Convenient Asymmetric Synthesis of Fmoc-(S)-6,6,6-Trifluoro-Norleucine

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
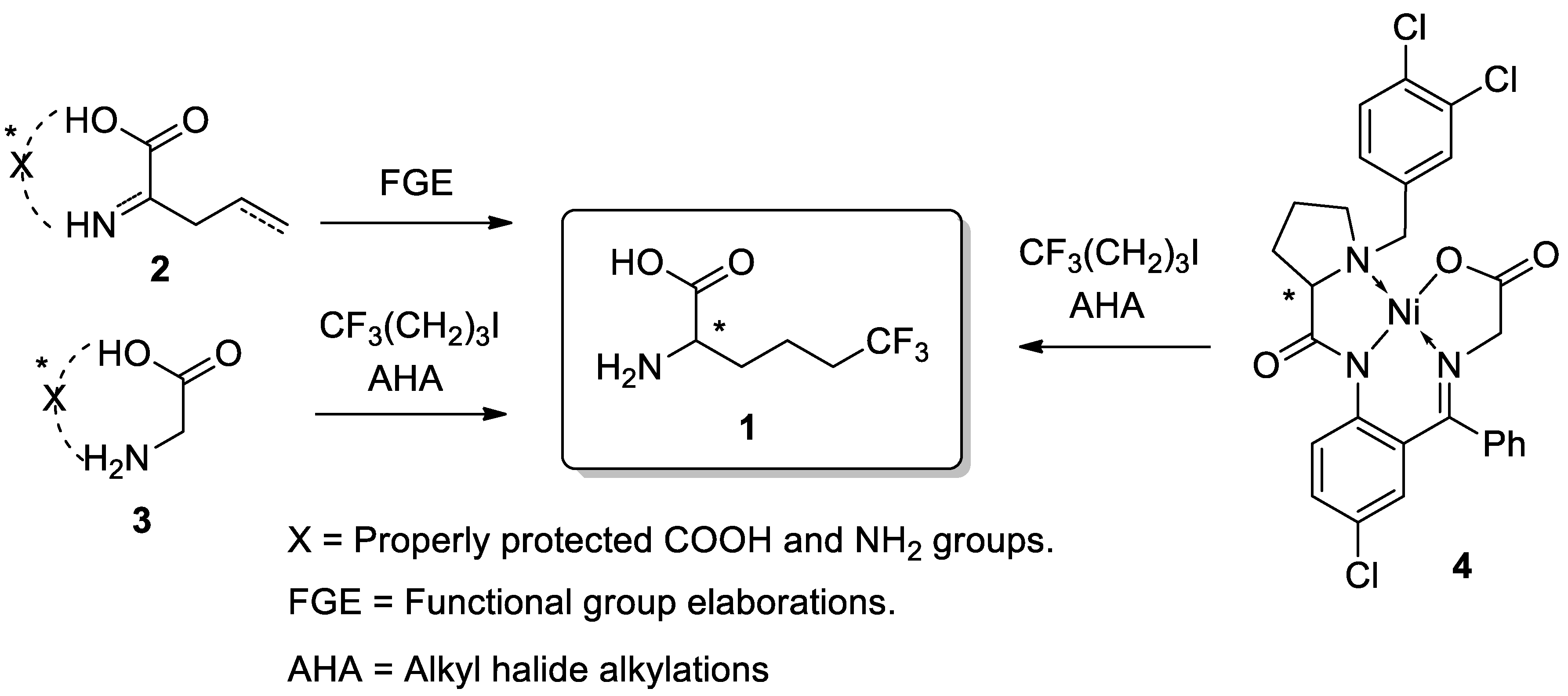
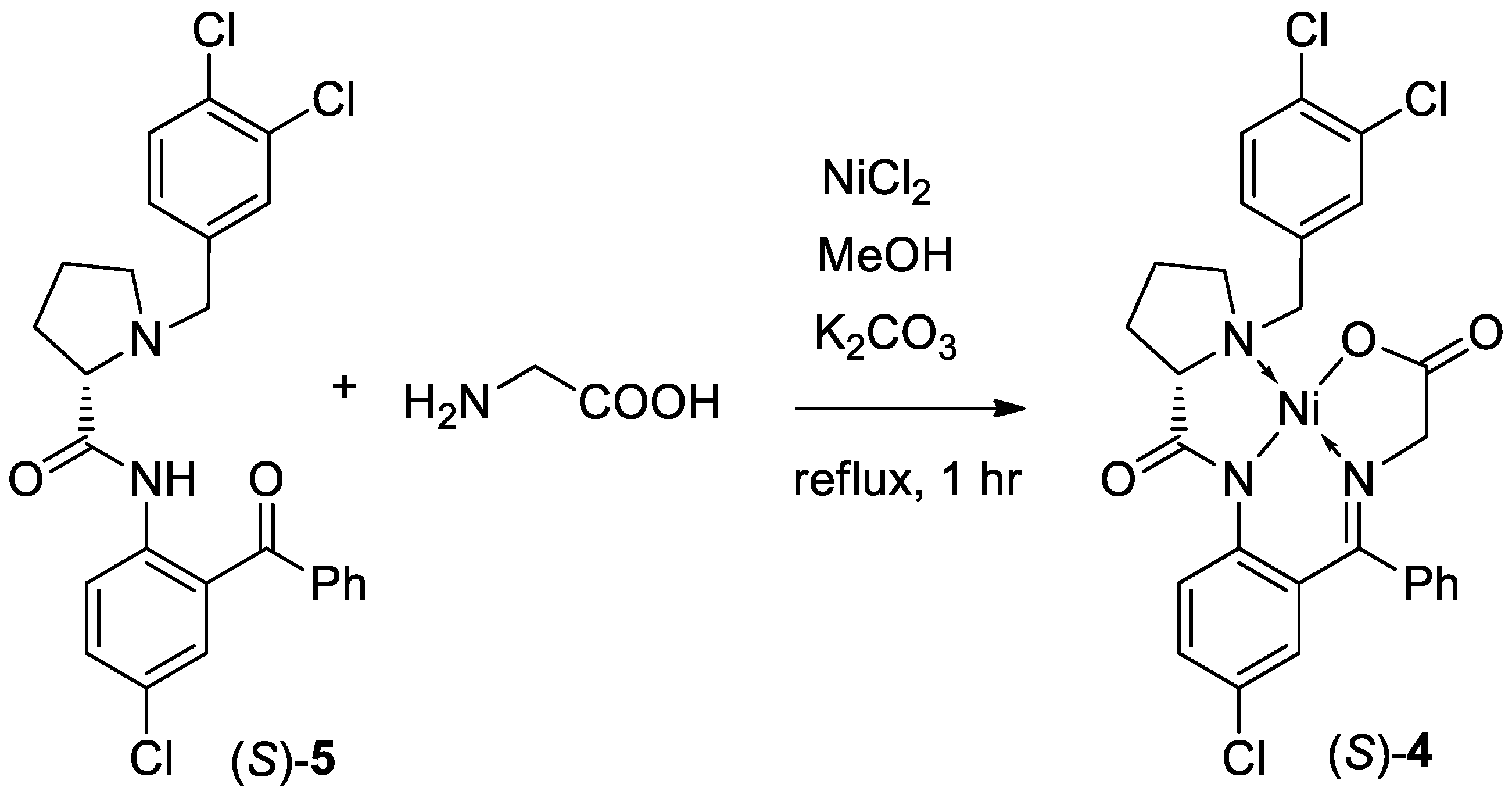
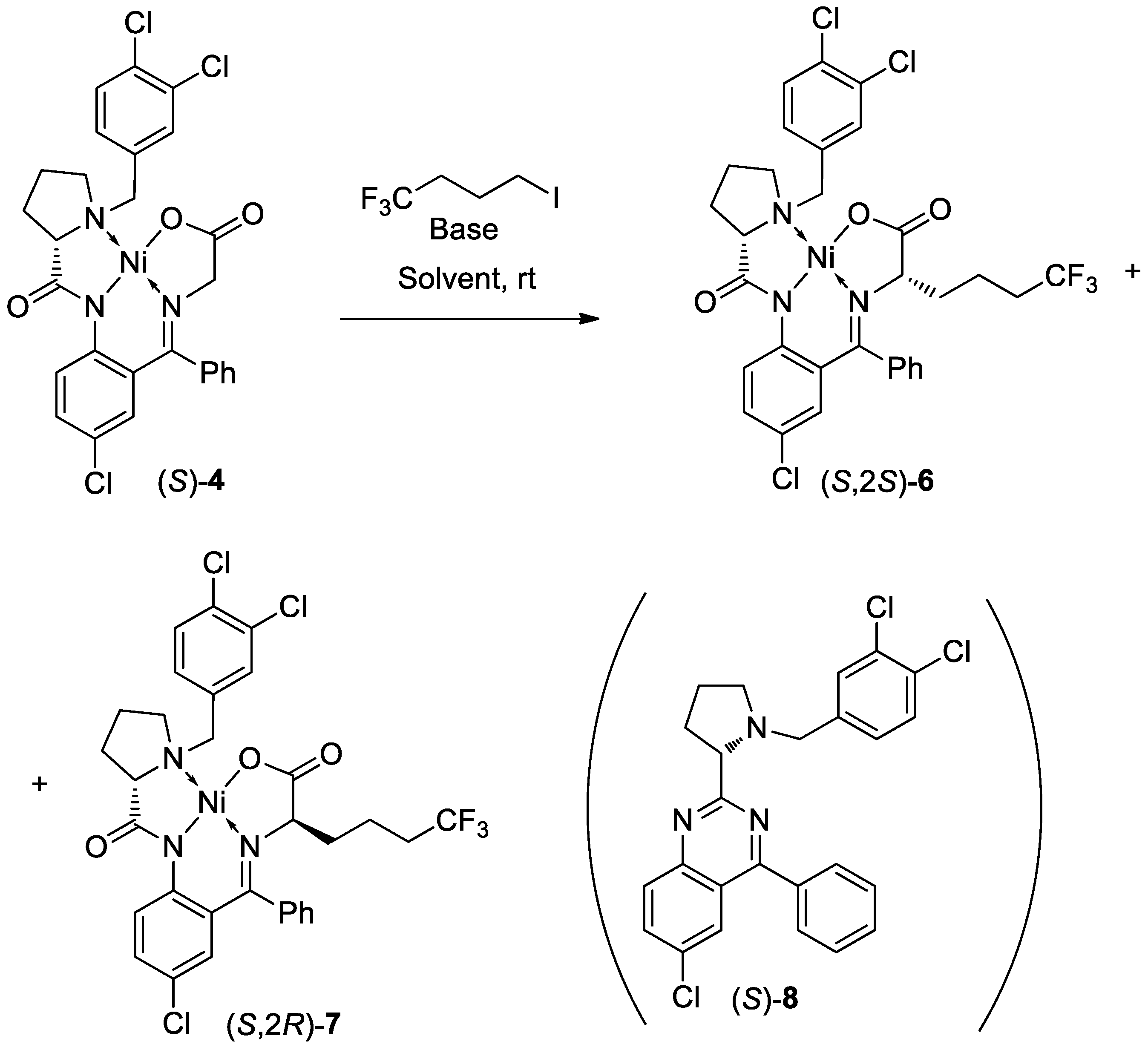
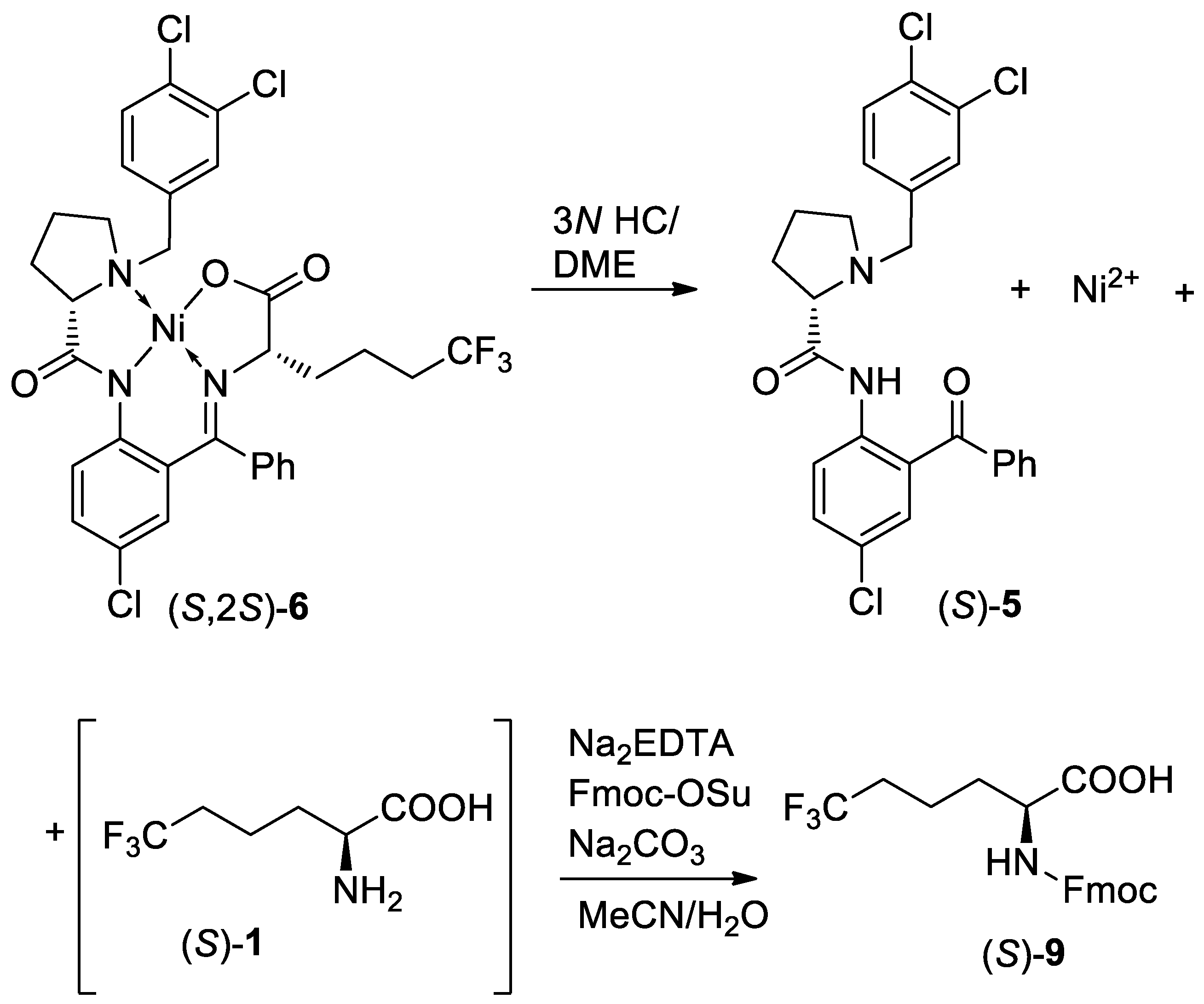
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Henninot, A.; Collins, J.C.; Nuss, J.M. The current state of peptide drug discovery: Back to the future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef]
- Blaskovich, M.A.T. Unusual amino acids in medicinal chemistry. J. Med. Chem. 2016, 59, 10807–10836. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.S. Unnatural amino acids in drug discovery. Chim. Oggi 2003, 21, 65–68. [Google Scholar]
- Hodgson, D.R.W.; Sanderson, J.M. The synthesis of peptides and proteins containing non-natural amino acids. Chem. Soc. Rev. 2004, 33, 422–430. [Google Scholar] [CrossRef]
- Sato, T.; Izawa, K.; Aceña, J.L.; Liu, H.; Soloshonok, V.A. Tailor-Made α-Amino Acids in the Pharmaceutical Industry: Synthetic Approaches to (1R, 2S)-1-Amino-2-vinylcyclopropane-1-carboxylic Acid (Vinyl-ACCA). Eur. J. Org. Chem. 2016, 2757–2774. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Aceña, J.L.; Moriwaki, H.; Sato, T.; Soloshonok, V.A. Asymmetric synthesis of α-amino acids via homologation of Ni (II) complexes of glycine Schiff bases; Part 1: Alkyl halide alkylations. Amino Acids 2013, 45, 691–718. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Izawa, K. Asymmetric Synthesis and Application of alpha-Amino Acids; ACS Symposium Series 1009; Oxford University Press: Oxford, UK, 2009. [Google Scholar]
