IgE in the Pathogenesis of SLE: From Pathogenic Role to Therapeutic Target


Abstract
:1. Introduction
2. IgE Receptors and IgE in SLE
3. Autoantibodies of IgE Isotype in SLE: Specificities and Prevalence
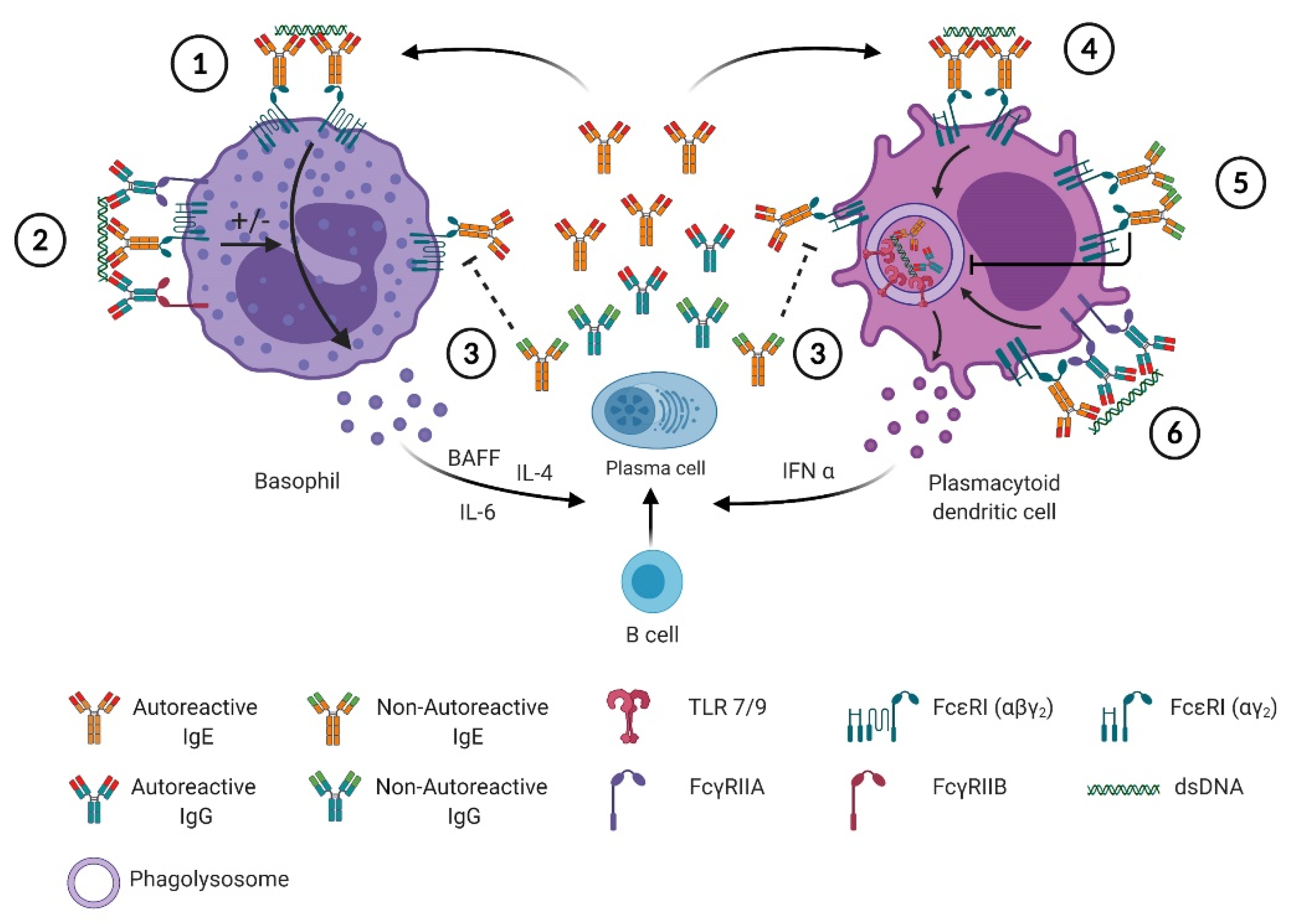
4. Autoreactive IgE and FcεRI-Bearing Cells in SLE
4.1. Mast Cells
4.2. Basophils
4.3. Plasmacytoid Dendritic Cells
4.4. Other under-Investigated Actors
5. IgE-Oriented Therapies
5.1. Omalizumab
5.2. Ligelizumab
5.3. Quilizumab
5.4. CSL362
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Kaul, A.; Gordon, C.; Crow, M.K.; Touma, Z.; Urowitz, M.B.; van Vollenhoven, R.; Ruiz-Irastorza, G.; Hughes, G. Systemic lupus erythematosus. Nat. Rev. Dis. Primers 2016, 2, 16039. [Google Scholar] [CrossRef]
- Dema, B.; Charles, N. Autoantibodies in SLE: Specificities, Isotypes and Receptors. Antibodies 2016, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.A.; Hsu, H.; Mountz, J.D. Autoreactive B cells in SLE, villains or innocent bystanders? Immunol. Rev. 2019, 292, 120–138. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Park, J.; Takamatsu, H.; Konaka, H.; Aoki, W.; Aburaya, S.; Ueda, M.; Nishide, M.; Koyama, S.; Hayama, Y.; et al. Apoptosis-derived membrane vesicles drive the cGAS–STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann. Rheum. Dis. 2018, 77, 1507–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dema, B.; Charles, N. Advances in mechanisms of systemic lupus erythematosus. Discov. Med. 2014, 17, 247–255. [Google Scholar] [PubMed]
- Charles, N.; Hardwick, D.; Daugas, E.; Illei, G.G.; Rivera, J. Basophils and the T helper 2 environment can promote the development of lupus nephritis. Nat. Med. 2010, 16, 701–707. [Google Scholar] [CrossRef] [Green Version]
- Pellefigues, C.; Dema, B.; Lamri, Y.; Saidoune, F.; Chavarot, N.; Lohéac, C.; Pacreau, E.; Dussiot, M.; Bidault, C.; Marquet, F.; et al. Prostaglandin D2 amplifies lupus disease through basophil accumulation in lymphoid organs. Nat. Commun. 2018, 9, 725. [Google Scholar] [CrossRef] [Green Version]
- Linge, P.; Fortin, P.R.; Lood, C.; Bengtsson, A.A.; Boilard, E. The non-haemostatic role of platelets in systemic lupus erythematosus. Nat. Rev. Rheumatol. 2018, 14, 195–213. [Google Scholar] [CrossRef]
- Koelsch, K.; Zheng, N.-Y.; Zhang, Q.; Duty, A.; Helms, C.; Mathias, M.D.; Jared, M.; Smith, K.; Capra, J.D.; Wilson, P.C. Mature B cells class switched to IgD are autoreactive in healthy individuals. J. Clin. Investig. 2007, 117, 1558–1565. [Google Scholar] [CrossRef] [Green Version]
- Villalta, D.; Bizzaro, N.; Bassi, N.; Zen, M.; Gatto, M.; Ghirardello, A.; Iaccarino, L.; Punzi, L.; Doria, A. Anti-dsDNA Antibody Isotypes in Systemic Lupus Erythematosus: IgA in Addition to IgG Anti-dsDNA Help to Identify Glomerulonephritis and Active Disease. PLoS ONE 2013, 8, e71458. [Google Scholar] [CrossRef] [PubMed]
- Miyawaki, S.; Ritchie, R.F. Heterogeneity of antinucleolar antibody and IgE antinuclear antibody in patients with systemic rheumatic diseases. J. Immunol. 1974, 113, 1346–1352. [Google Scholar] [PubMed]
