Monoclonal Antibodies in Cancer Therapy



Abstract
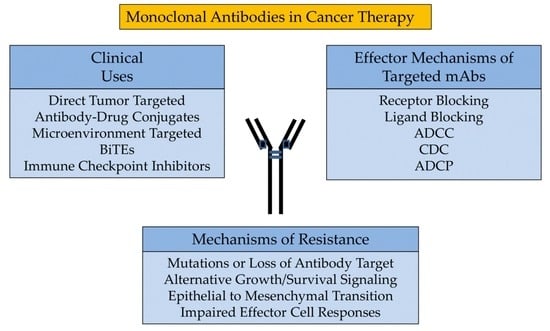
:
1. Introduction
2. Antibody Structure and Function
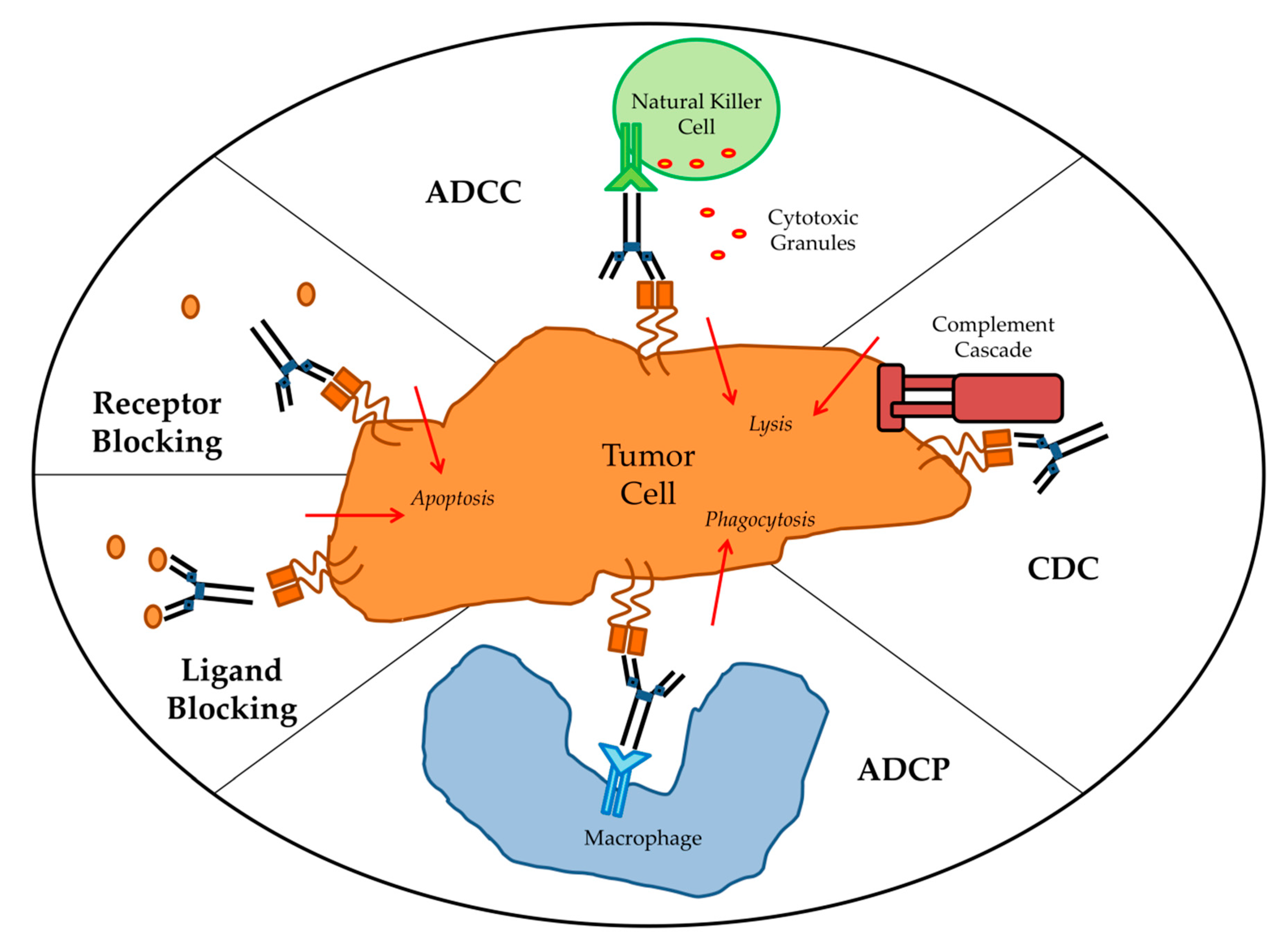
3. Effector Mechanisms of Targeted mAbs
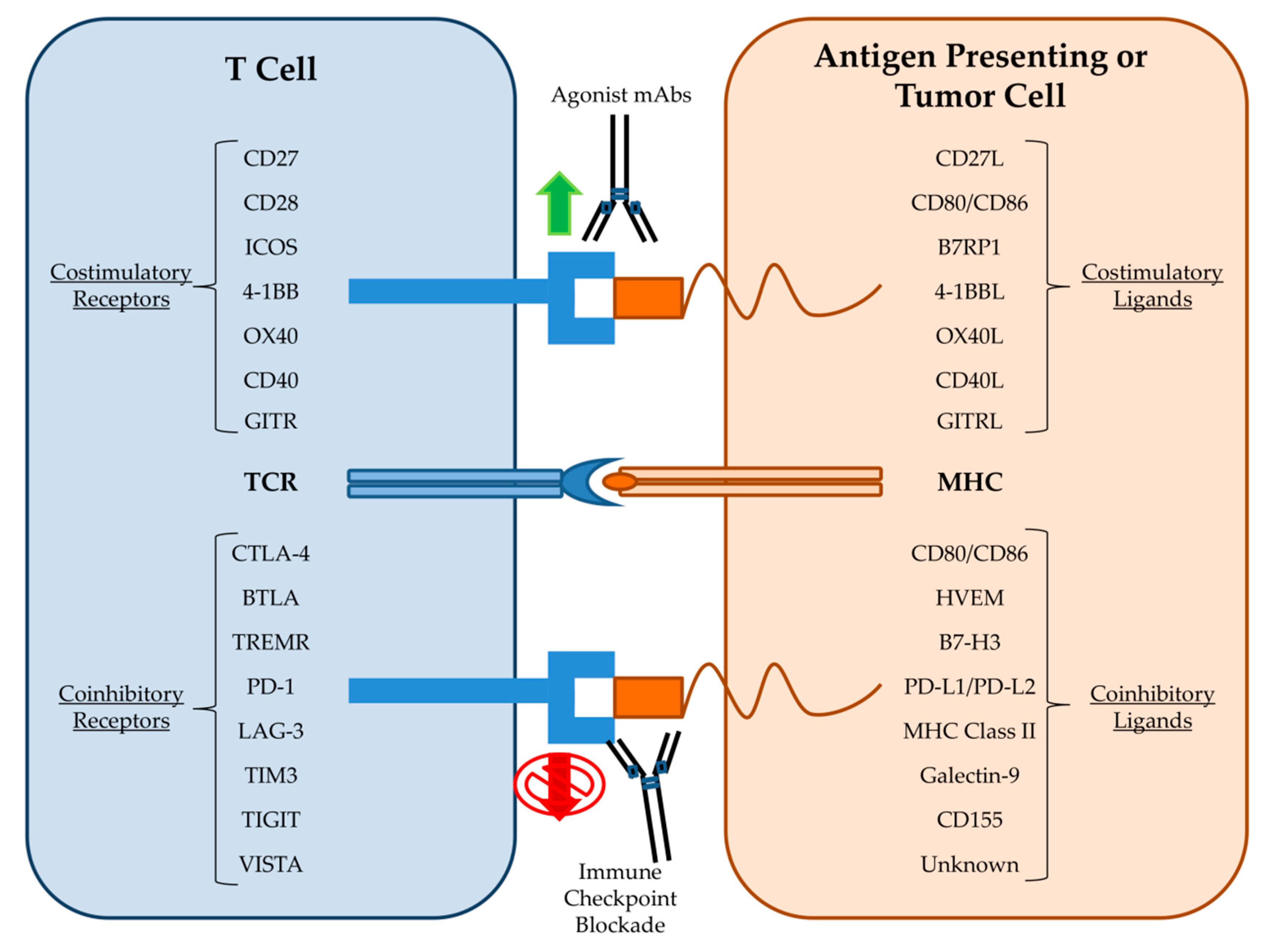
4. Clinical Uses
5. Mechanisms of Resistance
6. Combination Therapies
7. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Behring, E.V. Ueber das Zustandekommen der Diphtherie-Immunitüt und der Tetanus-Immunitüt bei Thieren. Dtsch. Med. Wochenschr. 1890. [Google Scholar] [CrossRef]
- Van Epps, H.L. How Heidelberger and Avery Sweetened Immunology. J. Exp. Med. 2005, 202, 1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagraeus, A. Plasma Cellular Reaction and Its Relation to the Formation of Antibodies in Vitro. Nature 1947, 159, 499. [Google Scholar] [CrossRef] [PubMed]
- Nossal, G.J.V.; Lederberg, J. Antibody Production by Single Cells. Nature 1958. [CrossRef] [Green Version]
- Schwaber, J.; Cohen, E.P. Human × Mouse Somatic Cell Hybrid Clone Secreting Immunoglobulins of Both Parental Types. Nature 1973, 244, 444–447. [Google Scholar] [CrossRef]
- Köhler, G.; Milstein, C. Continuous Cultures of Fused Cells Secreting Antibody of Predefined Specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Koprowski, H.; Steplewski, Z.; Herlyn, D.; Herlyn, M. Study of Antibodies against Human Melanoma Produced by Somatic Cell Hybrids. Proc. Natl. Acad. Sci. USA 1978, 75, 3405–3409. [Google Scholar] [CrossRef] [Green Version]
- Stashenko, P.; Antman, K.H.; Schlossman, S.F. Serotherapy of a Patient with a Monoclonal Antibody Directed against a Human Lymphoma-Associated Antigen. Cancer Res. 1980, 40, 3147–3154. [Google Scholar]
- Shin, S.U.; Morrison, S.L. Production and Properties of Chimeric Antibody Molecules. Methods Enzymol. 1989, 178, 459–476. [Google Scholar] [CrossRef]
- Riechmann, L.; Clark, M.; Waldmann, H.; Winter, G. Reshaping Human Antibodies for Therapy. Nature 1988, 332, 323–327. [Google Scholar] [CrossRef]
- Nelson, A.L.; Dhimolea, E.; Reichert, J.M. Development Trends for Human Monoclonal Antibody Therapeutics. Nat. Rev. Drug Discov. 2010, 9, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K. Immunobiology, 9th ed.; Garland Science: New York, NY, USA, 2017. [Google Scholar] [CrossRef]
- Weiner, L.M.; Surana, R.; Wang, S. Monoclonal Antibodies: Versatile Platforms for Cancer Immunotherapy. Nat. Rev. Immunol. 2010, 10, 317–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Schmitz, K.R.; Jeffrey, P.D.; Wiltzius, J.J.W.; Kussie, P.; Ferguson, K.M. Structural Basis for Inhibition of the Epidermal Growth Factor Receptor by Cetuximab. Cancer Cell 2005, 7, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, D.; Bassi, R.; Hooper, A.; Prewett, M.; Hicklin, D.J.; Kang, X. Anti-Epidermal Growth Factor Receptor Monoclonal Antibody Cetuximab Inhibits EGFR/HER-2 Heterodimerization and Activation. Int. J. Oncol. 2009, 34, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; et al. Studies of the HER-2/Neu Proto-Oncogene in Human Breast and Ovarian Cancer. Science 1989, 244, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Lan, K.; Hung, M.C. Strategies to Target HER2/Neu Overexpression for Cancer Therapy. Drug Resist. Updates 2003, 6, 129–136. [Google Scholar] [CrossRef]
- Di Gaetano, N.; Cittera, E.; Nota, R.; Vecchi, A.; Grieco, V.; Scanziani, E.; Botto, M.; Introna, M.; Golay, J. Complement Activation Determines the Therapeutic Activity of Rituximab in Vivo. J. Immunol. 2003, 171, 1581–1587. [Google Scholar] [CrossRef] [Green Version]
- Racila, E.; Link, B.K.; Weng, W.K.; Witzig, T.E.; Ansell, S.; Maurer, M.J.; Huang, J.; Dahle, C.; Halwani, A.; Levy, R.; et al. A Polymorphism in the Complement Component C1qA Correlates with Prolonged Response Following Rituximab Therapy of Follicular Lymphoma. Clin. Cancer Res. 2008, 14, 6697–6703. [Google Scholar] [CrossRef] [Green Version]
- Coiffier, B.; Lepretre, S.; Pedersen, L.M.; Gadeberg, O.; Fredriksen, H.; Van Oers, M.H.J.; Wooldridge, J.; Kloczko, J.; Holowiecki, J.; Hellmann, A.; et al. Safety and Efficacy of Ofatumumab, a Fully Human Monoclonal Anti-CD20 Antibody, in Patients with Relapsed or Refractory B-Cell Chronic Lymphocytic Leukemia: A Phase 1-2 Study. Blood 2008, 111, 1094–1100. [Google Scholar] [CrossRef] [Green Version]
- Gül, N.; Babes, L.; Siegmund, K.; Korthouwer, R.; Bögels, M.; Braster, R.; Vidarsson, G.; Ten Hagen, T.L.M.; Kubes, P.; Van Egmond, M. Macrophages Eliminate Circulating Tumor Cells after Monoclonal Antibody Therapy. J. Clin. Investig. 2014, 124, 812–823. [Google Scholar] [CrossRef]
- Möller, E. Contact-Induced Cytotoxicity by Lymphoid Cells Containing Foreign Isoantigens. Science 1965, 147, 873–879. [Google Scholar] [CrossRef] [PubMed]