- Soloshonok, V.A.; Sorochinsky, A.E. Practical Methods for the Synthesis of Symmetrically α,α-Disubstituted-α-Amino Acids. Synthesis 2010, 14, 2319–2344. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Cai, C.; Hruby, V.J.; Meervelt, L.V. Asymmetric Synthesis of Novel Highly Sterically Constrained (2S,3S)-3-Methyl-3-Trifluoromethyl- and (2S,3S,4R)-3-Trifluoromethyl-4-Methylpyroglutamic Acids. Tetrahedron 1999, 55, 12045–12058. [Google Scholar] [CrossRef]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef]
- Zhu, W.; Wang, J.; Wang, S.; Gu, Z.; Aceña, J.L.; Izawa, K.; Liu, H.; Soloshonok, V.A. Recent advances in the trifluoromethylation methodology and new CF3-containing drugs. J. Fluorine Chem. 2014, 167, 37–54. [Google Scholar] [CrossRef]
- Smits, R.; Cadicamo, C.D.; Burger, K.; Koksch, B. Synthetic strategies to α-trifluoromethyl and α-difluoromethyl substituted α-amino acids. Chem. Soc. Rev. 2008, 37, 1727–1739. [Google Scholar] [CrossRef] [PubMed]
- Kukhar, V.P.; Sorochinsky, A.E.; Soloshonok, V.A. Practical synthesis of fluorine containing alpha- and beta-amino acids: Recipes from Kiev, Ukraine. Future Med. Chem. 2009, 1, 793–819. [Google Scholar] [CrossRef] [PubMed]
- Sorochinsky, A.E.; Soloshonok, V.A. Asymmetric synthesis of fluorine-containing amines, amino alcohols, α- and β-amino acids mediated by chiral sulfinyl group. J. Fluorine Chem. 2010, 131, 127–139. [Google Scholar] [CrossRef]