- Camussi, G.; Tetta, C.; Benveniste, J. Detection of Basophil Sensitization by IgE Antibodies to Nuclear Antigens in Connective Tissue Diseases. Int. Arch. Allergy Immunol. 1982, 69, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Egido, J.; Crespo, M.S.; Lahoz, C.; Garcia, R.; Lopez-Trascasa, M.; Hernando, L. Evidence of an immediate hypersensitivity mechanism in systemic lupus erythematosus. Ann. Rheum. Dis. 1980, 39, 312–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishizaka, K.; Ishizaka, T. Identification of IgE. J. Allergy Clin. Immunol. 2016, 137, 1646–1650. [Google Scholar] [CrossRef] [Green Version]
- Lambrecht, B.N.; Hammad, H.; Fahy, J.V. The Cytokines of Asthma. Immunity 2019, 50, 975–991. [Google Scholar] [CrossRef]
- Kinet, J.-P. THE HIGH-AFFINITY IgE RECEPTOR (FcεRI): From Physiology to Pathology. Annu. Rev. Immunol. 1999, 17, 931–972. [Google Scholar] [CrossRef]
- Oettgen, H.C. Fifty years later: Emerging functions of IgE antibodies in host defense, immune regulation, and allergic diseases. J. Allergy Clin. Immunol. 2016, 137, 1631–1645. [Google Scholar] [CrossRef] [Green Version]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef]
- Maurer, M.; Altrichter, S.; Schmetzer, O.; Scheffel, J.; Church, M.K.; Metz, M. Immunoglobulin E-Mediated Autoimmunity. Front. Immunol. 2018, 9, 689. [Google Scholar] [CrossRef] [Green Version]
- Fitzsimmons, C.M.; Falcone, F.H.; Dunne, D.W. Helminth Allergens, Parasite-Specific IgE, and Its Protective Role in Human Immunity. Front. Immunol. 2014, 5, 61. [Google Scholar] [CrossRef] [Green Version]
- Karasuyama, H.; Tabakawa, Y.; Ohta, T.; Wada, T.; Yoshikawa, S. Crucial Role for Basophils in Acquired Protective Immunity to Tick Infestation. Front. Physiol. 2018, 9, 1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starkl, P.; Marichal, T.; Gaudenzio, N.; Reber, L.L.; Sibilano, R.; Tsai, M.; Galli, S.J. IgE antibodies, FcεRIα, and IgE-mediated local anaphylaxis can limit snake venom toxicity. J. Allergy Clin. Immunol. 2016, 137, 246–257.e11. [Google Scholar] [CrossRef] [Green Version]
- Orengo, J.M.; Radin, A.R.; Kamat, V.; Badithe, A.; Ben, L.H.; Bennett, B.L.; Zhong, S.; Birchard, D.; Limnander, A.; Rafique, A.; et al. Treating cat allergy with monoclonal IgG antibodies that bind allergen and prevent IgE engagement. Nat. Commun. 2018, 9, 1421. [Google Scholar] [CrossRef]
- Malbec, O.; Cassard, L.; Albanesi, M.; Ejönsson, F.; Mancardi, D.; Chicanne, G.; Payrastre, B.; Dubreuil, P.; Vivier, E.; Daëron, M. Trans-inhibition of activation and proliferation signals by Fc receptors in mast cells and basophils. Sci. Signal. 2016, 9, ra126. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Zhong, L.; Huang, C.; Long, J.; Ye, X.; Wu, J.; Dai, W.; Lv, W.; Xie, C.; Zhang, J. Cell-bound IgE and plasma IgE as a combined clinical diagnostic indicator for allergic patients. Sci. Rep. 2020, 10, 4700. [Google Scholar] [CrossRef] [PubMed]
