Cytokines in Cancer Immunotherapy. Cold Spring Harb. Perspect. Biol. 2018, 10, a028472. [Google Scholar] [CrossRef]
- Neri, D. Antibody-Cytokine Fusions: Versatile Products for the Modulation of Anticancer Immunity. Cancer Immunol. Res. 2019, 7, 348–354. [Google Scholar] [CrossRef]
- Webb, E.S.; Liu, P.; Baleeiro, R.; Lemoine, N.R.; Yuan, M.; Wang, Y. Immune checkpoint inhibitors in cancer therapy. J. Biomed. Res. 2018, 32, 317–326. [Google Scholar]
- Li, X.; Shao, C.; Shi, Y.; Han, W. Lessons learned from the blockade of immune checkpoints in cancer immunotherapy. J. Hematol. Oncol. 2018, 11, 31. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.B.; Salama, A.K.S. A review of cancer immunotherapy toxicity. CA Cancer J. Clin. 2020, 70, 86–104. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Johnson, D.B. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 306. [Google Scholar] [CrossRef] [PubMed]
- McGonagle, D.; Bragazzi, N.L.; Amital, H.; Watad, A. Mechanistic classification of immune checkpoint inhibitor toxicity as a pointer to minimal treatment strategies to further improve survival. Autoimmun. Rev. 2020, 19, 102456. [Google Scholar] [CrossRef]
- Saunders, K.O. Conceptual Approaches to Modulating Antibody Effector Functions and Circulation Half-Life. Front. Immunol. 2019, 10, 1296. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.H.; Jung, S.T. Boosting therapeutic potency of antibodies by taming Fc domain functions. Exp. Mol. Med. 2019, 51, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salfeld, J.G. Isotype selection in antibody engineering. Nat. Biotechnol. 2007, 25, 1369–1372. [Google Scholar] [CrossRef]
- De Aguiar, R.B.; de Moraes, J.Z. Exploring the Immunological Mechanisms Underlying the Anti-vascular Endothelial Growth Factor Activity in Tumors. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Bournazos, S.; Wang, T.T.; Dahan, R.; Maamary, J.; Ravetch, J.V. Signaling by Antibodies: Recent Progress. Annu. Rev. Immunol. 2017, 35, 285–311. [Google Scholar] [CrossRef] [Green Version]
- Davies, A.M.; Sutton, B.J. Human IgG4: A structural perspective. Immunol. Rev. 2015, 268, 139–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef]
- Chen, X.; Song, X.; Li, K.; Zhang, T. FcγR-Binding Is an Important Functional Attribute for Immune Checkpoint Antibodies in Cancer Immunotherapy. Front. Immunol. 2019, 10, 292. [Google Scholar] [CrossRef] [PubMed]
- Challa, D.K.; Velmurugan, R.; Ober, R.J.; Sally Ward, E. FcRn: From molecular interactions to regulation of IgG pharmacokinetics and functions. Curr. Top. Microbiol. Immunol. 2014, 382, 249–272. [Google Scholar] [PubMed]
- Stapleton, N.M.; Einarsdóttir, H.K.; Stemerding, A.M.; Vidarsson, G. The multiple facets of FcRn in immunity. Immunol. Rev. 2015, 268, 253–268. [Google Scholar] [CrossRef]
- Spiess, C.; Zhai, Q.; Carter, P.J. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol. Immunol. 2015, 67, 95–106. [Google Scholar] [CrossRef]
- Carter, P.J.; Lazar, G.A. Next generation antibody drugs: Pursuit of the “high-hanging fruit”. Nat. Rev. Drug. Discov. 2018, 17, 197–223. [Google Scholar] [CrossRef]
- Brinkmann, U.; Kontermann, R.E. The making of bispecific antibodies. MAbs 2017, 9, 182–212. [Google Scholar] [CrossRef]
- Goebeler, M.-E.; Bargou, R.C. T cell-engaging therapies—BiTEs and beyond. Nat. Rev. Clin. Oncol. 2020. [Google Scholar] [CrossRef]
- Bargou, R.; Leo, E.; Zugmaier, G.; Klinger, M.; Goebeler, M.; Knop, S.; Noppeney, R.; Viardot, A.; Hess, G.; Schuler, M.; et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 2008, 321, 974–977. [Google Scholar] [CrossRef]
- Dobosz, P.; Dzieciątkowski, T. The Intriguing History of Cancer Immunotherapy. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritz, J.M.; Lenardo, M.J. Development of immune checkpoint therapy for cancer. J. Exp. Med. 2019, 216, 1244–1254. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D. The concept of immune surveillance against tumors: The first theories. Oncotarget 2016, 8, 7175–7180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P.; Ohlsson, R.; Henikoff, S. The epigenetic progenitor origin of human cancer. Nat. Rev. Genet. 2006, 7, 21–33. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef]
- Reeves, E.; James, E. Antigen processing and immune regulation in the response to tumours. Immunology 2017, 150, 16–24. [Google Scholar] [CrossRef] [Green Version]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Driessens, G.; Kline, J.; Gajewski, T.F. Costimulatory and coinhibitory receptors in anti-tumor immunity. Immunol. Rev. 2009, 229, 126–144. [Google Scholar] [CrossRef]
- Bonavida, B.; Chouaib, S. Resistance to anticancer immunity in cancer patients: Potential strategies to reverse resistance. Ann. Oncol. 2017, 28, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korman, A.J.; Peggs, K.S.; Allison, J.P. Checkpoint Blockade in Cancer Immunotherapy. Adv. Immunol. 2006, 90, 297–339. [Google Scholar]
- Walunas, T.L.; Lenschow, D.J.; Bakker, C.Y.; Linsley, P.S.; Freeman, G.J.; Green, J.M.; Thompson, C.B.; Bluestone, J.A. CTLA-4 can function as a negative regulator of T cell activation. Immunity 1994, 1, 405–413. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H.; Lao, C.D.; et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Saenger, Y. The mechanism of anti-CTLA-4 activity and the negative regulation of T-cell activation. Oncologist 2008, 13 (Suppl. 4), 2–9. [Google Scholar] [CrossRef] [Green Version]
- Wing, J.B.; Tanaka, A.; Sakaguchi, S. Human FOXP3+ Regulatory T Cell Heterogeneity and Function in Autoimmunity and Cancer. Immunity 2019, 50, 302–316. [Google Scholar] [CrossRef] [Green Version]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Boussiotis, V.A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N. Engl. J. Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes Cancer 2018, 9, 176–189. [Google Scholar] [PubMed] [Green Version]
- Sedy, J.R.; Gavrieli, M.; Potter, K.G.; Hurchla, M.A.; Lindsley, R.C.; Hildner, K.; Scheu, S.; Pfeffer, K.; Ware, C.F.; Murphy, T.L.; et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat. Immunol. 2005, 6, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef] [Green Version]
- Friedlaender, A.; Addeo, A.; Banna, G. New emerging targets in cancer immunotherapy: The role of TIM3. ESMO Open 2019, 4 (Suppl. 3), e000497. [Google Scholar] [CrossRef] [Green Version]
- Lines, J.L.; Sempere, L.F.; Wang, L.; Pantazi, E.; Mak, J.; O’Connell, S.; Ceeraz, S.; Suriawinata, A.A.; Yan, S.; Ernstoff, M.S.; et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014, 74, 1924–1932. [Google Scholar] [CrossRef] [Green Version]
- Solomon, B.L.; Garrido-Laguna, I. TIGIT: A novel immunotherapy target moving from bench to bedside. Cancer Immunol. Immunother. 2018, 67, 1659–1667. [Google Scholar] [CrossRef]
- Manieri, N.A.; Chiang, E.Y.; Grogan, J.L. TIGIT: A Key Inhibitor of the Cancer Immunity Cycle. Trends Immunol. 2017, 38, 20–28. [Google Scholar] [CrossRef]