- Teillaud, J.-L. Antibody-Dependent Cellular Cytotoxicity (ADCC). In eLS; John Wiley & Sons, Ltd: Hoboken, NJ, USA, 2012. [Google Scholar] [CrossRef]
- Fanger, M.W.; Shen, L.; Graziano, R.F.; Guyre, P.M. Cytotoxicity Mediated by Human Fc Receptors for IgG. Immunol. Today 1989, 10, 92–99. [Google Scholar] [CrossRef]
- Wallace, P.K.; Howell, A.L.; Fanger, M.W. Role of Fcγ Receptors in Cancer and Infectious Disease. J. Leukoc. Biol. 1994, 55, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Ravetch, J.V. Fcγ Receptors as Regulators of Immune Responses. Nat. Rev. Immunol. 2008, 8. [Google Scholar] [CrossRef] [PubMed]
- De Saint Basile, G.; Ménasché, G.; Fischer, A. Molecular Mechanisms of Biogenesis and Exocytosis of Cytotoxic Granules. Nat. Rev. Immunol. 2010, 10, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, F.; Ravetch, J.V. Analyzing Antibody-Fc-Receptor Interactions. Methods Mol. Biol. 2008, 415, 151–162. [Google Scholar] [CrossRef]
- Eischen, C.M.; Leibson, P.J. Role for NK-Cell-Associated Fas Ligand in Cell-Mediated Cytotoxicity and Apoptosis. Res. Immunol. 1997, 148, 164–169. [Google Scholar] [CrossRef]
- Sondel, P.M.; Alderson, K.L. Clinical Cancer Therapy by NK Cells via Antibody-Dependent Cell-Mediated Cytotoxicity. J. Biomed. Biotechnol. 2011, 2011, 379123. [Google Scholar] [CrossRef]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc Receptors Modulate in Vivo Cytoxicity against Tumor Targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef]
- Minard-colin, V.; Xiu, Y.; Poe, J.C.; Horikawa, M.; Magro, C.M.; Hamaguchi, Y.; Haas, K.M.; Tedder, T.F.; Elisa, C. Lymphoma Depletion during CD20 Immunotherapy in Mice Is Mediated By. Blood 2008, 112, 1205–1213. [Google Scholar] [CrossRef]
- De Haij, S.; Jansen, J.H.M.; Boross, P.; Beurskens, F.J.; Bakema, J.E.; Bos, D.L.; Martens, A.; Verbeek, J.S.; Parren, P.W.H.I.; Van De Winkel, J.G.J.; et al. In Vivo Cytotoxicity of Type I CD20 Antibodies Critically Depends on Fc Receptor ITAM Signaling. Cancer Res. 2010, 70, 3209–3217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubert, P.; Heitzmann, A.; Viel, S.; Nicolas, A.; Sastre-Garau, X.; Oppezzo, P.; Pritsch, O.; Osinaga, E.; Amigorena, S. Antibody-Dependent Cell Cytotoxicity Synapses Form in Mice during Tumor-Specific Antibody Immunotherapy. Cancer Res. 2011, 71, 5134–5143. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Edberg, J.C.; Redecha, P.B.; Bansal, V.; Guyre, P.M.; Coleman, K.; Salmon, J.E.; Kimberly, R.P. A Novel Polymorphism of FcγRIIIa (CD16) Alters Receptor Function and Predisposes to Autoimmune Disease. J. Clin. Investig. 1997, 100, 1059–1070. [Google Scholar] [CrossRef]
- Bibeau, F.; Lopez-Crapez, E.; Di Fiore, F.; Thezenas, S.; Ychou, M.; Blanchard, F.; Lamy, A.; Penault-Llorca, F.; Frébourg, T.; Michel, P.; et al. Impact of FcγRIIa-FcγRIIIa Polymorphisms and KRAS Mutations on the Clinical Outcome of Patients with Metastatic Colorectal Cancer Treated with Cetuximab plus Irinotecan. J. Clin. Oncol. 2009, 27, 1122–1129. [Google Scholar] [CrossRef]
- Cartron, G.; Dacheux, L.; Salles, G.; Solal-Celigny, P.; Bardos, P.; Colombat, P.; Watier, H. Therapeutic Activity of Humanized Anti-CD20 Monoclonal Antibody and Polymorphism in IgG Fc Receptor FcγrIIIa Gene. Blood 2002, 99, 754–758. [Google Scholar] [CrossRef] [Green Version]
- Weng, W.K.; Levy, R. Two Immunoglobulin G Fragment C Receptor Polymorphisms Independently Predict Response to Rituximab in Patients with Follicular Lymphoma. J. Clin. Oncol. 2003, 21, 3940–3947. [Google Scholar] [CrossRef]
- Hatjiharissi, E.; Xu, L.; Santos, D.D.; Hunter, Z.R.; Ciccarelli, B.T.; Verselis, S.; Modica, M.; Cao, Y.; Manning, R.J.; Leleu, X.; et al. Increased Natural Killer Cell Expression of CD16, Augmented Binding and ADCC Activity to Rituximab among Individuals Expressing the FcγRIIIa-158 v/v and V/F Polymorphism. Blood 2007, 110, 2561–2564. [Google Scholar] [CrossRef]
- Rodríguez, J.; Zarate, R.; Bandres, E.; Boni, V.; Hernández, A.; Sola, J.J.; Honorato, B.; Bitarte, N.; García-Foncillas, J. Fc Gamma Receptor Polymorphisms as Predictive Markers of Cetuximab Efficacy in Epidermal Growth Factor Receptor Downstream-Mutated Metastatic Colorectal Cancer. Eur. J. Cancer 2012, 48, 1774–1780. [Google Scholar] [CrossRef]
- Musolino, A.; Naldi, N.; Bortesi, B.; Pezzuolo, D.; Capelletti, M.; Missale, G.; Laccabue, D.; Zerbini, A.; Camisa, R.; Bisagni, G.; et al. Immunoglobulin g Fragment c Receptor Polymorphisms and Clinical Efficacy of Trastuzumab-Based Therapy in Patients with HER-2/Neu-Positive Metastatic Breast Cancer. J. Clin. Oncol. 2008, 26, 1789–1796. [Google Scholar] [CrossRef]