- Atta, A.; Sousa, C.; Carvalho, E.; Sousa-Atta, M. Immunoglobulin E and systemic lupus erythematosus. Braz. J. Med Biol. Res. 2004, 37, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Parks, C.; Biagini, R.; Cooper, G.; Gilkeson, G.; Dooley, M.; Parks, C.G. Total serum IgE levels in systemic lupus erythematosus and associations with childhood onset allergies. Lupus 2010, 19, 1614–1622. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.A.; Klimek, G.A.; Ali, R. Allergy in systemic lupus erythematosus. IgE levels and reaginic phenomenon. Arthritis Rheum. 1976, 19, 669–676. [Google Scholar] [CrossRef]
- Wozniacka, A.; Sysa-Jedrzejowska, A.; Robak, E.; Samochocki, Z.; Zak-Prelich, M. Allergic diseases, drug adverse reactions and total immunoglobulin E levels in lupus erythematosus patients. Mediat. Inflamm. 2003, 12, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Sequeira, J.F.; Cesic, D.; Keser, G.; Bukelica, M.; Karanagnostis, S.; Khamashta, M.A.; Hughes, G.R. Allergic Disorders in Systemic Lupus Erythematosus. Lupus 1993, 2, 187–191. [Google Scholar] [CrossRef]
- Guo, R.; Zhou, Y.; Lu, L.-J.; Cao, L.; Cao, J. Atopy in children with juvenile systemic lupus erythematosus is associated with severe disease. PLoS ONE 2017, 12, e0177774. [Google Scholar] [CrossRef] [PubMed]
- Dema, B.; Charles, N.; Pellefigues, C.; Ricks, T.K.; Suzuki, R.; Jiang, C.; Scheffel, J.; Hasni, S.; Hoffman, V.; Jablonski, M.; et al. Immunoglobulin E plays an immunoregulatory role in lupus. J. Exp. Med. 2014, 211, 2159–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, M.J.; Calatayud, I.; Urquizu-Padilla, M.; Wijetilleka, S.; Kiani-Alikhan, S.; Karim, M.Y. Immunoglobulin abnormalities are frequent in patients with lupus nephritis. BMC Rheumatol. 2019, 3, 1–5. [Google Scholar] [CrossRef]
- Ter Borg, E.J.; Horst, G.; Hummel, E.J.; Limburg, P.C.; Kallenberg, C.G. Measurement of increases in anti-double-stranded DNA antibody levels as a predictor of disease exacerbation in systemic lupus erythematosus. A long-term, prospective study. Arthritis Rheum. 1990, 33, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Charles, N.; Watford, W.T.; Ramos, H.L.; Hellman, L.; Oettgen, H.C.; Gomez, G.; Ryan, J.J.; O’Shea, J.J.; Rivera, J. Lyn Kinase Controls Basophil GATA-3 Transcription Factor Expression and Induction of Th2 Cell Differentiation. Immunity 2009, 30, 533–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fish, S.C.; Donaldson, D.D.; Goldman, S.J.; Williams, C.M.M.; Kasaian, M.T. IgE Generation and Mast Cell Effector Function in Mice Deficient in IL-4 and IL-13. J. Immunol. 2005, 174, 7716–7724. [Google Scholar] [CrossRef] [Green Version]
- Atta, A.M.; Santiago, M.B.; Guerra, F.G.; Pereira, M.M.; De Sousa-Atta, M.L.B. Autoimmune Response of IgE Antibodies to Cellular Self-Antigens in Systemic Lupus Erythematosus. Int. Arch. Allergy Immunol. 2010, 152, 401–406. [Google Scholar] [CrossRef]