- Chao, M.P.; Weissman, I.L.; Majeti, R. The CD47-SIRPα pathway in cancer immune evasion and potential therapeutic implications. Curr. Opin. Immunol. 2012, 24, 225–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, K.E. Revisiting CD28 Superagonist TGN1412 as Potential Therapeutic for Pediatric B Cell Leukemia: A Review. Diseases 2018, 6, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amatore, F.; Gorvel, L.; Olive, D. Inducible Co-Stimulator (ICOS) as a potential therapeutic target for anti-cancer therapy. Expert Opin. Ther. Targets 2018, 22, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Knee, D.A.; Hewes, B.; Brogdon, J.L. Rationale for anti-GITR cancer immunotherapy. Eur. J. Cancer 2016, 67, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Soto, A.; Huergo-Zapico, L.; Acebes-Huerta, A.; Villa-Alvarez, M.; Gonzalez, S. NKG2D signaling in cancer immunosurveillance. Int. J. Cancer 2015, 136, 1741–1750. [Google Scholar] [CrossRef]
- Aspeslagh, S.; Postel-Vinay, S.; Rusakiewicz, S.; Soria, J.-C.; Zitvogel, L.; Marabelle, A. Rationale for anti-OX40 cancer immunotherapy. Eur. J. Cancer 2016, 52, 50–66. [Google Scholar] [CrossRef]
- Chester, C.; Sanmamed, M.F.; Wang, J.; Melero, I. Immunotherapy targeting 4-1BB: Mechanistic rationale, clinical results, and future strategies. Blood 2018, 131, 49–57. [Google Scholar] [CrossRef]
- Ribas, A.; Kefford, R.; Marshall, M.A.; Punt, C.J.A.; Haanen, J.B.; Marmol, M.; Garbe, C.; Gogas, H.; Schachter, J.; Linette, G.; et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J. Clin. Oncol. 2013, 31, 616–622. [Google Scholar] [CrossRef] [Green Version]
- Maio, M.; Scherpereel, A.; Calabrò, L.; Aerts, J.; Cedres Perez, S.; Bearz, A.; Nackaerts, K.; Fennell, D.A.; Kowalski, D.; Tsao, A.S.; et al. Tremelimumab as second-line or third-line treatment in relapsed malignant mesothelioma (DETERMINE): A multicentre, international, randomised, double-blind, placebo-controlled phase 2b trial. Lancet Oncol. 2017, 18, 1261–1273. [Google Scholar] [CrossRef]
- Maio, M.; Grob, J.-J.; Aamdal, S.; Bondarenko, I.; Robert, C.; Thomas, L.; Garbe, C.; Chiarion-Sileni, V.; Testori, A.; Chen, T.-T.; et al. Five-year survival rates for treatment-naive patients with advanced melanoma who received ipilimumab plus dacarbazine in a phase III trial. J. Clin. Oncol. 2015, 33, 1191–1196. [Google Scholar] [CrossRef]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.-F.; Testori, A.; Grob, J.-J.; et al. Ipilimumab plus Dacarbazine for Previously Untreated Metastatic Melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geoerger, B.; Bergeron, C.; Gore, L.; Sender, L.; Dunkel, I.J.; Herzog, C.; Brochez, L.; Cruz, O.; Nysom, K.; Berghorn, E.; et al. Phase II study of ipilimumab in adolescents with unresectable stage III or IV malignant melanoma. Eur. J. Cancer 2017, 86, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Eggermont, A.M.M.; Chiarion-Sileni, V.; Grob, J.-J.; Dummer, R.; Wolchok, J.D.; Schmidt, H.; Hamid, O.; Robert, C.; Ascierto, P.A.; Richards, J.M.; et al. Prolonged Survival in Stage III Melanoma with Ipilimumab Adjuvant Therapy. N. Engl. J. Med. 2016, 375, 1845–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascierto, P.A.; Del Vecchio, M.; Robert, C.; Mackiewicz, A.; Chiarion-Sileni, V.; Arance, A.; Lebbé, C.; Bastholt, L.; Hamid, O.; Rutkowski, P.; et al. Ipilimumab 10 mg/kg versus ipilimumab 3 mg/kg in patients with unresectable or metastatic melanoma: A randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2017, 18, 611–622. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Cho, J.Y.; Kim, Y.H.; Kim, J.W.; Di Bartolomeo, M.; Ajani, J.A.; Yamaguchi, K.; Balogh, A.; Sanchez, T.; Moehler, M. Efficacy of Sequential Ipilimumab Monotherapy versus Best Supportive Care for Unresectable Locally Advanced/Metastatic Gastric or Gastroesophageal Junction Cancer. Clin. Cancer Res. 2017, 23, 5671–5678. [Google Scholar] [CrossRef] [Green Version]
- Grywalska, E.; Sobstyl, M.; Putowski, L.; Roliński, J. Current Possibilities of Gynecologic Cancer Treatment with the Use of Immune Checkpoint Inhibitors. Int. J. Mol. Sci. 2019, 20, 4705. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Minor, D.; D’Angelo, S.; Neyns, B.; Smylie, M.; Miller, W.H.; Gutzmer, R.; Linette, G.; Chmielowski, B.; Lao, C.D.; et al. Overall Survival in Patients With Advanced Melanoma Who Received Nivolumab Versus Investigator’s Choice Chemotherapy in CheckMate 037: A Randomized, Controlled, Open-Label Phase III Trial. J. Clin. Oncol. 2018, 36, 383–390. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Long, G.V.; Robert, C.; Brady, B.; Dutriaux, C.; Di Giacomo, A.M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; et al. Survival Outcomes in Patients With Previously Untreated BRAF Wild-Type Advanced Melanoma Treated With Nivolumab Therapy. JAMA Oncol. 2019, 5, 187–194. [Google Scholar] [CrossRef] [Green Version]
- Herrera, A.F.; Goy, A.; Mehta, A.; Ramchandren, R.; Pagel, J.M.; Svoboda, J.; Guan, S.; Hill, J.S.; Kwei, K.; Liu, E.A.; et al. Safety and activity of ibrutinib in combination with durvalumab in patients with relapsed or refractory follicular lymphoma or diffuse large B-cell lymphoma. Am. J. Hematol. 2020, 95, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Ansell, S.M.; Minnema, M.C.; Johnson, P.; Timmerman, J.M.; Armand, P.; Shipp, M.A.; Rodig, S.J.; Ligon, A.H.; Roemer, M.G.M.; Reddy, N.; et al. Nivolumab for Relapsed/Refractory Diffuse Large B-Cell Lymphoma in Patients Ineligible for or Having Failed Autologous Transplantation: A Single-Arm, Phase II Study. J. Clin. Oncol. 2019, 37, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Santoro, A.; Shipp, M.; Zinzani, P.L.; Timmerman, J.M.; Ansell, S.; Armand, P.; Fanale, M.; Ratanatharathorn, V.; Kuruvilla, J.; et al. Nivolumab for classical Hodgkin’s lymphoma after failure of both autologous stem-cell transplantation and brentuximab vedotin: A multicentre, multicohort, single-arm phase 2 trial. Lancet Oncol. 2016, 17, 1283–1294. [Google Scholar] [CrossRef] [Green Version]
- Armand, P.; Engert, A.; Younes, A.; Fanale, M.; Santoro, A.; Zinzani, P.L.; Timmerman, J.M.; Collins, G.P.; Ramchandren, R.; Cohen, J.B.; et al. Nivolumab for Relapsed/Refractory Classic Hodgkin Lymphoma After Failure of Autologous Hematopoietic Cell Transplantation: Extended Follow-Up of the Multicohort Single-Arm Phase II CheckMate 205 Trial. J. Clin. Oncol. 2018, 36, 1428–1439. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Retz, M.; Siefker-Radtke, A.; Baron, A.; Necchi, A.; Bedke, J.; Plimack, E.R.; Vaena, D.; Grimm, M.-O.; Bracarda, S.; et al. Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2017, 18, 312–322. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef]
- Ma, B.B.Y.; Lim, W.-T.; Goh, B.-C.; Hui, E.P.; Lo, K.-W.; Pettinger, A.; Foster, N.R.; Riess, J.W.; Agulnik, M.; Chang, A.Y.C.; et al. Antitumor Activity of Nivolumab in Recurrent and Metastatic Nasopharyngeal Carcinoma: An International, Multicenter Study of the Mayo Clinic Phase 2 Consortium (NCI-9742). J. Clin. Oncol. 2018, 36, 1412–1418. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Mazières, J.; Planchard, D.; Stinchcombe, T.E.; Dy, G.K.; Antonia, S.J.; Horn, L.; Lena, H.; Minenza, E.; Mennecier, B.; et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): A phase 2, single-arm trial. Lancet Oncol. 2015, 16, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-L.; Lu, S.; Cheng, Y.; Zhou, C.; Wang, J.; Mok, T.; Zhang, L.; Tu, H.-Y.; Wu, L.; Feng, J.; et al. Nivolumab Versus Docetaxel in a Predominantly Chinese Patient Population With Previously Treated Advanced NSCLC: CheckMate 078 Randomized Phase III Clinical Trial. J. Thorac. Oncol. 2019, 14, 867–875. [Google Scholar] [CrossRef]