- Boero, S.; Morabito, A.; Banelli, B.; Cardinali, B.; Dozin, B.; Lunardi, G.; Piccioli, P.; Lastraioli, S.; Carosio, R.; Salvi, S.; et al. Analysis of in Vitro ADCC and Clinical Response to Trastuzumab: Possible Relevance of FcγRIIIA/FcγRIIA Gene Polymorphisms and HER-2 Expression Levels on Breast Cancer Cell Lines. J. Transl. Med. 2015, 13, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siebert, N.; Jensen, C.; Troschke-Meurer, S.; Zumpe, M.; Jüttner, M.; Ehlert, K.; Kietz, S.; Müller, I.; Lode, H.N. Neuroblastoma Patients with High-Affinity FCGR2A, -3A and Stimulatory KIR 2DS2 Treated by Long-Term Infusion of Anti-GD2 Antibody Ch14.18/CHO Show Higher ADCC Levels and Improved Event-Free Survival. Oncoimmunology 2016, 5, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnould, L.; Gelly, M.; Penault-Llorca, F.; Benoit, L.; Bonnetain, F.; Migeon, C.; Cabaret, V.; Fermeaux, V.; Bertheau, P.; Garnier, J.; et al. Trastuzumab-Based Treatment of HER2-Positive Breast Cancer: An Antibody-Dependent Cellular Cytotoxicity Mechanism? Br. J. Cancer 2006, 94, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Vermi, W.; Micheletti, A.; Finotti, G.; Tecchio, C.; Calzetti, F.; Costa, S.; Bugatti, M.; Calza, S.; Agostinelli, C.; Pileri, S.; et al. Slan + Monocytes and Macrophages Mediate CD20-Dependent B Cell Lymphoma Elimination via ADCC and ADCP. Cancer Res. 2018, 78, 3544–3559. [Google Scholar] [CrossRef] [Green Version]
- de Weers, M.; Tai, Y.-T.; van der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.H.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a Novel Therapeutic Human CD38 Monoclonal Antibody, Induces Killing of Multiple Myeloma and Other Hematological Tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef]
- Ferris, R.L.; Jaffee, E.M.; Ferrone, S. Tumor Antigen-Targeted, Monoclonal Antibody-Based Immunotherapy: Clinical Response, Cellular Immunity, and Immunoescape. J. Clin. Oncol. 2010, 28, 4390–4399. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Gunasekaran, K.; Wang, W.; Razinkov, V.; Sekirov, L.; Leng, E.; Sweet, H.; Foltz, I.; Howard, M.; Rousseau, A.M.; et al. Asymmetrical Fc Engineering Greatly Enhances Antibodydependent Cellular Cytotoxicity (ADCC) Effector Function and Stability of the Modified Antibodies. J. Biol. Chem. 2014, 289, 3571–3590. [Google Scholar] [CrossRef] [Green Version]
- Umaña, P.; Jean-Mairet, J.; Moudry, R.; Amstutz, H.; Bailey, J.E. Engineered Glycoforms of an Antineuroblastoma IgG1 with Optimized Antibody-Dependent Cellular Cytotoxic Activity. Nat. Biotechnol. 1999, 17, 176–180. [Google Scholar] [CrossRef]
- Davies, J.; Jiang, L.; Pan, L.Z.; Labarre, M.J.; Anderson, D.; Reff, M. Expression of GnTIII in a Recombinant Anti-CD20 CHO Production Cell Line: Expression of Antibodies with Altered Glycoforms Leads to an Increase in ADCC through Higher Affinity for FcγRIII. Biotechnol. Bioeng. 2001, 74, 288–294. [Google Scholar] [CrossRef]
- Shields, R.L.; Lai, J.; Keck, R.; O’Connell, L.Y.; Hong, K.; Gloria Meng, Y.; Weikert, S.H.A.; Presta, L.G. Lack of Fucose on Human IgG1 N-Linked Oligosaccharide Improves Binding to Human FcγRIII and Antibody-Dependent Cellular Toxicity. J. Biol. Chem. 2002, 277, 26733–26740. [Google Scholar] [CrossRef] [Green Version]
- Ishida, T.; Joh, T.; Uike, N.; Yamamoto, K.; Utsunomiya, A.; Yoshida, S.; Saburi, Y.; Miyamoto, T.; Takemoto, S.; Suzushima, H.; et al. Defucosylated Anti-CCR4 Monoclonal Antibody (KW-0761) for Relapsed Adult T-Cell Leukemia-Lymphoma: A Multicenter Phase II Study. J. Clin. Oncol. 2012, 30, 837–842. [Google Scholar] [CrossRef]
- Finn, O.J. Human Tumor Antigens Yesterday, Today, and Tomorrow. Cancer Immunol. Res. 2017, 5, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maloney, D.G.; Grillo-López, A.J.; White, C.A.; Bodkin, D.; Schilder, R.J.; Neidhart, J.A.; Janakiraman, N.; Foon, K.A.; Liles, T.M.; Dallaire, B.K.; et al. IDEC-C2B8 (Rituximab) Anti-CD20 Monoclonal Antibody Therapy in Patients with Relapsed Low-Grade Non-Hodgkin’s Lymphoma. Blood 1997, 90, 2188–2195. [Google Scholar] [CrossRef] [PubMed]
- Rimawi, M.F.; Schiff, R.; Osborne, C.K. Targeting HER2 for the Treatment of Breast Cancer. Annu. Rev. Med. 2015, 66, 111–128. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, J. The Epidermal Growth Factor Receptor as a Target for Therapy with Antireceptor Monoclonal Antibodies. Semin. Cancer Biol. 1990, 1, 339–344. [Google Scholar]
- Indications and Year of First Approval for Each Antibody Were Accessed Using the FDA Drug Database. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/ (accessed on 21 May 2020).
- Chari, R.V.J. Targeted Cancer Therapy: Conferring Specificity to Cytotoxic Drugs. Acc. Chem. Res. 2008, 41, 98–107. [Google Scholar] [CrossRef]