- Dema, B.; Pellefigues, C.; Hasni, S.; Gault, N.; Jiang, C.; Ricks, T.K.; Bonelli, M.M.; Scheffel, J.; Sacré, K.; Jablonski, M.; et al. Autoreactive IgE is Prevalent in Systemic Lupus Erythematosus and is Associated with Increased Disease Activity and Nephritis. PLoS ONE 2014, 9, e90424. [Google Scholar] [CrossRef]
- Sanjuan, M.; Henault, J.; Riggs, J.; Karnell, J.; Liarski, V.; Shirinian, L.; Xu, L.; Casey, K.; Smith, M.; Khatry, D.; et al. Self-reactive IgE exacerbates interferon responses associated with autoimmunity. Nat. Immunol. 2016, 17, 196–203. [Google Scholar] [CrossRef]
- Pan, Q.; Gong, L.; Xiao, H.; Feng, Y.; Li, L.; Deng, Z.; Ye, L.; Zheng, J.; Dickerson, C.A.; An, N.; et al. Basophil Activation-Dependent Autoantibody and Interleukin-17 Production Exacerbate Systemic Lupus Erythematosus. Front. Immunol. 2017, 8, 348. [Google Scholar] [CrossRef] [Green Version]
- Khoryati, L.; Augusto, J.-F.; Shipley, E.; Contin-Bordes, C.; Douchet, I.; Mitrovic, S.; Truchetet, M.-E.; Lazaro, E.; Duffau, P.; Couzi, L.; et al. IgE Inhibits Toll-like Receptor 7- and Toll-like Receptor 9-Mediated Expression of Interferon-α by Plasmacytoid Dendritic Cells in Patients With Systemic Lupus Erythematosus. Arthritis Rheumatol. 2016, 68, 2221–2231. [Google Scholar] [CrossRef] [PubMed]
- Wągrowska-Danilewicz, M. Quantitative analysis of interstitial mast cells in lupus and non-lupus membranous glomerulopathy. Pol. J. Pathol. 2001, 52, 211–217. [Google Scholar]
- Hiromura, K.; Kurosawa, M.; Yano, S.; Naruse, T. Tubulointerstitial mast cell infiltration in glomerulonephritis. Am. J. Kidney Dis. 1998, 32, 593–599. [Google Scholar] [CrossRef]
- Rascio, F.; Pontrelli, P.; Netti, G.S.; Manno, E.; Infante, B.; Simone, S.; Castellano, G.; Ranieri, E.; Seveso, M.; Cozzi, E.; et al. IgE-Mediated Immune Response and Antibody-Mediated Rejection. Clin. J. Am. Soc. Nephrol. 2020, 15, 1474–1483. [Google Scholar] [CrossRef]
- Inaba, Y.; Kanazawa, N.; Yoshimasu, T.; Shimokawa, T.; Nosaka, M.; Kondo, T.; Furukawa, F. Severer lupus erythematosus-like skin lesions in MRL/lpr mice with homozygous Kitwsh/wsh mutation. Mod. Rheumatol. 2018, 28, 319–326. [Google Scholar] [CrossRef]
- Lin, L.; Gerth, A.J.; Peng, S.L. Susceptibility of mast cell-deficient W/Wv mice to pristane-induced experimental lupus nephritis. Immunol. Lett. 2004, 91, 93–97. [Google Scholar] [CrossRef]
- Van Nieuwenhuijze, A.E.M.; Cauwe, B.; Klatt, D.; Humblet-Baron, S.; Liston, A. Lpr-induced systemic autoimmunity is unaffected by mast cell deficiency. Immunol. Cell Biol. 2015, 93, 841–848. [Google Scholar] [CrossRef]
- Cassard, L.; Jönsson, F.; Arnaud, S.; Daëron, M. Fcgamma receptors inhibit mouse and human basophil activation. J. Immunol. 2012, 189, 2995–3006. [Google Scholar] [CrossRef]
- Dema, B.; Lamri, Y.; Pellefigues, C.; Pacreau, E.; Saidoune, F.; Bidault, C.; Karasuyama, H.; Sacré, K.; Daugas, E.; Charles, N. Basophils contribute to pristane-induced Lupus-like nephritis model. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Wu, L.C.; Zarrin, A.A. The production and regulation of IgE by the immune system. Nat. Rev. Immunol. 2014, 14, 247–259. [Google Scholar] [CrossRef]