- Vokes, E.E.; Ready, N.; Felip, E.; Horn, L.; Burgio, M.A.; Antonia, S.J.; Arén Frontera, O.; Gettinger, S.; Holgado, E.; Spigel, D.; et al. Nivolumab versus docetaxel in previously treated advanced non-small-cell lung cancer (CheckMate 017 and CheckMate 057): 3-year update and outcomes in patients with liver metastases. Ann. Oncol. 2018, 29, 959–965. [Google Scholar] [CrossRef]
- Reck, M.; Brahmer, J.; Bennett, B.; Taylor, F.; Penrod, J.R.; DeRosa, M.; Dastani, H.; Spigel, D.R.; Gralla, R.J. Evaluation of health-related quality of life and symptoms in patients with advanced non-squamous non-small cell lung cancer treated with nivolumab or docetaxel in CheckMate 057. Eur. J. Cancer 2018, 102, 23–30. [Google Scholar] [CrossRef]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; van den Heuvel, M.M.; Ciuleanu, T.-E.; Badin, F.; et al. First-Line Nivolumab in Stage IV or Recurrent Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Sharma, P.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. CheckMate 025 Randomized Phase 3 Study: Outcomes by Key Baseline Factors and Prior Therapy for Nivolumab Versus Everolimus in Advanced Renal Cell Carcinoma. Eur. Urol. 2017, 72, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Redman, B.G.; Kuzel, T.M.; Harrison, M.R.; Vaishampayan, U.N.; Drabkin, H.A.; George, S.; Logan, T.F.; et al. Nivolumab for Metastatic Renal Cell Carcinoma: Results of a Randomized Phase II Trial. J. Clin. Oncol. 2015, 33, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Roemer, M.G.M.; Redd, R.A.; Cader, F.Z.; Pak, C.J.; Abdelrahman, S.; Ouyang, J.; Sasse, S.; Younes, A.; Fanale, M.; Santoro, A.; et al. Major Histocompatibility Complex Class II and Programmed Death Ligand 1 Expression Predict Outcome After Programmed Death 1 Blockade in Classic Hodgkin Lymphoma. J. Clin. Oncol. 2018, 36, 942–950. [Google Scholar] [CrossRef]
- Ratner, L.; Waldmann, T.A.; Janakiram, M.; Brammer, J.E. Rapid Progression of Adult T-Cell Leukemia-Lymphoma after PD-1 Inhibitor Therapy. N. Engl. J. Med. 2018, 378, 1947–1948. [Google Scholar] [CrossRef]
- Rauch, D.A.; Conlon, K.C.; Janakiram, M.; Brammer, J.E.; Harding, J.C.; Ye, B.H.; Zang, X.; Ren, X.; Olson, S.; Cheng, X.; et al. Rapid progression of adult T-cell leukemia/lymphoma as tumor-infiltrating Tregs after PD-1 blockade. Blood 2019, 134, 1406–1414. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S.; et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [CrossRef]
- Loi, S.; Giobbie-Hurder, A.; Gombos, A.; Bachelot, T.; Hui, R.; Curigliano, G.; Campone, M.; Biganzoli, L.; Bonnefoi, H.; Jerusalem, G.; et al. Pembrolizumab plus trastuzumab in trastuzumab-resistant, advanced, HER2-positive breast cancer (PANACEA): A single-arm, multicentre, phase 1b-2 trial. Lancet Oncol. 2019, 20, 371–382. [Google Scholar] [CrossRef]
- Shah, M.A.; Kojima, T.; Hochhauser, D.; Enzinger, P.; Raimbourg, J.; Hollebecque, A.; Lordick, F.; Kim, S.-B.; Tajika, M.; Kim, H.T.; et al. Efficacy and Safety of Pembrolizumab for Heavily Pretreated Patients With Advanced, Metastatic Adenocarcinoma or Squamous Cell Carcinoma of the Esophagus. JAMA Oncol. 2019, 5, 546–550. [Google Scholar] [CrossRef] [Green Version]
- Shitara, K.; Özgüroğlu, M.; Bang, Y.-J.; Di Bartolomeo, M.; Mandalà, M.; Ryu, M.-H.; Fornaro, L.; Olesiński, T.; Caglevic, C.; Chung, H.C.; et al. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): A randomised, open-label, controlled, phase 3 trial. Lancet 2018, 392, 123–133. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Soulières, D.; Le Tourneau, C.; Dinis, J.; Licitra, L.; Ahn, M.-J.; Soria, A.; Machiels, J.-P.; Mach, N.; Mehra, R.; et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): A randomised, open-label, phase 3 study. Lancet 2019, 393, 156–167. [Google Scholar] [CrossRef]
- Herbst, R.S.; Baas, P.; Kim, D.-W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.-Y.; Molina, J.; Kim, J.-H.; Arvis, C.D.; Ahn, M.-J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, T.S.K.; Wu, Y.-L.; Kudaba, I.; Kowalski, D.M.; Cho, B.C.; Turna, H.Z.; Castro, G.; Srimuninnimit, V.; Laktionov, K.K.; Bondarenko, I.; et al. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): A randomised, open-label, controlled, phase 3 trial. Lancet 2019, 393, 1819–1830. [Google Scholar] [CrossRef]
- Balar, A.V.; Castellano, D.; O’Donnell, P.H.; Grivas, P.; Vuky, J.; Powles, T.; Plimack, E.R.; Hahn, N.M.; de Wit, R.; Pang, L.; et al. First-line pembrolizumab in cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer (KEYNOTE-052): A multicentre, single-arm, phase 2 study. Lancet Oncol. 2017, 18, 1483–1492. [Google Scholar] [CrossRef]
- Fradet, Y.; Bellmunt, J.; Vaughn, D.J.; Lee, J.L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; Necchi, A.; et al. Randomized phase III KEYNOTE-045 trial of pembrolizumab versus paclitaxel, docetaxel, or vinflunine in recurrent advanced urothelial cancer: Results of >2 years of follow-up. Ann Oncol. 2019, 30, 970–976. [Google Scholar] [CrossRef]
- Chen, R.; Zinzani, P.L.; Fanale, M.A.; Armand, P.; Johnson, N.A.; Brice, P.; Radford, J.; Ribrag, V.; Molin, D.; Vassilakopoulos, T.P.; et al. Phase II Study of the Efficacy and Safety of Pembrolizumab for Relapsed/Refractory Classic Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 2125–2132. [Google Scholar] [CrossRef]
- Migden, M.R.; Khushalani, N.I.; Chang, A.L.S.; Lewis, K.D.; Schmults, C.D.; Hernandez-Aya, L.; Meier, F.; Schadendorf, D.; Guminski, A.; Hauschild, A.; et al. Cemiplimab in locally advanced cutaneous squamous cell carcinoma: Results from an open-label, phase 2, single-arm trial. Lancet Oncol. 2020, 21, 294–305. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, W.; Xu, Z.P.; Gu, W. PD-L1 Distribution and Perspective for Cancer Immunotherapy—Blockade, Knockdown, or Inhibition. Front. Immunol. 2019, 10, 2022. [Google Scholar] [CrossRef] [Green Version]
- Greillier, L.; Tomasini, P.; Barlesi, F. Necitumumab for non-small cell lung cancer. Expert Opin. Biol. Ther. 2015, 15, 1231–1239. [Google Scholar] [CrossRef]
- Weiner, L.M.; Surana, R.; Wang, S. Antibodies and cancer therapy: Versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 2010, 10, 317–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheuer, W.; Friess, T.; Burtscher, H.; Bossenmaier, B.; Endl, J.; Hasmann, M. Strongly enhanced antitumor activity of trastuzumab and pertuzumab combination treatment on HER2-positive human xenograft tumor models. Cancer Res. 2009, 69, 9330–9336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyerinas, B.; Jochems, C.; Fantini, M.; Heery, C.R.; Gulley, J.L.; Tsang, K.Y.; Schlom, J. Antibody-dependent cellular cytotoxicity (ADCC) activity of a novel anti-PD-L1 antibody avelumab (MSB0010718C) on human tumor cells. Cancer Immunol. Res. 2015, 3, 1148–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jardim, D.L.; de Melo Gagliato, D.; Giles, F.J.; Kurzrock, R. Analysis of Drug Development Paradigms for Immune Checkpoint inhibitors. Clin. Cancer Res. 2018, 24, 1785–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, Y.-J.; Ruiz, E.Y.; Van Cutsem, E.; Lee, K.-W.; Wyrwicz, L.; Schenker, M.; Alsina, M.; Ryu, M.-H.; Chung, H.-C.; Evesque, L.; et al. Phase III, randomised trial of avelumab versus physician’s choice of chemotherapy as third-line treatment of patients with advanced gastric or gastro-oesophageal junction cancer: Primary analysis of JAVELIN Gastric 300. Ann. Oncol. 2018, 29, 2052–2060. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.L.; Russell, J.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbé, C.; Linette, G.P.; Milella, M.; et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: A multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 1374–1385. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, H.L.; Russell, J.S.; Hamid, O.; Bhatia, S.; Terheyden, P.; D’Angelo, S.P.; Shih, K.C.; Lebbé, C.; Milella, M.; Brownell, I.; et al. Updated efficacy of avelumab in patients with previously treated metastatic Merkel cell carcinoma after ≥1 year of follow-up: JAVELIN Merkel 200, a phase 2 clinical trial. J. Immunother. Cancer 2018, 6, 7. [Google Scholar] [CrossRef] [Green Version]