- Younes, A.; Bartlett, N.L.; Leonard, J.P.; Kennedy, D.A.; Lynch, C.M.; Sievers, E.L.; Forero-Torres, A. Brentuximab Vedotin (SGN-35) for Relapsed CD30-Positive Lymphomas. N. Engl. J. Med. 2010, 363, 1812–1821. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab Emtansine for HER2-Positive Advanced Breast Cancer. N. Engl. J. Med. 2012, 18, 732–742. [Google Scholar] [CrossRef] [Green Version]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody–Drug Conjugates for Cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef]
- Thomas, A.; Teicher, B.A.; Hassan, R. Antibody–Drug Conjugates for Cancer Therapy. Lancet Oncol. 2016, 17, e254–e262. [Google Scholar] [CrossRef]
- Becker, N.; Benhar, I. Antibody-Based Immunotoxins for the Treatment of Cancer. Antibodies 2012, 1, 39–69. [Google Scholar] [CrossRef] [Green Version]
- Dhillon, S. Moxetumomab Pasudotox: First Global Approval. Drugs 2018, 78, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Steiner, M.; Neri, D. Antibody-Radionuclide Conjugates for Cancer Therapy: Historical Considerations and New Trends. Clin. Cancer Res. 2011, 17, 6406–6416. [Google Scholar] [CrossRef] [Green Version]
- Ellis, L.M.; Hicklin, D.J. VEGF-Targeted Therapy: Mechanisms of Anti-Tumour Activity. Nat. Rev. Cancer 2008, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Krupitskaya, Y.; Wakelee, H.A. Ramucirumab, a Fully Human MAb to the Transmembrane Signaling Tyrosine Kinase VEGFR-2 for the Potential Treatment of Cancer. Curr. Opin. Investig. Drugs 2009, 10, 597–605. [Google Scholar] [PubMed]
- Wu, Y.; Zhong, Z.; Huber, J.; Bassi, R.; Finnerty, B.; Corcoran, E.; Li, H.; Navarro, E.; Balderes, P.; Jimenez, X.; et al. Anti-Vascular Endothelial Growth Factor Receptor-1 Antagonist Antibody as a Therapeutic Agent for Cancer. Clin. Cancer Res. 2006, 12, 6573–6584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Vil, M.D.; Prewett, M.; Damoci, C.; Zhang, H.; Li, H.; Jimenez, X.; Deevi, D.S.; Iacolina, M.; Kayas, A.; et al. Development of a Fully Human Anti-PDGFRβ Antibody That Suppresses Growth of Human Tumor Xenografts and Enhances Antitumor Activity of an Anti-VEGFR2 Antibody. Neoplasia 2009, 11, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Colak, S.; ten Dijke, P. Targeting TGF-β Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Grütter, C.; Wilkinson, T.; Turner, R.; Podichetty, S.; Finch, D.; McCourt, M.; Loning, S.; Jermutus, L.; Grütter, M.G. A Cytokine-Neutralizing Antibody as a Structural Mimetic of 2 Receptor Interactions. Proc. Natl. Acad. Sci. USA 2008, 105, 20251–20256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutterbuese, R.; Raum, T.; Kischel, R.; Hoffmann, P.; Mangold, S.; Rattel, B.; Friedrich, M.; Thomas, O.; Lorenczewski, G.; Rau, D.; et al. T Cell-Engaging BiTE Antibodies Specific for EGFR Potently Eliminate KRAS- and BRAF-Mutated Colorectal Cancer Cells. Proc. Natl. Acad. Sci. USA 2010, 107, 12605–12610. [Google Scholar] [CrossRef] [Green Version]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.-M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Redman, J.M.; Hill, E.M.; AlDeghaither, D.; Weiner, L.M. Mechanisms of Action of Therapeutic Antibodies for Cancer. Mol. Immunol. 2015, 67, 28–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rech, A.J.; Vonderheide, R.H. Clinical Use of Anti-CD25 Antibody Daclizumab to Enhance Immune Responses to Tumor Antigen Vaccination by Targeting Regulatory T Cells. Ann. N. Y. Acad. Sci. 2009, 1174, 99–106. [Google Scholar] [CrossRef]
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell-Extrinsic Function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of Lupus-like Autoimmune Diseases by Disruption of the PD-1 Gene Encoding an ITIM Motif-Carrying Immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti-PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Sznol, M.; Chen, L. Antagonist Antibodies to PD-1 and B7-H1 (PD-L1) in the Treatment of Advanced Human Cancer. Clin. Cancer Res. 2013, 19, 1021–1034. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H.; Lao, C.D.; et al. Nivolumab versus Chemotherapy in Patients with Advanced Melanoma Who Progressed after Anti-CTLA-4 Treatment (CheckMate 037): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Puzanov, I.; Dummer, R.; Schadendorf, D.; Hamid, O.; Robert, C.; Hodi, F.S.; Schachter, J.; Pavlick, A.C.; Lewis, K.D.; et al. Pembrolizumab versus Investigator-Choice Chemotherapy for Ipilimumab-Refractory Melanoma (KEYNOTE-002): A Randomised, Controlled, Phase 2 Trial. Lancet Oncol. 2015, 16, 908–918. [Google Scholar] [CrossRef]
- Abdin, S.M.; Zaher, D.M.; Arafa, E.S.A.; Omar, H.A. Tackling Cancer Resistance by Immunotherapy: Updated Clinical Impact and Safety of PD-1/PD-L1 Inhibitors. Cancers 2018, 10, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune Checkpoint Blockade Therapy for Cancer: An Overview of FDA-Approved Immune Checkpoint Inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-Inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [Green Version]