- Chasset, F.; Arnaud, L. Targeting interferons and their pathways in systemic lupus erythematosus. Autoimmun. Rev. 2018, 17, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Pascual, V.; Palucka, A.K. Autoimmunity through cytokine-induced dendritic cell activation. Immunity 2004, 20, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Gill, M.A.; Bajwa, G.; George, T.A.; Dong, C.C.; Dougherty, I.I.; Jiang, N.; Gan, V.N.; Gruchalla, R.S. Counterregulation between the FcepsilonRI pathway and antiviral responses in human plasmacytoid dendritic cells. J. Immunol. 2010, 184, 5999–6006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, J.T.; Bieneman, A.P.; Xiao, H.; Chichester, K.L.; Vasagar, K.; Saini, S.; Liu, M.C. TLR9- and FcepsilonRI-mediated responses oppose one another in plasmacytoid dendritic cells by down-regulating receptor expression. J. Immunol. 2005, 175, 5724–5731. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, J.T.; Chichester, K.L.; Bieneman, A.P. Toll-like receptor 9 suppression in plasmacytoid dendritic cells after IgE-dependent activation is mediated by autocrine TNF-α. J. Allergy Clin. Immunol. 2008, 121, 486–491. [Google Scholar] [CrossRef]
- Laurent, J.; Lagrue, G.; Sobel, A. Increased Serum IgE Levels in Patients with Lupus nephritis. Am. J. Nephrol. 1986, 6, 413–414. [Google Scholar] [CrossRef]
- James, K.M.; Peebles, R.S., Jr.; Hartert, T.V. Response to infections in patients with asthma and atopic disease: An epiphenomenon or reflection of host susceptibility? J. Allergy Clin. Immunol. 2012, 130, 343–351. [Google Scholar] [CrossRef]
- De Bandt, M. Anti-TNF-alpha-induced lupus. Arthritis Res. 2019, 21, 235. [Google Scholar] [CrossRef] [Green Version]
- Gruber, B.L.; Kaufman, L.D.; Marchese, M.J.; Roth, W.; Kaplan, A.P. Anti-ige autoantibodies in systemic lupus erythematosus. Arthritis Rheum. 1988, 31, 1000–1006. [Google Scholar] [CrossRef]
- Fiebiger, E.; Hammerschmid, F.; Stingl, G.; Maurer, D. Anti-FcepsilonRIalpha autoantibodies in autoimmune-mediated disorders. Identification of a structure-function relationship. J. Clin. Investig. 1998, 101, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Yu, H.; Jin, X.; Li, J.; Guo, H.; Shi, Q.; Yin, Z.; Xu, Y.; Wang, X.; Liu, R.; et al. Human Blood CD1c+ Dendritic Cells Encompass CD5high and CD5low Subsets That Differ Significantly in Phenotype, Gene Expression, and Functions. J. Immunol. 2017, 198, 1553–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammers, C.M.; Stanley, J.R. Mechanisms of Disease: Pemphigus and Bullous Pemphigoid. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 175–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, M.; Giménez-Arnau, A.M.; Sussman, G.; Metz, M.; Baker, D.R.; Bauer, A.; Bernstein, J.A.; Brehler, R.; Chu, C.-Y.; Chung, W.-H.; et al. Ligelizumab for Chronic Spontaneous Urticaria. N. Engl. J. Med. 2019, 381, 1321–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messingham, K.N.; Crowe, T.P.; Fairley, J.A. The Intersection of IgE Autoantibodies and Eosinophilia in the Pathogenesis of Bullous Pemphigoid. Front. Immunol. 2019, 10, 2331. [Google Scholar] [CrossRef] [PubMed]