- Tarui, A.; Sato, K.; Omote, M.; Kumadaki, I.; Ando, A. Stereoselective synthesis of α-fluorinated amino acid derivatives. Adv. Synth. Catal. 2010, 352, 2733–2744. [Google Scholar] [CrossRef]
- Czekelius, C.; Tzschucke, C.C. Synthesis of halogenated carboxylic acids and amino acids. Synthesis 2010, 4, 543–566. [Google Scholar] [CrossRef]
- Qiu, X.-L.; Qing, F.-L. Recent advances in the synthesis of fluorinated amino acids. Eur. J. Org. Chem. 2011, 2011, 3261–3278. [Google Scholar] [CrossRef]
- Turcheniuk, K.V.; Kukhar, V.P.; Roeschenthaler, G.-V.; Acena, J.L.; Soloshonok, V.A.; Sorochinsky, A.E. Recent advances in the synthesis of fluorinated aminophosphonates and aminophosphonic acids. RSC Adv. 2013, 3, 6693–6716. [Google Scholar] [CrossRef]
- Aceña, J.L.; Sorochinsky, A.E.; Soloshonok, V.A. Recent Advances in the Asymmetric Synthesis of α-(Trifluoromethyl)-Containing α-Amino Acids. Synthesis 2012, 44, 1591–1602. [Google Scholar] [CrossRef]
- Aceña, J.L.; Sorochinsky, A.E.; Moriwaki, H.; Sato, T.; Soloshonok, V.A. Synthesis of fluorine containing α-amino acids in enantiomerically pure form via homologation of Ni(II) complexes of glycine and alanine Schiff bases. J. Fluorine Chem. 2013, 155, 21–38. [Google Scholar] [CrossRef]
- Mikami, K.; Fustero, S.; Sánchez-Roselló, M.; Aceña, J.L.; Soloshonok, V.A.; Sorochinsky, A.E. Synthesis of fluorinated beta-amino acids. Synthesis 2011, 19, 3045–3079. [Google Scholar]
- Han, J.; Sorochinsky, A.E.; Ono, T.; Soloshonok, V.A. Biomimetic Transamination—A Metal-Free Alternative to the Reductive Amination. Application for Generalized Preparation of Fluorine-Containing Amines and Amino Acids. Curr. Org. Synth. 2011, 8, 281–294. [Google Scholar] [CrossRef]