- Ni, L.; Dong, C. New Checkpoints in Cancer Immunotherapy. Immunol. Rev. 2017, 276, 52–65. [Google Scholar] [CrossRef]
- Kim, N.; Kim, H.S. Targeting Checkpoint Receptors and Molecules for Therapeutic Modulation of Natural Killer Cells. Front. Immunol. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Kohrt, H.E.; Thielens, A.; Marabelle, A.; Sagiv-Barfi, I.; Sola, C.; Chanuc, F.; Fuseri, N.; Bonnafous, C.; Czerwinski, D.; Rajapaksa, A.; et al. Anti-KIR Antibody Enhancement of Anti-Lymphoma Activity of Natural Killer Cells as Monotherapy and in Combination with Anti-CD20 Antibodies. Blood 2014, 123, 678–686. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, P.; Grillo-López, A.J.; Link, B.K.; Levy, R.; Czuczman, M.S.; Williams, M.E.; Heyman, M.R.; Bence-Bruckler, I.; White, C.A.; Cabanillas, F.; et al. Rituximab Chimeric Anti-CD20 Monoclonal Antibody Therapy for Relapsed Indolent Lymphoma: Half of Patients Respond to a Four-Dose Treatment Program. J. Clin. Oncol. 1998, 16, 2825–2833. [Google Scholar] [CrossRef]
- Benavente, S.; Huang, S.; Armstrong, E.A.; Chi, A.; Hsu, K.T.; Wheeler, D.L.; Harari, P.M. Establishment and Characterization of a Model of Acquired Resistance to Epidermal Growth Factor Receptor Targeting Agents in Human Cancer Cells. Clin. Cancer Res. 2009, 15, 1585–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, A. Current Updates on Trastuzumab Resistance in HER2 Overexpressing Breast Cancers. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2019; Volume 1152, pp. 217–228. [Google Scholar] [CrossRef]
- Mishima, Y.; Terui, Y.; Takeuchi, K.; Matsumoto-Mishima, Y.; Matsusaka, S.; Utsubo-Kuniyoshi, R.; Hatake, K. The Identification of Irreversible Rituximab-Resistant Lymphoma Caused by CD20 Gene Mutations. Blood Cancer J. 2011, 1, e15–e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sickmier, E.A.; Kurzeja, R.J.M.; Michelsen, K.; Vazir, M.; Yang, E.; Tasker, A.S. The Panitumumab EGFR Complex Reveals a Binding Mechanism That Overcomes Cetuximab Induced Resistance. PLoS ONE 2016, 11, e0163366. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Lahiji, A.; Patel, S.; Franklin, M.; Jimenez, X.; Hicklin, D.J.; Kang, X. Monoclonal Antibody Cetuximab Binds to and Down-Regulates Constitutively Activated Epidermal Growth Factor Receptor VIII on the Cell Surface. Anticancer Res. 2007, 27, 3355–3366. [Google Scholar] [PubMed]
- Czuczman, M.S.; Olejniczak, S.; Gowda, A.; Kotowski, A.; Binder, A.; Kaur, H.; Knight, J.; Starostik, P.; Deans, J.; Hernandez-Ilizaliturri, F.J. Acquirement of Rituximab Resistance in Lymphoma Cell Lines Is Associated with Both Global CD20 Gene and Protein Down-Regulation Regulated at the Pretranscriptional and Posttranscriptional Levels. Clin. Cancer Res. 2008, 14, 1561–1570. [Google Scholar] [CrossRef] [Green Version]
- Nijhof, I.S.; Casneuf, T.; Van Velzen, J.; Van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.J.; Van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 Expression and Complement Inhibitors Affect Response and Resistance to Daratumumab Therapy in Myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.; Ishii, Y.; Ochiai, H.; Fukuda, K.; Akimoto, S.; Hayashida, T.; Okabayashi, K.; Tsuruta, M.; Hasegawa, H.; Kitagawa, Y. Cetuximab-Mediated ADCC Activity Is Correlated with the Cell Surface Expression Level of EGFR but Not with the KRAS/BRAF Mutational Status in Colorectal Cancer. Oncol. Rep. 2014, 31, 2115–2122. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.C.; López-Albaitero, A.; Ferris, R.L. Immunotherapy of Head and Neck Cancer Using Tumor Antigen-Specific Monoclonal Antibodies. Curr. Oncol. Rep. 2009, 11, 156–162. [Google Scholar] [CrossRef]
- Braig, F.; Kriegs, M.; Voigtlaender, M.; Habel, B.; Grob, T.; Biskup, K.; Blanchard, V.; Sack, M.; Thalhammer, A.; Batalla, I.B.; et al. Cetuximab Resistance in Head and Neck Cancer Is Mediated by EGFR-K521 Polymorphism. Cancer Res. 2017, 77, 1188–1199. [Google Scholar] [CrossRef] [Green Version]
- Nakadate, Y.; Kodera, Y.; Kitamura, Y.; Shirasawa, S.; Tachibana, T.; Tamura, T.; Koizumi, F. KRAS Mutation Confers Resistance to Antibody-Dependent Cellular Cytotoxicity of Cetuximab against Human Colorectal Cancer Cells. Int. J. Cancer 2014, 134, 2146–2155. [Google Scholar] [CrossRef] [PubMed]
- Valabrega, G.; Montemurro, F.; Aglietta, M. Trastuzumab: Mechanism of Action, Resistance and Future Perspectives in HER2-Overexpressing Breast Cancer. Ann. Oncol. 2007, 18, 977–984. [Google Scholar] [CrossRef]
- Gennari, R.; Menard, S.; Fagnoni, F.; Ponchio, L.; Scelsi, M.; Tagliabue, E.; Castiglioni, F.; Villani, L.; Magalotti, C.; Gibelli, N.; et al. Pilot Study of the Mechanism of Action of Preoperative Trastuzumab in Patients with Primary Operable Breast Tumors Overexpressing HER2. Clin. Cancer Res. 2004, 10, 5650–5655. [Google Scholar] [CrossRef] [Green Version]