- Barlesi, F.; Vansteenkiste, J.; Spigel, D.; Ishii, H.; Garassino, M.; de Marinis, F.; Özgüroğlu, M.; Szczesna, A.; Polychronis, A.; Uslu, R.; et al. Avelumab versus docetaxel in patients with platinum-treated advanced non-small-cell lung cancer (JAVELIN Lung 200): An open-label, randomised, phase 3 study. Lancet Oncol. 2018, 19, 1468–1479. [Google Scholar] [CrossRef]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef]
- Peters, S.; Gettinger, S.; Johnson, M.L.; Jänne, P.A.; Garassino, M.C.; Christoph, D.; Toh, C.K.; Rizvi, N.A.; Chaft, J.E.; Carcereny Costa, E.; et al. Phase II Trial of Atezolizumab As First-Line or Subsequent Therapy for Patients With Programmed Death-Ligand 1-Selected Advanced Non-Small-Cell Lung Cancer (BIRCH). J. Clin. Oncol. 2017, 35, 2781–2789. [Google Scholar] [CrossRef] [PubMed]
- Spigel, D.R.; Chaft, J.E.; Gettinger, S.; Chao, B.H.; Dirix, L.; Schmid, P.; Chow, L.Q.M.; Hicks, R.J.; Leon, L.; Fredrickson, J.; et al. FIR: Efficacy, Safety, and Biomarker Analysis of a Phase II Open-Label Study of Atezolizumab in PD-L1-Selected Patients With NSCLC. J. Thorac. Oncol. 2018, 13, 1733–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balar, A.V.; Galsky, M.D.; Rosenberg, J.E.; Powles, T.; Petrylak, D.P.; Bellmunt, J.; Loriot, Y.; Necchi, A.; Hoffman-Censits, J.; Perez-Gracia, J.L.; et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: A single-arm, multicentre, phase 2 trial. Lancet 2017, 389, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Powles, T.; Durán, I.; van der Heijden, M.S.; Loriot, Y.; Vogelzang, N.J.; De Giorgi, U.; Oudard, S.; Retz, M.M.; Castellano, D.; Bamias, A.; et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma (IMvigor211): A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2018, 391, 748–757. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef]
- Garassino, M.C.; Cho, B.-C.; Kim, J.-H.; Mazières, J.; Vansteenkiste, J.; Lena, H.; Corral Jaime, J.; Gray, J.E.; Powderly, J.; Chouaid, C.; et al. Durvalumab as third-line or later treatment for advanced non-small-cell lung cancer (ATLANTIC): An open-label, single-arm, phase 2 study. Lancet Oncol. 2018, 19, 521–536. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef]
- Picardo, S.L.; Doi, J.; Hansen, A.R. Structure and Optimization of Checkpoint Inhibitors. Cancers 2019, 12, 38. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Subudhi, S.K.; Blando, J.; Scutti, J.; Vence, L.; Wargo, J.; Allison, J.P.; Ribas, A.; Sharma, P. Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3+ Regulatory T Cells (Tregs) in Human Cancers. Clin. Cancer Res. 2019, 25, 1233–1238. [Google Scholar] [CrossRef] [Green Version]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Nivolumab and Ipilimumab versus Ipilimumab in Untreated Melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodi, F.S.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.F.; McDermott, D.F.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2016, 17, 1558–1568. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Previously Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Frontera, O.A.; Hammers, H.J.; Carducci, M.A.; Salman, P.; Escudier, B.; Beuselinck, B.; Amin, A.; et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: Extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet Oncol. 2019, 20, 1370–1385. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.-J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Bendell, J.; Calvo, E.; Kim, J.W.; Ascierto, P.A.; Sharma, P.; Ott, P.A.; Peltola, K.; Jaeger, D.; Evans, J.; et al. CheckMate-032 Study: Efficacy and Safety of Nivolumab and Nivolumab Plus Ipilimumab in Patients With Metastatic Esophagogastric Cancer. J. Clin. Oncol. 2018, 36, 2836–2844. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Mahoney, M.R.; Van Tine, B.A.; Atkins, J.; Milhem, M.M.; Jahagirdar, B.N.; Antonescu, C.R.; Horvath, E.; Tap, W.D.; Schwartz, G.K.; et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): Two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018, 19, 416–426. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Ciuleanu, T.-E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.-W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef] [PubMed]
- Ready, N.E.; Ott, P.A.; Hellmann, M.D.; Zugazagoitia, J.; Hann, C.L.; de Braud, F.; Antonia, S.J.; Ascierto, P.A.; Moreno, V.; Atmaca, A.; et al. Nivolumab Monotherapy and Nivolumab Plus Ipilimumab in Recurrent Small Cell Lung Cancer: Results From the CheckMate 032 Randomized Cohort. J. Thorac. Oncol. 2020, 15, 426–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonia, S.J.; López-Martin, J.A.; Bendell, J.; Ott, P.A.; Taylor, M.; Eder, J.P.; Jäger, D.; Pietanza, M.C.; Le, D.T.; de Braud, F.; et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): A multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 883–895. [Google Scholar] [CrossRef] [Green Version]
- Scherpereel, A.; Mazieres, J.; Greillier, L.; Lantuejoul, S.; Dô, P.; Bylicki, O.; Monnet, I.; Corre, R.; Audigier-Valette, C.; Locatelli-Sanchez, M.; et al. Nivolumab or nivolumab plus ipilimumab in patients with relapsed malignant pleural mesothelioma (IFCT-1501 MAPS2): A multicentre, open-label, randomised, non-comparative, phase 2 trial. Lancet Oncol. 2019, 20, 239–253. [Google Scholar] [CrossRef]
- Syed, Y.Y. Durvalumab: First global approval. Drugs 2017, 77, 1369–1376. [Google Scholar] [CrossRef] [Green Version]
- Siu, L.L.; Even, C.; Mesía, R.; Remenar, E.; Daste, A.; Delord, J.-P.; Krauss, J.; Saba, N.F.; Nabell, L.; Ready, N.E.; et al. Safety and Efficacy of Durvalumab With or Without Tremelimumab in Patients With PD-L1–Low/Negative Recurrent or Metastatic HNSCC: The Phase 2 CONDOR Randomized Clinical Trial. JAMA Oncol. 2019, 5, 195–203. [Google Scholar] [CrossRef]
- Ferris, R.L.; Haddad, R.; Even, C.; Tahara, M.; Dvorkin, M.; Ciuleanu, T.E.; Clement, P.M.; Mesia, R.; Kutukova, S.; Zholudeva, L.; et al. Durvalumab with or without Tremelimumab in Patients with Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma: EAGLE, a Randomized, Open-Label Phase III Study. Available online: https://www.sciencedirect.com/science/article/pii/S0923753420364346 (accessed on 20 April 2020).