- Aceña, J.L.; Simon-Fuentes, A.; Santos, F. Recent Developments in the Synthesis of Fluorinated β-Amino Acids. Curr. Org. Chem. 2010, 14, 928–949. [Google Scholar] [CrossRef]
- Dhillon, S. Ivosidenib: First Global Approval. Drugs 2018, 78, 1509–1516. [Google Scholar] [CrossRef]
- Urquhart, L. FDA new drug approvals in Q3 2018. Nat. Rev. Drug Discov. 2018, 17, 799. [Google Scholar]
- Scott, L.J. Eravacycline: A Review in Complicated Intra-Abdominal Infections. Drugs 2019, 79, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Dacomitinib: First Global Approval. Drugs 2018, 78, 1947–1953. [Google Scholar] [CrossRef] [PubMed]
- Tsushima, T.; Kawada, K.; Ishihara, S.; Uchida, N.; Shiratori, O.; Higaki, J.; Hirata, M. Fluorine containing amino acids and their derivatives. 7. Synthesis and antitumor activity of α- and γ-substituted methotrexate analogs. Tetrahedron 1988, 44, 5375–5387. [Google Scholar] [CrossRef]
- Shu, M.; Yu, R.; Zhang, Y.; Wang, J.; Yang, L.; Wang, L.; Lin, Z. Predicting the activity of antimicrobial peptides with amino acid topological information. Med. Chem. 2013, 9, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Ojima, I.; Jameison, F.A.; Pete, B.; Radunz, H.; Schittenhelm, C.; Lindner, H.J.; Emith, A.E. Design, synthesis and enzyme inhibitory activities of new trifluoromethyl-containing inhibitors for angiotensin converting enzyme. Drug Des. Discov. 1994, 11, 91–113. [Google Scholar] [PubMed]