- Kominsky, S.L.; Hobeika, A.C.; Lake, F.A.; Torres, B.A.; Johnson, H.M. Down-Regulation of Neu/HER-2 by Interferon-γ in Prostate Cancer Cells. Cancer Res. 2000, 60, 3904–3908. [Google Scholar]
- Shi, Y.; Fan, X.; Meng, W.; Deng, H.; Zhang, N.; An, Z. Engagement of Immune Effector Cells by Trastuzumab Induces HER2/ERBB2 Downregulation in Cancer Cells through STAT1 Activation. Breast Cancer Res. 2014, 16, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellucci, R.; Martin, A.; Bommarito, D.; Wang, K.; Hansen, S.H.; Freeman, G.J.; Ritz, J. Interferon-γ-Induced Activation of JAK1 and JAK2 Suppresses Tumor Cell Susceptibility to NK Cells through Upregulation of PD-L1 Expression. Oncoimmunology 2015, 4, 1–10. [Google Scholar] [CrossRef]
- Sforza, V.; Martinelli, E.; Ciardiello, F.; Gambardella, V.; Napolitano, S.; Martini, G.; Corte, C.D.; Cardone, C.; Ferrara, M.L.; Reginelli, A.; et al. Mechanisms of Resistance to Anti-Epidermal Growth Factor Receptor Inhibitors in Metastatic Colorectal Cancer. World J. Gastroenterol. 2016, 22, 6345–6361. [Google Scholar] [CrossRef] [PubMed]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS Mutations and Acquired Resistance to Anti-EGFR Therapy in Colorectal Cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [Green Version]
- Kasper, S.; Reis, H.; Ziegler, S.; Nothdurft, S.; Mueller, A.; Goetz, M.; Wiesweg, M.; Phasue, J.; Ting, S.; Wieczorek, S.; et al. Molecular Dissection of Effector Mechanisms of RAS-Mediated Resistance to Anti-EGFR Antibody Therapy. Oncotarget 2017, 8, 45898–45917. [Google Scholar] [CrossRef] [Green Version]
- Cuyàs, E.; Queralt, B.; Martin-Castillo, B.; Bosch-Barrera, J.; Menendez, J.A. EphA2 Receptor Activation with Ephrin-A1 Ligand Restores Cetuximab Efficacy in NRAS-Mutant Colorectal Cancer Cells. Oncol. Rep. 2017, 38, 263–270. [Google Scholar] [CrossRef]
- Ozawa, H.; Ranaweera, R.S.; Izumchenko, E.; Makarev, E.; Zhavoronkov, A.; Fertig, E.J.; Howard, J.D.; Markovic, A.; Bedi, A.; Ravi, R.; et al. SMAD4 Loss Is Associated with Cetuximab Resistance and Induction of MAPK/JNK Activation in Head and Neck Cancer Cells. Clin. Cancer Res. 2017, 23, 5162–5175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, F.; Liu, Y.; Chu, H.; Wen, Y.; Yan, L.; Tang, Q.; Xiao, E.; Zhang, D.; Zhang, H. EIF5A2 Is an Alternative Pathway for Cell Proliferation in Cetuximab-Treated Epithelial Hepatocellular Carcinoma. Am. J. Transl. Res. 2016, 8, 4670–4681. [Google Scholar] [PubMed]
- Kong, X.; Zhang, K.; Wang, X.; Yang, X.; Li, Y.; Zhai, J.; Xing, Z.; Qi, Y.; Gao, R.; Feng, X.; et al. Mechanism of Trastuzumab Resistance Caused by HER-2 Mutation in Breast Carcinomas. Cancer Manag. Res. 2019, 11, 5971–5982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandarlapaty, S.; Sakr, R.A.; Giri, D.; Patil, S.; Heguy, A.; Morrow, M.; Modi, S.; Norton, L.; Rosen, N.; Hudis, C.; et al. Frequent Mutational Activation of the PI3K-AKT Pathway in Trastuzumab-Resistant Breast Cancer. Clin. Cancer Res. 2012, 18, 6784–6791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritter, C.A.; Perez-Torres, M.; Rinehart, C.; Guix, M.; Dugger, T.; Engelman, J.A.; Arteaga, C.L. Human Breast Cancer Cells Selected for Resistance to Trastuzumab in Vivo Overexpress Epidermal Growth Factor Receptor and ErbB Ligands and Remain Dependent on the ErbB Receptor Network. Clin. Cancer Res. 2007, 13, 4909–4919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernieri, C.; Milano, M.; Brambilla, M.; Mennitto, A.; Maggi, C.; Cona, M.S.; Prisciandaro, M.; Fabbroni, C.; Celio, L.; Mariani, G.; et al. Resistance Mechanisms to Anti-HER2 Therapies in HER2-Positive Breast Cancer: Current Knowledge, New Research Directions and Therapeutic Perspectives. Crit. Rev. Oncol. Hematol. 2019, 139, 53–66. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Kono, K.; Mizukami, Y.; Mimura, K.; Fujii, H. Mechanisms of Escape from Trastuzumab-Mediated ADCC in Esophageal Squamous Cell Carcinoma: Relation to Susceptibility to Perforin-Granzyme. Anticancer Res. 2009, 29, 2137–2146. [Google Scholar]
- Evans, M.K.; Sauer, S.J.; Nath, S.; Robinson, T.J.; Morse, M.A.; Devi, G.R. X-Linked Inhibitor of Apoptosis Protein Mediates Tumor Cell Resistance to Antibody-Dependent Cellular Cytotoxicity. Cell Death Dis. 2016, 7, e2073. [Google Scholar] [CrossRef] [Green Version]
- Jazirehi, A.R.; Vega, M.I.; Bonavida, B. Development of Rituximab-Resistant Lymphoma Clones with Altered Cell Signaling and Cross-Resistance to Chemotherapy. Cancer Res. 2007, 67, 1270–1281. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, S.; Bindea, G.; Albu, R.I.; Mlecnik, B.; Machiels, J.P. Cetuximab Promotes Epithelial to Mesenchymal Transition and Cancer Associated Fibroblasts in Patients with Head and Neck Cancer. Oncotarget 2015, 6, 34288–34299. [Google Scholar] [CrossRef] [Green Version]