- O’Reilly, E.M.; Oh, D.-Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.A.; Hezel, A.F.; Chang, S.-C.; Vlahovic, G.; et al. A randomized phase 2 study of durvalumab monotherapy and in combination with tremelimumab in patients with metastatic pancreatic ductal adenocarcinoma (mPDAC): ALPS study. JCO 2018, 36, 217. [Google Scholar] [CrossRef]
- Cho, B.C.; Lee, K.H.; Ahn, M.-J.; Geater, S.L.; Ngoc, T.V.; Wang, C.-C.; Cho, E.K.; Lee, J.S.; Sriuranpong, V.; Bui, Q.; et al. 474O—Efficacy and safety of first-line durvalumab (D) ± tremelimumab (T) vs chemotherapy (CT) in Asian patients with metastatic NSCLC: Results from MYSTIC. Ann. Oncol. 2019, 30, ix157–ix158. [Google Scholar] [CrossRef]
- Andrews, L.P.; Yano, H.; Vignali, D.A.A. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: Breakthroughs or backups. Nat. Immunol. 2019, 20, 1425–1434. [Google Scholar] [CrossRef]
- Tundo, G.R.; Sbardella, D.; Lacal, P.M.; Graziani, G.; Marini, S. On the Horizon: Targeting Next-Generation Immune Checkpoints for Cancer Treatment. Chemotherapy 2019, 64, 62–80. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Harjunpää, H.; Guillerey, C. TIGIT as an emerging immune checkpoint. Clin. Exp. Immunol. 2020, 200, 108–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Correa, B.; Valhondo, I.; Hassouneh, F.; Lopez-Sejas, N.; Pera, A.; Bergua, J.M.; Arcos, M.J.; Bañas, H.; Casas-Avilés, I.; Durán, E.; et al. DNAM-1 and the TIGIT/PVRIG/TACTILE Axis: Novel Immune Checkpoints for Natural Killer Cell-Based Cancer Immunotherapy. Cancers 2019, 11, 877. [Google Scholar] [CrossRef] [Green Version]
- Whelan, S.; Ophir, E.; Kotturi, M.F.; Levy, O.; Ganguly, S.; Leung, L.; Vaknin, I.; Kumar, S.; Dassa, L.; Hansen, K.; et al. PVRIG and PVRL2 Are Induced in Cancer and Inhibit CD8+ T-cell Function. Cancer Immunol. Res. 2019, 7, 257–268. [Google Scholar] [CrossRef] [Green Version]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743. [Google Scholar] [CrossRef] [Green Version]
- Mingari, M.C.; Pietra, G.; Moretta, L. Immune Checkpoint Inhibitors: Anti-NKG2A Antibodies on Board. Trends Immunol. 2019, 40, 83–85. [Google Scholar] [CrossRef]
- Leone, R.D.; Emens, L.A. Targeting adenosine for cancer immunotherapy. J. Immunother. Cancer 2018, 6, 57. [Google Scholar] [CrossRef] [Green Version]
- Hammami, A.; Allard, D.; Allard, B.; Stagg, J. Targeting the adenosine pathway for cancer immunotherapy. Semin. Immunol. 2019, 42, 101304. [Google Scholar] [CrossRef]
- Folkes, A.S.; Feng, M.; Zain, J.M.; Abdulla, F.; Rosen, S.T.; Querfeld, C. Targeting CD47 as a cancer therapeutic strategy: The cutaneous T-cell lymphoma experience. Curr. Opin. Oncol. 2018, 30, 332–337. [Google Scholar] [CrossRef]
- Russ, A.; Hua, A.B.; Montfort, W.R.; Rahman, B.; Riaz, I.B.; Khalid, M.U.; Carew, J.S.; Nawrocki, S.T.; Persky, D.; Anwer, F. Blocking “don’t eat me” signal of CD47-SIRPα in hematological malignancies, an in-depth review. Blood Rev. 2018, 32, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Tsao, L.-C.; Crosby, E.J.; Trotter, T.N.; Agarwal, P.; Hwang, B.-J.; Acharya, C.; Shuptrine, C.W.; Wang, T.; Wei, J.; Yang, X.; et al. CD47 blockade augmentation of trastuzumab antitumor efficacy dependent on antibody-dependent cellular phagocytosis. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treffers, L.W.; ten Broeke, T.; Rösner, T.; Jansen, J.H.M.; van Houdt, M.; Kahle, S.; Schornagel, K.; Verkuijlen, P.J.J.H.; Prins, J.M.; Franke, K.; et al. IgA-Mediated Killing of Tumor Cells by Neutrophils Is Enhanced by CD47–SIRPα Checkpoint Inhibition. Cancer Immunol. Res. 2020, 8, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Weinmann, S.C.; Pisetsky, D.S. Mechanisms of immune-related adverse events during the treatment of cancer with immune checkpoint inhibitors. Rheumatology 2019, 58, vii59–vii67. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Vesely, M.D. Stimulating T Cells Against Cancer With Agonist Immunostimulatory Monoclonal Antibodies. Int. Rev. Cell. Mol. Biol. 2019, 342, 1–25. [Google Scholar]
- Chu, D.-T.; Bac, N.D.; Nguyen, K.-H.; Tien, N.L.B.; Thanh, V.V.; Nga, V.T.; Ngoc, V.T.N.; Anh Dao, D.T.; Hoan, L.N.; Hung, N.P.; et al. An Update on Anti-CD137 Antibodies in Immunotherapies for Cancer. Int. J. Mol. Sci. 2019, 20, 1822. [Google Scholar] [CrossRef] [Green Version]
- Polesso, F.; Sarker, M.; Weinberg, A.D.; Murray, S.E.; Moran, A.E. OX40 Agonist Tumor Immunotherapy Does Not Impact Regulatory T Cell Suppressive Function. J. Immunol. 2019, 203, 2011–2019. [Google Scholar] [CrossRef]
- Kvarnhammar, A.M.; Veitonmäki, N.; Hägerbrand, K.; Dahlman, A.; Smith, K.E.; Fritzell, S.; von Schantz, L.; Thagesson, M.; Werchau, D.; Smedenfors, K.; et al. The CTLA-4 x OX40 bispecific antibody ATOR-1015 induces anti-tumor effects through tumor-directed immune activation. J. Immunother. Cancer 2019, 7, 103. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, A.; Rodríguez, J.; Martínez, J.; Amezaga, R.; Ramos, R.; Galmes, B.; Bea, M.D.; Ferrer, J.; Pons, J.; Sampol, A.; et al. Pathogenic study of anti-CD20 infusion-related severe refractory shock in diffuse large B-cell lymphoma. Leuk. Lymphoma 2006, 47, 111–115. [Google Scholar] [CrossRef]
- Facciabene, A.; De Sanctis, F.; Pierini, S.; Reis, E.S.; Balint, K.; Facciponte, J.; Rueter, J.; Kagabu, M.; Magotti, P.; Lanitis, E.; et al. Local endothelial complement activation reverses endothelial quiescence, enabling t-cell homing, and tumor control during t-cell immunotherapy. Oncoimmunology 2017, 6, e1326442. [Google Scholar] [CrossRef]
- Hb, J.; Rm, S.; Argiris, A.; Je, B.; Lp, K.; Rl, F. Increased PD-1+ and TIM-3+ TILs during Cetuximab Therapy Inversely Correlate with Response in Head and Neck Cancer Patients. Cancer Immunol. Res. 2017, 5, 408–416. [Google Scholar]
- Chaganty, B.K.R.; Qiu, S.; Gest, A.; Lu, Y.; Ivan, C.; Calin, G.A.; Weiner, L.M.; Fan, Z. Trastuzumab upregulates PD-L1 as a potential mechanism of trastuzumab resistance through engagement of immune effector cells and stimulation of IFNγ secretion. Cancer Lett. 2018, 430, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Okita, R.; Maeda, A.; Shimizu, K.; Nojima, Y.; Saisho, S.; Nakata, M. PD-L1 overexpression is partially regulated by EGFR/HER2 signaling and associated with poor prognosis in patients with non-small-cell lung cancer. Cancer Immunol. Immunother. 2017, 66, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zeng, Y.; Du, W.; Zhu, J.; Shen, D.; Liu, Z.; Huang, J.-A. The EGFR pathway is involved in the regulation of PD-L1 expression via the IL-6/JAK/STAT3 signaling pathway in EGFR-mutated non-small cell lung cancer. Int. J. Oncol. 2016, 49, 1360–1368. [Google Scholar] [CrossRef] [Green Version]
- Bylicki, O.; Paleiron, N.; Margery, J.; Guisier, F.; Vergnenegre, A.; Robinet, G.; Auliac, J.-B.; Gervais, R.; Chouaid, C. Targeting the PD-1/PD-L1 Immune Checkpoint in EGFR-Mutated or ALK-Translocated Non-Small-Cell Lung Cancer. Target. Oncol. 2017, 12, 563–569. [Google Scholar] [CrossRef]
- Taberna, M.; Oliva, M.; Mesía, R. Cetuximab-Containing Combinations in Locally Advanced and Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2019, 9, 383. [Google Scholar] [CrossRef]