- Borozan, S.Z.; Zlatović, M.V.; Stojanović, S.Đ. Anion–π interactions in complexes of proteins and halogen-containing amino acids. J. Biol. Inorg. Chem. 2016, 21, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.; Eriksson, L.; Jonsson, J.; Sjöström, M.; Wold, S. New Chemical Descriptors Relevant for the Design of Biologically Active Peptides. A Multivariate Characterization of 87 Amino Acids. J. Med. Chem. 1998, 41, 2481–2491. [Google Scholar] [CrossRef] [PubMed]
- van Hest, J.C.M.; Kiick, K.L.; Tirrell, D.A. Efficient Incorporation of Unsaturated Methionine Analogues into Proteins in Vivo. J. Am. Chem. Soc. 2000, 122, 1282–1288. [Google Scholar] [CrossRef]
- Kiick, K.L.; Tirrell, D.A. Protein Engineering by In Vivo Incorporation of Non-Natural Amino Acids: Control of Incorporation of Methionine Analogues by Methionyl-tRNA Synthetase. Tetrahedron 2000, 56, 9487–9493. [Google Scholar] [CrossRef]
- Borozan, S.Z.; Stojanović, S.Đ. Halogen bonding in complexes of proteins and non-natural amino acids. Comput. Biol. Chem. 2013, 47, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Wadhwani, P.; Strandberg, E.; Heidenreich, N.; Bürck, J.; Fanghänel, S.; Ulrich, A.S. Self-Assembly of Flexible β-Strands into Immobile Amyloid-Like β-Sheets in Membranes As Revealed by Solid-State 19F NMR. J. Am. Chem. Soc. 2012, 134, 6512–6515. [Google Scholar] [CrossRef]
- Tkachenko, A.N.; Mykhailiuk, P.K.; Afonin, S.; Radchenko, D.S.; Kubyshkin, V.S.; Ulrich, A.S.; Komarov, I.V. 19F NMR Label to Substitute Polar Amino Acids in Peptides: A CF3-Substituted Analogue of Serine and Threonine. Angew. Chem. Int. Ed. 2013, 52, 1486–1489. [Google Scholar] [CrossRef]