- Pellefigues, C. IgE Autoreactivity in Atopic Dermatitis: Paving the Road for Autoimmune Diseases? Antibodies 2020, 9, 47. [Google Scholar] [CrossRef]
- Beck, L.A.; Marcotte, G.V.; MacGlashan, D.; Togias, A.; Saini, S.S. Omalizumab-induced reductions in mast cell Fce psilon RI expression and function. J. Allergy Clin. Immunol. 2004, 114, 527–530. [Google Scholar] [CrossRef]
- Lin, H.; Boesel, K.M.; Griffith, D.T.; Prussin, C.; Foster, B.; Romero, F.; Townley, R.; Casale, T.B. Omalizumab rapidly decreases nasal allergic response and FcεRI on basophils☆. J. Allergy Clin. Immunol. 2004, 113, 297–302. [Google Scholar] [CrossRef]
- Prussin, C.; Griffith, D.T.; Boesel, K.M.; Lin, H.; Foster, B.; Casale, T.B. Omalizumab treatment downregulates dendritic cell FcepsilonRI expression. J. Allergy Clin. Immunol. 2003, 112, 1147–1154. [Google Scholar] [CrossRef] [Green Version]
- Logsdon, S.L.; Oettgen, H.C. Anti-IgE therapy: Clinical utility and mechanistic insights. Curr. Top Microbiol. Immunol. 2015, 388, 39–61. [Google Scholar]
- Hasni, S.A.; Gupta, S.; Davis, M.; Poncio, E.; Bsn, Y.T.; Joyal, E.; Fike, A.; Manna, Z.; Auh, S.; Shi, Y.; et al. Safety and Tolerability of Omalizumab: A Randomized Clinical Trial of Humanized Anti-IgE Monoclonal Antibody in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019, 71, 1135–1140. [Google Scholar] [CrossRef]
- Gladman, D.D.; Ibañez, M.; Urowitz, M.B. Systemic lupus erythematosus disease activity index 2000. J. Rheumatol. 2002, 29, 288–291. [Google Scholar] [PubMed]
- Yee, C.-S.; Farewell, V.; Isenberg, D.A.; Prabu, A.; Sokoll, K.; Teh, L.-S.; Rahman, A.; Bruce, I.N.; Griffiths, B.; Akil, M.; et al. Revised British Isles Lupus Assessment Group 2004 index: A reliable tool for assessment of systemic lupus erythematosus activity. Arthritis Rheum. 2006, 54, 3300–3305. [Google Scholar] [CrossRef]
- Gasser, P.; Tarchevskaya, S.S.; Guntern, P.; Brigger, D.; Ruppli, R.; Zbären, N.; Kleinboelting, S.; Heusser, C.; Jardetzky, T.S.; Eggel, A. The mechanistic and functional profile of the therapeutic anti-IgE antibody ligelizumab differs from omalizumab. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arm, J.P.; Bottoli, I.; Skerjanec, A.; Floch, D.; Groenewegen, A.; Maahs, S.; Owen, C.E.; Jones, I.; Lowe, P.J. Pharmacokinetics, pharmacodynamics and safety of QGE 031 (ligelizumab), a novel high-affinity anti-IgE antibody, in atopic subjects. Clin. Exp. Allergy 2014, 44, 1371–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauvreau, G.M.; Harris, J.M.; Boulet, L.