- Cioroianu, A.I.; Stinga, P.I.; Sticlaru, L.; Cioplea, M.D.; Nichita, L.; Popp, C.; Staniceanu, F. Tumor Microenvironment in Diffuse Large B-Cell Lymphoma: Role and Prognosis. Anal. Cell. Pathol. (Amst.) 2019, 2019, 8586354. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Huang, X.; Ye, X.; Qian, W. Prognostic and clinicopathological significance of PD-1/PD-L1 expression in the tumor microenvironment and neoplastic cells for lymphoma. Int. Immunopharmacol. 2019, 77, 105999. [Google Scholar] [CrossRef]
- Timmerman, J.; Herbaux, C.; Ribrag, V.; Zelenetz, A.D.; Houot, R.; Neelapu, S.S.; Logan, T.; Lossos, I.S.; Urba, W.; Salles, G.; et al. Urelumab alone or in combination with rituximab in patients with relapsed or refractory B-cell lymphoma. Am. J. Hematol. 2020. [Google Scholar] [CrossRef]
- Zhao, J.; Song, Y.; Liu, D. Recent advances on blinatumomab for acute lymphoblastic leukemia. Exp. Hematol. Oncol. 2019, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Gökbuget, N. Clinical Experience with Bispecific T Cell Engagers. Recent Results Cancer Res. 2020, 214, 71–91. [Google Scholar] [PubMed]
- Van Dam, P.A.; Verhoeven, Y.; Trinh, X.B.; Wouters, A.; Lardon, F.; Prenen, H.; Smits, E.; Baldewijns, M.; Lammens, M. RANK/RANKL signaling inhibition may improve the effectiveness of checkpoint blockade in cancer treatment. Crit. Rev. Oncol. Hematol. 2019, 133, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Suarez, E.; Jacob, A.P.; Jones, J.; Miller, R.; Roudier-Meyer, M.P.; Erwert, R.; Pinkas, J.; Branstetter, D.; Dougall, W.C. RANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature 2010, 468, 103–107. [Google Scholar] [CrossRef]
- Palafox, M.; Ferrer, I.; Pellegrini, P.; Vila, S.; Hernandez-Ortega, S.; Urruticoechea, A.; Climent, F.; Soler, M.T.; Muñoz, P.; Viñals, F.; et al. RANK induces epithelial-mesenchymal transition and stemness in human mammary epithelial cells and promotes tumorigenesis and metastasis. Cancer Res. 2012, 72, 2879–2888. [Google Scholar] [CrossRef] [Green Version]
- Smyth, E.C.; Fassan, M.; Cunningham, D.; Allum, W.H.; Okines, A.F.C.; Lampis, A.; Hahne, J.C.; Rugge, M.; Peckitt, C.; Nankivell, M.; et al. Effect of Pathologic Tumor Response and Nodal Status on Survival in the Medical Research Council Adjuvant Gastric Infusional Chemotherapy Trial. J. Clin. Oncol. 2016, 34, 2721–2727. [Google Scholar] [CrossRef]
- Afzal, M.Z.; Shirai, K. Immune checkpoint inhibitor (anti-CTLA-4, anti-PD-1) therapy alone versus immune checkpoint inhibitor (anti-CTLA-4, anti-PD-1) therapy in combination with anti-RANKL denosumuab in malignant melanoma: A retrospective analysis at a tertiary care center. Melanoma Res. 2018, 28, 341–347. [Google Scholar] [CrossRef]
- Ahern, E.; Harjunpää, H.; Barkauskas, D.; Allen, S.; Takeda, K.; Yagita, H.; Wyld, D.; Dougall, W.C.; Teng, M.W.L.; Smyth, M.J. Co-administration of RANKL and CTLA4 Antibodies Enhances Lymphocyte-Mediated Antitumor Immunity in Mice. Clin. Cancer Res. 2017, 23, 5789–5801. [Google Scholar] [CrossRef] [Green Version]
- Haibe, Y.; Kreidieh, M.; El Hajj, H.; Khalifeh, I.; Mukherji, D.; Temraz, S.; Shamseddine, A. Resistance Mechanisms to Anti-angiogenic Therapies in Cancer. Front. Oncol. 2020, 10, 221. [Google Scholar] [CrossRef] [Green Version]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef] [Green Version]
- Reis, E.S.; Mastellos, D.C.; Hajishengallis, G.; Lambris, J.D. New insights into the immune functions of complement. Nat. Rev. Immunol. 2019, 19, 503–516. [Google Scholar] [CrossRef]
- Pio, R.; Ajona, D.; Ortiz-Espinosa, S.; Mantovani, A.; Lambris, J.D. Complementing the Cancer-Immunity Cycle. Front Immunol 2019, 10, 774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajona, D.; Ortiz-Espinosa, S.; Pio, R.; Lecanda, F. Complement in Metastasis: A Comp in the Camp. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajona, D.; Ortiz-Espinosa, S.; Moreno, H.; Lozano, T.; Pajares, M.J.; Agorreta, J.; Bértolo, C.; Lasarte, J.J.; Vicent, S.; Hoehlig, K.; et al. A Combined PD-1/C5a Blockade Synergistically Protects against Lung Cancer Growth and Metastasis. Cancer Discov. 2017, 7, 694–703. [Google Scholar] [CrossRef] [Green Version]
- Zha, H.; Han, X.; Zhu, Y.; Yang, F.; Li, Y.; Li, Q.; Guo, B.; Zhu, B. Blocking C5aR signaling promotes the anti-tumor efficacy of PD-1/PD-L1 blockade. OncoImmunology 2017, 6, e1349587. [Google Scholar] [CrossRef] [PubMed]
- Lind, H.; Gameiro, S.R.; Jochems, C.; Donahue, R.N.; Strauss, J.; MD Gulley, J.L.; Palena, C.; Schlom, J. Dual targeting of TGF-β and PD-L1 via a bifunctional anti-PD-L1/TGF-βRII agent: Status of preclinical and clinical advances. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dougall, W.C.; Roman Aguilera, A.; Smyth, M.J. Dual targeting of RANKL and PD-1 with a bispecific antibody improves anti-tumor immunity. Clin. Transl. Immunol. 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera-Camacho, I.; Anaya-Ruiz, M.; Perez-Santos, M.; Millán-Pérez Peña, L.; Bandala, C.; Landeta, G. Cancer immunotherapy using anti-TIM3/PD-1 bispecific antibody: A patent evaluation of EP3356411A1. Expert Opin. Ther. Pat. 2019, 29, 587–593. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Pishvaian, M.J.; Shepard, D.R.; Wang, D.; Weiss, J.; Johnson, M.L.; Chung, C.H.; Chen, Y.; Huang, B.; Davis, C.B.; et al. A phase Ib study of utomilumab (PF-05082566) in combination with mogamulizumab in patients with advanced solid tumors. J Immunother. Cancer 2019, 7, 1–11. [Google Scholar] [CrossRef]
- Claus, C.; Ferrara, C.; Xu, W.; Sam, J.; Lang, S.; Uhlenbrock, F.; Albrecht, R.; Herter, S.; Schlenker, R.; Hüsser, T.; et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Koopmans, I.; Hendriks, D.; Samplonius, D.F.; van Ginkel, R.J.; Heskamp, S.; Wierstra, P.J.; Bremer, E.; Helfrich, W. A novel bispecific antibody for EGFR-directed blockade of the PD-1/PD-L1 immune checkpoint. Oncoimmunology 2018, 7, e1466016. [Google Scholar] [CrossRef]
- Buatois, V.; Johnson, Z.; Salgado-Pires, S.; Papaioannou, A.; Hatterer, E.; Chauchet, X.; Richard, F.; Barba, L.; Daubeuf, B.; Cons, L.; et al. Preclinical Development of a Bispecific Antibody that Safely and Effectively Targets CD19 and CD47 for the Treatment of B-Cell Lymphoma and Leukemia. Mol. Cancer Ther. 2018, 17, 1739–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golay, J.; Regazzi, M. Key Features Defining the Disposition of Bispecific Antibodies and Their Efficacy In Vivo. Ther. Drug Monit. 2020, 42, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Golay, J.; Choblet, S.; Iwaszkiewicz, J.; Cérutti, P.; Ozil, A.; Loisel, S.; Pugnière, M.; Ubiali, G.; Zoete, V.; Michielin, O.; et al. Design and Validation of a Novel Generic Platform for the Production of Tetravalent IgG1-like Bispecific Antibodies. J. Immunol. 2016, 196, 3199–3211. [Google Scholar] [CrossRef] [Green Version]
- Klein, C.; Schaefer, W.; Regula, J.T. The use of CrossMAb technology for the generation of bi- and multispecific antibodies. MAbs 2016, 8, 1010–1020. [Google Scholar] [CrossRef] [Green Version]
- Davda, J.; Declerck, P.; Hu-Lieskovan, S.; Hickling, T.P.; Jacobs, I.A.; Chou, J.; Salek-Ardakani, S.; Kraynov, E. Immunogenicity of immunomodulatory, antibody-based, oncology therapeutics. J. Immunother. Cancer 2019, 7, 105. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Referring to | Abbreviation | Referring to |
|---|---|---|---|
| A2a | Adenosine A2a receptor | HMGB1 | High-mobility group box 1 protein |
| A2b | Adenosine A2b receptor | HNSCC | Head and neck squamous cell carcinoma |