- Gfeller, D.; Michielin, O.; Zoete, V. Expanding molecular modeling and design tools to non-natural sidechains. J. Comput. Chem. 2012, 33, 1525–1535. [Google Scholar] [CrossRef]
- Li, S.G.; Portela-Cubillo, F.; Zard, S.Z. A Convergent Synthesis of Enantiopure Open-Chain, Cyclic, and Fluorinated α-Amino Acids. Org. Lett. 2016, 18, 1888–1891. [Google Scholar] [CrossRef]
- Ojima, I.; Kato, K.; Nakahashi, K.; Fuchikami, T.; Fujita, M. New and effective routes to fluoro analogs of aliphatic and aromatic amino acids. J. Org. Chem. 1989, 54, 4511–4522. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Kukhar, V.P. Biomimetic Transamination of α-Keto Perfluorocarboxylic Esters. An Efficient Preparative Synthesis of β,β,β-Trifluoroalanine. Tetrahedron 1997, 53, 8307–8314. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Kirilenko, A.G.; Kukhar, V.P.; Resnati, G. Transamination of Fluorinated β-Keto Carboxylic Esters. A Biomimetic Approach to β-Polyfluoroalkyl-β-Amino Acids. Tetrahedron Lett. 1993, 34, 3621–3624. [Google Scholar] [CrossRef]
- Peng, W.; Wan, J.; Xie, B.; Ma, X. 9-Amino-(9-deoxy) cinchona alkaloid-derived new chiral phase-transfer catalysts. Org. Biomol. Chem. 2014, 12, 8336–8345. [Google Scholar] [CrossRef] [PubMed]
- Scott, W.L.; Alsina, J.; Audu, C.O.; Babaev, E.; Cook, L.; Dage, J.L.; Goodwin, L.A.; Martynow, J.G.; Matosiuk, D.; Royo, M.; et al. Distributed Drug Discovery, Part 2: Global Rehearsal of Alkylating Agents for the Synthesis of Resin-Bound Unnatural Amino Acids and Virtual D3 Catalog Construction. J. Comb. Chem. 2009, 11, 14–33. [Google Scholar] [CrossRef]