- Kimura, I.; Kitahara, H.; Ooi, K.; Kato, K.; Noguchi, N.; Yoshizawa, K.; Nakamura, H.; Kawashiri, S. Loss of Epidermal Growth Factor Receptor Expression in Oral Squamous Cell Carcinoma Is Associated with Invasiveness and Epithelial-Mesenchymal Transition. Oncol. Lett. 2016, 11, 201–207. [Google Scholar] [CrossRef] [Green Version]
- Hsu, D.S.S.; Hwang, W.L.; Yuh, C.H.; Chu, C.H.; Ho, Y.H.; Chen, P.B.; Lin, H.S.; Lin, H.K.; Wu, S.P.; Lin, C.Y.; et al. Lymphotoxin-β Interacts with Methylated EGFR to Mediate Acquired Resistance to Cetuximab in Head and Neck Cancer. Clin. Cancer Res. 2017, 23, 4388–4401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Fertig, E.J.; Ozawa, H.; Hatakeyama, H.; Howard, J.D.; Perez, J.; Considine, M.; Thakar, M.; Ranaweera, R.; Krigsfeld, G.; et al. Decreased SMAD4 Expression Is Associated with Induction of Epithelial-to-Mesenchymal Transition and Cetuximab Resistance in Head and Neck Squamous Cell Carcinoma. Cancer Biol. Ther. 2015, 16, 1252–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveras-Ferraros, C.; Vazquez-Martin, A.; Cufí, S.; Queralt, B.; Báez, L.; Guardeño, R.; Hernández-Yagü e, X.; Martin-Castillo, B.; Brunet, J.; Menendez, J.A. Stem Cell Property Epithelial-to-Mesenchymal Transition Is a Core Transcriptional Network for Predicting Cetuximab (ErbituxTM) Efficacy in KRAS Wild-Type Tumor Cells. J. Cell. Biochem. 2011, 112, 10–29. [Google Scholar] [CrossRef] [PubMed]
- Oliveras-Ferraros, C.; Corominas-Faja, B.; Vazquez-Martin, S.C.A.; Martin-Castillo, B.; Iglesias, J.M.; López-Bonet, E.; Martin, Á.G.; Menendez, J.A. Epithelial-to-Mesenchymal Transition (EMT) Confers Primary Resistance to Trastuzumab (Herceptin). Cell Cycle 2012, 11, 4020–4032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnett, J.P.; Korkaya, H.; Ouzounova, M.D.; Jiang, H.; Conley, S.J.; Newman, B.W.; Sun, L.; Connarn, J.N.; Chen, C.S.; Zhang, N.; et al. Trastuzumab Resistance Induces EMT to Transform HER2 + PTEN’ to a Triple Negative Breast Cancer That Requires Unique Treatment Options. Sci. Rep. 2015, 5, 15821. [Google Scholar] [CrossRef] [Green Version]
- Capuano, C.; Romanelli, M.; Pighi, C.; Cimino, G.; Rago, A.; Molfetta, R.; Paolini, R.; Santoni, A.; Galandrini, R. Anti-CD20 Therapy Acts via FcγRIIIA to Diminish Responsiveness of Human Natural Killer Cells. Cancer Res. 2015, 75, 4097–4108. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Sunderland, A.; Zhou, Y.; Schulick, R.D.; Edil, B.H.; Zhu, Y. Blockade of CD112R and TIGIT Signaling Sensitizes Human Natural Killer Cell Functions. Cancer Immunol. Immunother. 2017, 66, 1367–1375. [Google Scholar] [CrossRef]
- Cooley, S.; Burns, L.J.; Repka, T.; Miller, J.S. Natural Killer Cell Cytotoxicity of Breast Cancer Targets Is Enhanced by Two Distinct Mechanisms of Antibody-Dependent Cellular Cytotoxicity against LFA-3 and HER2/Neu. Exp. Hematol. 1999, 27, 1533–1541. [Google Scholar] [CrossRef]
- Sordo-Bahamonde, C.; Vitale, M.; Lorenzo-Herrero, S.; López-Soto, A.; Gonzalez, S. Mechanisms of Resistance to NK Cell Immunotherapy. Cancers 2020, 12, 893. [Google Scholar] [CrossRef] [Green Version]
- Aldeghaither, D.S.; Zahavi, D.J.; Murray, J.C.; Fertig, E.J.; Graham, G.T.; Zhang, Y.-W.; O’Connell, A.; Ma, J.; Jablonski, S.A.; Weiner, L.M. A Mechanism of Resistance to Antibody-Targeted Immune Attack. Cancer Immunol. Res. 2019, 7, 230–243. [Google Scholar] [CrossRef] [Green Version]
- Corraliza-Gorjón, I.; Somovilla-Crespo, B.; Santamaria, S.; Garcia-Sanz, J.A.; Kremer, L. New Strategies Using Antibody Combinations to Increase Cancer Treatment Effectiveness. Front. Immunol. 2017, 8, 1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linares, J.; Rullan, A.; Taberna, M.; Vazquez, S.; Mesia, R. Emergence of Long-Term Surviving Patients with the Introduction of Cetuximab in Recurrent/Metastatic Disease of Squamous Cell Carcinoma of Head and Neck. Oral Oncol. 2016, 100, e4. [Google Scholar] [CrossRef] [PubMed]
- Melero, I.; Berman, D.M.; Aznar, M.A.; Korman, A.J.; Gracia, J.L.P.; Haanen, J. Evolving Synergistic Combinations of Targeted Immunotherapies to Combat Cancer. Nat. Rev. Cancer 2015, 15, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Stagg, J.; Loi, S.; Divisekera, U.; Ngiow, S.F.; Duret, H.; Yagita, H.; Teng, M.W.; Smyth, M.J. Anti-ErbB-2 MAb Therapy Requires Type I and II Interferons and Synergizes with Anti-PD-1 or Anti-CD137 MAb Therapy. Proc. Natl. Acad. Sci. USA 2011, 108, 7142–7147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griguolo, G.; Pascual, T.; Dieci, M.V.; Guarneri, V.; Prat, A. Interaction of Host Immunity with HER2-Targeted Treatment and Tumor Heterogeneity in HER2-Positive Breast Cancer. J. Immunother. Cancer 2019, 7, 90. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Giobbie-Hurder, A.; Gombos, A.; Bachelot, T.; Hui, R.; Curigliano, G.; Campone, M.; Biganzoli, L.; Bonnefoi, H.; Jerusalem, G.; et al. Pembrolizumab plus Trastuzumab in Trastuzumab-Resistant, Advanced, HER2-Positive Breast Cancer (PANACEA): A Single-Arm, Multicentre, Phase 1b–2 Trial. Lancet Oncol. 2019, 20, 371–382. [Google Scholar] [CrossRef]