| ADC | Antibody-drug conjugate | ICI | Immune checkpoint inhibitor |
| ADCC | Antibody dependent cellular cytotoxicity | ICOS | Inducible T cell costimulator |
| ADCP | Antibody dependent cellular phagocytosis | ITIM | Immunoreceptor typosine based motif |
| AML | Acute myeloid leukemia | LAG-3 | Lymphocyte activation gene 3 |
| APC | Antigen presenting cell | MAb | Monoclonal antibody |
| ATLL | Adult T-cell leukemia/lymphoma | MCC | Merkel Cell carcinoma |
| B-NHL | B-Non Hodgkin’s lymphoma | MDSC | Myeloid derived suppressor cell |
| BsAb | Bispecific antibody | NSCLC | Non-Small Cell Lung Cancer |
| BTLA | B and T lymphocyte attenuator | ORR | Overall response rate |
| cHL | Classical Hodgkin’s lymphoma | OS | Overall survival |
| CML | Chronic myelogenous leukemia | PD-1 | Programmed cell death protein 1 |
| CRC | Colorectal cancer | PD-L1/2 | PD-1 ligand 1 or 2 |
| CTLA-4 | Cytotoxic T-lymphocyte-associated protein 4 | PFS | Progression-free survival |
| DART | Dual affinity retargeting (BsAb format) | PMBCL | Primary mediastinal B cell lymphoma |
| DC | Dendritic cell | PTCL | Peripheral T cell lymphoma |
| DLBCL | Diffuse large B cell lymphoma | PVRIG | Poliovirus receptor -related Ig domain |
| DNAM-1 | DNAX accessory protein 1 | RANK(L) | Receptor activator of nuclear factor kappa-Β (ligand) |
| DOR | Duration of response | RCC | Renal cell carcinoma |
| EMA | European Medicines Agency | RFS | Relapse-free survival |
| EGFR | Epidermal growth factor receptor | scFv | Single chain fragment variable |
| EpCAM | Epithelial Cell Adhesion Molecule | SCLC | Small cell lung cancer |
| Fab | Fragment antigen binding | TAM | Tumor associated macrophage |
| FL | Follicular Lymphoma | TCR | T cell receptor |
| FDA | Food and Drug Administration | TE | T cell engaging |
| Fv | Fragment variable (domain) | TIGIT | T cell immunoreceptor with immunoglobulin and ITIM domains |
| FcγR | Fc gamma receptor | TIL | Tumor infiltrating lymphocyte |
| FcRn | Neonatal Fc receptor | Treg | regulatory T cell |
| GITR | Glucocorticoid-induced TNFR-related protein | UC | Urothelial carcinoma |
| HCC | Hepatocellular carcinoma | VEGFR2 | Vascular endothelial growth factor receptor 2 |
| HCL | Hairy cell leukemia | VISTA | V-domain immunoglobulin suppressor of T-cell activation |
| HER2 | Human epidermal growth factor receptor 2 | ||
| Name | Target Antigen | Antibody Type a | First Indication | Year of First Approval b |
|---|---|---|---|---|
| Rituximab | CD20 | Chimeric IgG1k | B-NHL | 1997 (US) 1998 (EU) |
| Ofatumumab | CD20 | Human IgG1k | CLL | 2009 (US) 2010 (EU) |
| Obinutuzumab | CD20 | Humanized IgG1k; Glycoengin | CLL | 2013 (US) 2014 (EU) |
| Trastuzumab | HER2 | Humanized IgG1k | Breast cancer | 1998 (US) 2000 (EU) |
| Pertuzumab | HER2 | Humanized IgG1k | Breast cancer | 2012 (US) 2013 (EU) |
| Cetuximab | EGFR | Chimeric IgG1k | CRC | 2004 (US/EU) |
| Panitumumab | EGFR | Human IgG2k | CRC | 2006 (US) 2007 (EU) |
| Necitumumab | EGFR | Human IgG1k | NSCLC | 2015 (US/EU) |
| Daratumumab | CD38 | Human IgG1k | MM | 2015 (US) 2016 (EU) |
| Isatuximab | CD38 | Chimeric IgG1k | MM | 2020 (US) |
| Alemtuzumab | CD52 | Humanized IgG1k | CLL | 2001 (US/EU) |
| Mogamulizumab | CCR4 | Humanized IgG1k | T leukemia/lymphoma | 2012 Japan 2018 EU |
| Elotuzumab | SLAMF7 | Humanized IgG1k | MM | 2015 (US) 2016 (EU) |
| Olaratumab | PDGRFα | Human IgG1k | Soft tissue sarcoma | 2016 (US/EU) |
| Dinutuximab-β | GD2 | Chimeric IgG1k | Neuroblastoma | 2015 (US/EU) |
| Ramucirumab | VEGFR2 | Human IgG1k | Gastric cancer | 2014 (US/EU) |
| Bevacizumab | VEGF | Humanized IgG1k | CRC | 2004 (US) 2005 (EU) |
| Tremelimumab | CTLA-4 | Human IgG2k | Melanoma | Orphan 2006 |
| Ipilimumab | CTLA-4 | Human IgG1k | Melanoma | 2011 (US/EU) |
| Nivolumab | PD-1 | Human IgG4k S228P | Melanoma + Solid cancer | 2014 (US) 2015 (EU) |
| Pembrolizumab | PD-1 | Humanized IgG4k S228P | Melanoma + Solid cancer + HL + PMBCL | 2014 (US) 2015 (EU) |
| Cemiplimab | PD-1 | Human IgG4k S228P | Cutaneous squamous cell carcinoma | 2018 (US) 2019 (EU) |
| Avelumab | PD-L1 | Human IgG1k | MC, UC, RCC | 2017 (US/EU) |
| Atezolizumab | PD-L1 | Humanized IgG1k mut | UC, NSCLC | 2016 (US) 2017 (EU) |
| Durvalumab | PD-L1 | Human IgG1k mut | NSCLC | 2017 (US) 2018 (EU) |
| Name | Target Antigen | Antibody Type | First Indication | Year of Approval |
|---|---|---|---|---|
| Antidoby drug conjugates (ADCs) | ||||
| Gemtuzumab ozogamicin | CD33 | Humanized IgG4k calicheamicin | AML | 2000–2017 (US) 2018 (EU) |
| Brentuximab vedotin | CD30 | Chimeric IgG1k-MMAE | HD and CD30+ PTCL | 2011 (US) 2012 (EU) |
| Trastuzumab emtansine | HER2 | Humanized IgG1k | Breast | 2013 (US/EU) |
| Inotuzumab ozogamicin | CD22 | Humanized IgG4k-calicheamicin | pre-B ALL | 2017 (US/EU) |
| Moxetumomab pasudotox | CD22 | Murine IgG1 dsFv Pseudomonas exotoxin | HCL | 2018 (US) |
| Enfortumab vedotin | Nectin-4 | Human IgG1k-MMAE | UC | 2019 (US) |
| Polatuzumab vedotin | CD79b | Humanized IgG1k-MMAE | DLBCL | 2019 (US) 2020 (EU) |
| Sacituzumab govitecan | TROP2 | Humanized IgG1k-SN-38 | Triple-negative breast cancer | 2020 (US) |
| Radiolabelled Abs | ||||
| Ibritumomab tiuxetan | CD20 | Mouse IgG1-Y90 | B-NHL | 2002 (US) 2004 (EU) |
| Tositumomab-I131 | CD20 | Mouse IgG2a-I131 | B-NHL | 2003 (US) |
| Bispecific antibodies (BsAbs) | ||||
| Catumaxomab | EPCAM/CD3 | Rat/mouse bispecific mAb | Malignant ascites | 2009 (EU) |
| Blinatumomab | CD19/CD3 | Tandem scFv, Bispecifc | ALL | 2014 (US) 2015 (EU) |
| Immune Checkpoint Receptor | CD Number | Receptor Family | Cellular Expression of the Receptor | Ligand | CD Number | Cellular Expression of the Ligand |
|---|---|---|---|---|---|---|
| A. Immune Checkpoint Inhibitory Proteins | ||||||
| CTLA-4 | CD152 | CD28 | Activated T-cells and Tregs | CD80 | CD80 | APC |
| CD86 | CD86 | |||||
| PD-1 | CD279 | CD28 | Activated T and B-cells, NK cells and APCs | PD-L1 | CD274 | Activated DC, Macrophage and Tumors |
| PD-L2 | CD273 | APCs | ||||
| BTLA | CD272 | CD28 | T and B-cells, Macrophages, DCs and NK cells | HVEM | CD270 | T-cells and Macrophage |
| LAG3 | CD223 | - | Activated T-cells, Tregs, B cells, NK cells and Plasmacytoid DCs | MHC class II/Lectins | - | APC |
| TIGIT | - | CD28 | Activated T-cells, Tregs and NK cells | PVR | CD155 | DC, APCs and Tumors |
| Nectin-2 (PVRL2) | CD112 | |||||
| TIM3 | CD366 | - | Activated T-cells (Th1 cells), Treg | Gal9 | - | Variety of tissues |
| PtdSer | ||||||
| HMGB1 | ||||||
| CEACAM-1 | ||||||
| VISTA (B7-H5) | - | CD28 | Macrophages, DCs, Naïve CD4+ T-cells, Tregs, Circulating Neutrophils and Monocytes | VSIG-3 | - | Neurons and glial cells |
| NKG2A | CD94 | NKG2 | NK | HLA-E | - | - |
| ecto-5′NT | CD73 | Ecto-nucleotidase | Many cell types, upregulated in Treg | - | - | - |
| NTPDase1 | CD39 | Ecto-nucleotidase | Many cell types, upregulated in Treg | - | - | - |
| CD47 | CD47 | Ig superfamily | Ubiquitous | SIRPα | CD172α | Myeloid, neurons |
| THBS1 (TSP-1) | - | Extracellular matrix | ||||
| B. Immune Checkpoint Stimulatory Proteins | ||||||
| CD27 | CD27 | TNFR | Activated T-cells, B-cells and NK cells | CD70 | CD70 | Activated T, B-cells and DC |
| CD28 | CD28 | CD28 | T-cells | B7 | CD80 | APC |
| CD86 | ||||||
| GITR | CD357 | TNFR | Tregs and Naïve and Memory T-cells | GITRL | - | DC, Macrophage and Activated B-cells |