- Yajima, T.; Nagano, H. Photoinduced Diastereoselective Addition of Perfluoroalkyl Iodides to Acrylic Acid Derivatives for the Synthesis of Fluorinated Amino Acids. Org. Lett. 2007, 9, 2513–2515. [Google Scholar] [CrossRef]
- Larsson, U.; Carlson, R.; Leroy, J. Synthesis of amino acids with modified principal properties 1. Amino acids with fluorinated side chains. Acta Chem. Scand. 1993, 47, 380–390. [Google Scholar] [CrossRef]
- Wang, J.; Lin, D.; Zhou, S.; Soloshonok, V.A.; Liu, H. Asymmetric Synthesis of Sterically Constrained Linear Trifluoromethyl Containing Amino Acids via Alkylation of Chiral Equivalents of Nucleophilic Glycine and Alanine. J. Org. Chem. 2011, 76, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Gerus, I.I.; Yagupolskii, Y.L.; Kukhar, V.P. Fluorine-Containing Amino Acids. III. α-Trifluoromethyl-α-Amino Acids. Zh. Org. Khim. 1987, 23, 2308–2313. [Google Scholar]
- Soloshonok, V.A.; Ohkura, H.; Yasumoto, M. Operationally Convenient Asymmetric Synthesis of (S)- and (R)-3-Amino-4,4,4-trifluorobutanoic Acid. Part II: Enantioselective Biomimetic Transamination of 4,4,4-Trifluoro-3-oxo-N-[(R)-1-phenylethyl)butanamide. J. Fluorine Chem. 2006, 127, 930–935. [Google Scholar] [CrossRef]
- Röschenthaler, G.-V.; Kukhar, V.P.; Kulik, I.B.; Belik, M.Y.; Sorochinsky, A.E.; Rusanov, E.B.; Soloshonok, V.A. Asymmetric synthesis of phosphonotrifluoroalanine and its derivatives using N-tert-butanesulfinyl imine derived from fluoral. Tetrahedron Lett. 2012, 53, 539–542. [Google Scholar] [CrossRef]