-P.; Scheerens, H.; Fitzgerald, J.M.; Putnam, W.S.; Cockcroft, D.W.; Davis, B.E.; Leigh, R.; Zheng, Y.; et al. Targeting membrane-expressed IgE B cell receptor with an antibody to the M1 prime epitope reduces IgE production. Sci. Transl. Med. 2014, 6, 243ra85. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.M.; Maciuca, R.; Bradley, M.S.; Cabanski, C.R.; Scheerens, H.; Lim, J.J.; Cai, F.; Kishnani, M.; Liao, X.C.; Samineni, D.; et al. A randomized trial of the efficacy and safety of quilizumab in adults with inadequately controlled allergic asthma. Respir. Res. 2016, 17, 29. [Google Scholar] [CrossRef] [Green Version]
- Oon, S.; Huynh, H.; Tai, T.Y.; Ng, M.; Monaghan, K.; Biondo, M.; Vairo, G.; Maraskovsky, E.; Nash, A.D.; Wicks, I.P.; et al. A cytotoxic anti-IL-3Ralpha antibody targets key cells and cytokines implicated in systemic lupus erythematosus. JCI Insight 2016, 1, e86131. [Google Scholar] [CrossRef] [Green Version]
- Klavdianou, K.; Lazarini, A.; Fanouriakis, A. Targeted Biologic Therapy for Systemic Lupus Erythematosus: Emerging Pathways and Drug Pipeline. BioDrugs 2020, 34, 133–147. [Google Scholar] [CrossRef]
- Furie, R.; Werth, V.P.; Merola, J.F.; Stevenson, L.; Reynolds, T.L.; Naik, H.; Wang, W.; Christmann, R.; Gardet, A.; Pellerin, A.; et al. Monoclonal antibody targeting BDCA2 ameliorates skin lesions in systemic lupus erythematosus. J. Clin. Investig. 2019, 129, 1359–1371. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Autoreactive IgE Specificity | Prevalence (%) | Ref. | Activation of Basophils or pDC | Associated with | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| Basophils | Ref. | pDC | Ref. | |||||
| dsDNA | 48.3 | [37] | yes | [12] | yes | [39] | active LN and active disease | [6,38,39,40] |
| 35.4 | [38] | |||||||
| 54.4 | [39] | |||||||
| ssDNA | - | - | yes | [12] | NT | - | - | - |
| Sm | 48.3 | [37] | yes | [12] | NT | - | - | - |
| 7.5 | [38] | |||||||
| SSA/Ro | 48.3 | [37] | yes | [12] | NT | - | ||
| 8.5 | [38] | |||||||
| SSB/La | 6.9 | [37] | [12] | mild and active LN | [38] | |||
| 4.1 | [38] | |||||||
| nRNP | 62.1 | [37] | yes | [12] | NT | - | - | - |
| Nucleosome | 79.3 | [37] | ||||||
| CLIP4 | 10.0 | [38] | NT | - | NT | - | - | - |
| APEX nuclease 1 | 0.2 | [38] | NT | - | NT | - | - | - |
| MPG | 0.2 | [38] | NT | - | NT | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamri, Y.; Charles, N. IgE in the Pathogenesis of SLE: From Pathogenic Role to Therapeutic Target. Antibodies 2020, 9, 69. https://doi.org/10.3390/antib9040069
Lamri Y, Charles N. IgE in the Pathogenesis of SLE: From Pathogenic Role to Therapeutic Target. Antibodies. 2020; 9(4):69. https://doi.org/10.3390/antib9040069
Chicago/Turabian StyleLamri, Yasmine, and Nicolas Charles. 2020. "IgE in the Pathogenesis of SLE: From Pathogenic Role to Therapeutic Target" Antibodies 9, no. 4: 69. https://doi.org/10.3390/antib9040069
APA StyleLamri, Y., & Charles, N. (2020). IgE in the Pathogenesis of SLE: From Pathogenic Role to Therapeutic Target. Antibodies, 9(4), 69. https://doi.org/10.3390/antib9040069