- Korman, A.; Chen, B.; Wang, C.; Wu, L.; Cardarelli, P.; Selby, M. Activity of Anti-PD-1 in Murine Tumor Models: Role of “Host” PD-L1 and Synergistic Effect of Anti-PD-1 and Anti-CTLA-4 (48.37). J. Immunol. 2007, 178 (Suppl. 1), S82. [Google Scholar]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Siu, L.L.; Steeghs, N.; Meniawy, T.; Joerger, M.; Spratlin, J.L.; Rottey, S.; Nagrial, A.; Cooper, A.; Meier, R.; Guan, X.; et al. Preliminary Results of a Phase I/IIa Study of BMS-986156 (Glucocorticoid-Induced Tumor Necrosis Factor Receptor–Related Gene (GITR) Agonist), Alone and in Combination with Nivolumab in Pts with Advanced Solid Tumors. J. Clin. Oncol. 2017. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Name | Antigen | Format | Indications (Year of First Approval) 1 |
|---|---|---|---|
| Unconjugated Antibodies | |||
| Atezolizumab | PD-L1 | Humanized IgG1 | Bladder, Non-small cell lung (2016), and Triple-negative breast (2019) cancers (2019) |
| Avelumab | PD-L1 | Human IgG1 | Urothelial Carcinoma (2017) and Merkel Cell Carcinoma (2017) |
| Bevacizumab | VEGF | Humanized IgG1 | Colorectal (2004), Non-small cell lung (2006), Renal (2009), Glioblastoma (2009), and Ovarian (2018) Cancers |
| Cemiplimab | PD-1 | Human IgG4 | Cutaneous squamous-cell carcinoma (2018) |
| Cetuximab | EGFR | Chimeric IgG1 | Colorectal cancer (2004) and Head and neck squamous cell carcinoma (2006) |
| Daratumumab | CD38 | Human IgG1 | Multiple Myeloma (2015) |
| Dinutuximab | GD2 | Chimeric IgG1 | Neuroblastoma (2015) |
| Durvalumab | PD-L1 | Human IgG1 | Bladder Cancer (2017) |
| Elotuzumab | SLAMF7 | Humanized IgG1 | Multiple Myeloma (2015) |
| Ipilimumab | CTLA-4 | Human IgG1 | Melanoma (2011) and Renal cell carcinoma (2018) |
| Isatuximab | CD38 | Chimeric IgG1 | Multiple Myeloma (2020) |
| Mogamulizumab | CCR4 | Humanized IgG1 | Cutaneous T-cell lymphoma (2018) |
| Necitumumab | EGFR | Human IgG1 | Non-small cell lung cancer (2015) |
| Nivolumab | PD-1 | Human IgG4 | Melanoma (2014), Lung (2015), and Renal (2018) cancers |
| Obinutuzumab | CD20 | Humanized IgG2 | Chronic lymphocytic leukemia (2013) |
| Ofatumumab | CD20 | Human IgG1 | Chronic lymphocytic leukemia (2014) |
| Olaratumab | PDGFRα | Human IgG1 | Sarcoma (2016) |
| Panitumumab | EGFR | Human IgG2 | Colorectal Cancer (2006) |
| Pembrolizumab | PD-1 | Humanized IgG4 | Melanoma (2014), Various (2015-) |
| Pertuzumab | HER2 | Humanized IgG1 | Breast cancer (2012) |
| Ramucirumab | VEGFR2 | Human IgG1 | Gastric cancer (2014) |
| Rituximab | CD20 | Chimeric IgG1 | B-Cell Lymphoma (1997) |
| Trastuzumab | HER2 | Humanized IgG1 | Breast cancer (1998) |
| Antibody–Drug Conjugates (ADCs) | |||
| Gemtuzumab ozogamicin | CD33 | Humanized ADC | Acute myeloid leukemia (2000) |
| Brentuximab vedotin | CD30 | Chimeric ADC | Hodgkin’s lymphoma and Anaplastic large-cell lymphoma (2011) |
| Trastuzumab emtansine | HER2 | Humanized ADC | Breast cancer (2013) |
| Inotuzumab ozogamicin | CD22 | Humanized ADC | Acute lymphoblastic leukemia (2017) |
| Polatuzumab vedotin | CD79B | Humanized ADC | B-Cell Lymphoma (2019) |
| Enfortumab vedotin | Nectin-4 | Human ADC | Bladder cancer (2019) |
| Trastuzumab deruxtecan | HER2 | Humanized ADC | Breast cancer (2019) |
| Sacituzumab govitecan | TROP2 | Humanized ADC | Triple negative breast cancer (2020) |
| Moxetumomab pasudotox | CD22 | Mouse ADC | Hairy-cell leukemia (2018) |
| Ibritumomab tiuxetan | CD20 | Mouse IgG1-Y90 or In111 | Non-Hodgkin’s lymphoma (2002) |
| Iodine (I131) tositumomab | CD20 | Mouse IgG2-I131 | Non-Hodgkin’s lymphoma (2003) |
| Blinatumomab | CD19, CD3 | Mouse BiTE | Acute lymphoblastic leukemia (2014) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34. https://doi.org/10.3390/antib9030034
Zahavi D, Weiner L. Monoclonal Antibodies in Cancer Therapy. Antibodies. 2020; 9(3):34. https://doi.org/10.3390/antib9030034
Chicago/Turabian StyleZahavi, David, and Louis Weiner. 2020. "Monoclonal Antibodies in Cancer Therapy" Antibodies 9, no. 3: 34. https://doi.org/10.3390/antib9030034
APA StyleZahavi, D., & Weiner, L. (2020). Monoclonal Antibodies in Cancer Therapy. Antibodies, 9(3), 34. https://doi.org/10.3390/antib9030034