| ICOS | CD278 | B7/CD28 | Activated T-cells | ICOSLG | CD275 | B-cells, Macrophage and DC |
| NKG2D | CD314 | NKG2 | NK cells, CD8+ T-cells and γδ T-cells | MHC class I | - | Epithelial and endothelial cells |
| UL16-binding protein | ||||||
| OX40 | CD134 | TNFR | Activated T cells, Tregs and NK cells | OX40L | CD252 | DC, Macrophage, B-cell and Endothelial cells |
| 4-1BB | CD137 | TNFR | Activated T and NK cells | 4-1BBL | CD137L | DC, Macrophage and B-cells |
| Cancer Type | Trial | Number of Patients | Main Clinical Results | References | |
|---|---|---|---|---|---|
| Combination of nivolumab (anti-PD-1) and ipililumab (anti-CTLA-4) | |||||
| Metastatic and Unresectable Melanoma | Phase II NCT01927419 | 142 | Placebo + Ipilimumab (3 mg/kg) | 2-year OS: 53.6% ORR: 10.6% PFS: 3.0 months (mo) | [131,132] |
| Nivolumab (1 mg/kg) + Ipilimumab (3 mg/kg) | 2-year OS: 63.8% ORR: 55.9% PFS: Not reached | ||||
| Phase III NCT01844505 | 1296 | Nivolumab (3 mg/kg) | ORR: 45.0% OS: 36.9 mo PFS: 6.9 mo | [133,134] | |
| Ipilimumab (3 mg/kg) | ORR: 19.0% OS: 19.9 mo PFS: 2.9 mo | ||||
| Nivolumab (1 mg/kg) + Ipilimumab (3 mg/kg) | ORR: 58.0% OS: >60 mo PFS: 11.5 mo | ||||
| Renal Cell Carcinoma | Phase III NCT02231749 | 1390 | Nivolumab (3 mg/kg) + Ipilimumab (1 mg/kg) | ORR: 42.0% OS: Not reached PFS: 8.2 mo | [135,136] |
| Sunitinib (50 mg) | ORR: 29.0% OS: 26.6 mo PFS: 8.3 mo | ||||
| Non-Small-Cell Lung Cancer | Phase III NCT02477826 | 2220 | Nivolumab (3 mg/kg) + Ipilimumab (1 mg/kg) | ORR: 45.3% 1-year PFS: 42.6% PFS: 7.2 mo | [141,142] |
| Chemotherapy | ORR: 26.9% 1-year PFS: 13.2% PFS: 5.5 mo | ||||
| Sarcoma | Phase II NCT02500797 | 96 | Nivolumab (3 mg/kg) | ORR: 5.0% PFS: 1.7 mo OS: 10.7 mo | [140] |
| Nivolumab (3 mg/kg) + Ipilimumab (1 mg/kg) | ORR: 16.0% PFS: 4.1 mo OS: 14.3 mo | ||||
| Colorectal Cancer | Phase II NCT02060188 | 183 | Nivolumab (3 mg/kg) | ORR: 31.1% 1-year OS: 73.4% 1-year PFS: 50.4% | [137,138] |
| Nivolumab (3 mg/kg) + Ipilimumab (1 mg/kg) | ORR: 54.6% 1-year OS: 85.0% 1-year PFS: 71.0% | ||||
| Esophagogastric Cancer | Phase I/II NCT01928394 | 160 | Nivolumab (3 mg/kg) | ORR: 12.0% PFS: 1.4 mo OS: 6.2 mo | [139] |
| Nivolumab (1 mg/kg)+ Ipilimumab (3 mg/kg) | ORR: 24.0% PFS: 1.4 mo OS: 6.9 mo | ||||
| Nivolumab (3 mg/kg) + Ipilimumab (1 mg/kg) | ORR: 4.0% PFS: 1.6 mo OS: 4.8 mo | ||||
| Recurrent Small-Cell Lung Cancer | Phase I/II (NCT01928394) | 243 | Nivolumab (3 mg/kg) | ORR: 11.6% OS: 5.7 mo PFS: 1.4 mo | [143] |
| Nivolumab (1 mg/kg)+ Ipilimumab (3 mg/kg) | ORR: 21.9% OS: 4.7 mo PFS: 1.5 mo | ||||
| 216 | Nivolumab (3 mg/kg) | ORR: 10.0% | [144] | ||
| Nivolumab (1 mg/kg)+ Ipilimumab (3 mg/kg) | ORR: 23.0% | ||||
| Nivolumab (3 mg/kg)+ Ipilimumab (1 mg/kg) | ORR: 19.0% | ||||
| Relapsed Malignant Pleural Mesothelioma | Phase II NCT02716272 | 125 | Nivolumab (3 mg/kg) | 12-week DC: 40.0% ORR: 19.0% PFS: 4.0 mo OS: 11.9 mo | [145] |
| Nivolumab (3 mg/kg)+ Ipilimumab (1 mg/kg) | 12-week DC: 52.0% ORR: 28.0% PFS: 5.6 mo OS: 15.9 mo | ||||
| Combination of durvalumab (anti-PD-1) and tremelimumab (anti-CTLA-4) | |||||
| Squamous Cell Carcinoma of the Head and Neck | Phase II randomized NCT02319044 | 267 | Durvalumab (10 mg/kg) | ORR: 9.2% PFS: 1.9 mo OS: 6.0 mo | [146,147] |
| Tremelimumab (10 mg/kg) | ORR: 1.6% PFS: 1.9 mo OS: 5.5 mo | ||||
| Durvalumab (20 mg/kg) + Tremelimumab (1 mg/kg) | ORR: 7.8% PFS: 2.0 mo OS: 7.6 mo | ||||
| Phase III NCT02369874 | 736 | Durvalumab (10 mg/kg) | ORR: 17.9% PFS: 2.1 mo OS: 7.6 mo | [148] | |
| Durvalumab (20 mg/kg) + Tremelimumab (1 mg/kg) | ORR: 18.2% PFS: 2.0 mo OS: 6.5 mo | ||||
| Chemotherapy | ORR: 17.3% PFS: 3.7 mo OS: 8.3 mo | ||||
| NSCLC | Phase III NCT02453282 | 1118 | Durvalumab (20 mg/kg) | OS: 12.3 mo PFS: 2.8 mo | [150] |
| Durvalumab (20 mg/kg) + Tremelimumab (1 mg/kg) | OS: 11.2 mo PFS: 9.9 mo | ||||
| Chemotherapy | OS: 11.8 mo PFS: 5.4 mo | ||||
| Metastatic Pancreatic Ductal Adenocarcinoma | Phase IINCT02558894 | 65 | Durvalumab (1.5 g) | ORR: 0.0% PFS: 1.5 mo OS: 3.6 mo | [149] |
| Durvalumab (1.5 g) + Tremelimumab (75 mg) | ORR: 3.1% PFS: 1.5 mo OS: 3.1 mo | ||||
| Combination of pembrolizumab (anti-PD-1) and trastuzumab (anti-HER2) | |||||
| Advanced Metastatic Breast Cancer (trastuzumab resistant) | Phase I/II NCT02129556 | 52 (Onlyphase II: 40 PDL1+, 12 PDL1−) | Pembrolizumab (200 mg) + Trastuzumab (6 mg/kg) | ORR: PD-L1+: 15.0% PD-L1−: 0.0% | [98] |
| OS at 12 months: PD-L1+: 65.0% PD-L1−: 12.0% | |||||
| PFS: PD-L1+: 2.7 mo PD-L1−: 2.5 mo | |||||
| First Specificity | Combined with Antibodies Against | Category of Combination | Diseases | References |
|---|---|---|---|---|
| PD-1 or PD-L1 | TIGIT | 2 ICI | Solid tumors and hematological malignancies | [43] [154] [155] [160] [166] [177] [183] [193] |
| TIM3 | ||||
| LAG3 | ||||
| NKG2A | ||||
| CD73 | ||||
| PVRIG | ||||
| CD47 | ||||
| CD137 | ICI + immune stimulator | |||
| OX40 | ||||
| CD27 | ||||
| GITR | ||||
| EGFR | ICI + anti-tumor MAb | |||
| HER2 | ||||
| CD20 | ||||
| CD22 | ||||
| CCR4 | ||||
| FGFR3 | ||||
| CD19 × CD3 ± CTLA4 | ICI + TE BsAb | |||
| CD20 × CD3 | ||||
| gPA × CD3 | ||||
| VEGF × ANG2 | ICI + anti-angiogenic BsAb | |||
| C5aR | ICI + anti-complement receptor | |||
| TGF-β RII | Miscellaneous | |||
| RANKL ± CTLA4 | ||||
| CTLA4 | LAG3 | 2 ICI | Melanoma | [152] |
| OX40 | ICI + immune stimulator | Solid tumors | [169] | |
| EGFR | ICI + anti-tumor MAb | HNSCC | [177] | |
| OX40 | CTLA-4 ± PD-1 | 2 ICI + immune stimulator | Solid tumors | [166] |
| CD137 | 2 immune stimulator | Solid tumors | ||
| TLR4 ot TLR9 | ||||
| CD20 | immune stimulator + anti-tumor MAb | DLBCL | ||
| CD27 | GPNMB (ADC) | immune stimulator and ADC | Melanoma | [166] |
| CD137 | CCR4 | immune stimulator and anti-tumor MAb | Advanced solid tumors | [166,199] |
| CD20 ± PD-L1 | DLBCL | |||
| EGFR | CRC | |||
| HER2 (Mab or ADC) | Breast cancer | |||
| OX40+PD-L1 | 2 immune stimulators and 1 ICI antibody | Solid tumors | ||
| CD47 | EGFR | ICI + anti-tumor MAb | Colorectal | [162] |
| CD20 | B-NHL | |||
| NKG2A | EGFR | ICI + anti-tumor MAb | Squamous cell carcinoma of head and neck | [157] |
| CD73 | EGFR | ICI + anti-tumor MAb | Solid tumors | [160] |
| LAG3 | PD-1 + TIM3 | 3 ICI antibodies | Solid + Lymphoma | [152] |
| PD-1 + CTLA4 ± CD38 | 3 ICI MAbs + anti-tumor MAb | Advance tunors | ||
| PD-1 + CD137 | 2 ICI MAbs + 1 immune stimulator | Glioblastoma | ||
| GITR | PD-1 ± CTLA4 | 2 ICI MAbs + 1 immune stimulator | Solid tumors | [166] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golay, J.; Andrea, A.E. Combined Anti-Cancer Strategies Based on Anti-Checkpoint Inhibitor Antibodies. Antibodies 2020, 9, 17. https://doi.org/10.3390/antib9020017
Golay J, Andrea AE. Combined Anti-Cancer Strategies Based on Anti-Checkpoint Inhibitor Antibodies. Antibodies. 2020; 9(2):17. https://doi.org/10.3390/antib9020017
Chicago/Turabian StyleGolay, Josée, and Alain E. Andrea. 2020. "Combined Anti-Cancer Strategies Based on Anti-Checkpoint Inhibitor Antibodies" Antibodies 9, no. 2: 17. https://doi.org/10.3390/antib9020017
APA StyleGolay, J., & Andrea, A. E. (2020). Combined Anti-Cancer Strategies Based on Anti-Checkpoint Inhibitor Antibodies. Antibodies, 9(2), 17. https://doi.org/10.3390/antib9020017