- Turcheniuk, K.V.; Poliashko, K.O.; Kukhar, V.P.; Rozhenko, A.B.; Soloshonok, V.A.; Sorochinsky, A.E. Efficient asymmetric synthesis of trifluoromethylated β-aminophosphonates and their incorporation into dipeptides. Chem. Commun. 2012, 48, 11519–11521. [Google Scholar] [CrossRef] [PubMed]
- Soloshonok, V.A.; Cai, C.; Hruby, V.J. A Practical Asymmetric Synthesis of Enantiomerically Pure 3-Substituted Pyroglutamic Acids and Related Compounds. Angew. Chem. Int. Ed. 2000, 39, 2172–2175. [Google Scholar] [CrossRef]
- Yamada, T.; Okada, T.; Sakaguchi, K.; Ohfune, Y.; Ueki, H.; Soloshonok, V.A. Efficient Asymmetric Synthesis of Novel 4-Substituted and Configurationally Stable Analogs of Thalidomide. Org. Lett. 2006, 8, 5625–5628. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kitagawa, O.; Wzorek, A.; Klika, K.D.; Soloshonok, V.A. The self-disproportionation of enantiomers (SDE): A menace or an opportunity? Chem. Sci. 2018, 9, 1718–1739. [Google Scholar] [CrossRef]
- Han, J.; Nelson, D.J.; Sorochinsky, A.E.; Soloshonok, V.A. Self-Disproportionation of Enantiomers via Sublimation; New and Truly Green Dimension in Optical Purification. Curr. Org. Synth. 2011, 8, 310–317. [Google Scholar] [CrossRef]
- Han, J.; Wzorek, A.; Kwiatkowska, M.; Soloshonok, V.A.; Klika, K.D. The self-disproportionation of enantiomers (SDE) of amino acids and their derivatives. Amino Acids 2019. [Google Scholar] [CrossRef] [PubMed]
- Aceña, J.L.; Sorochinsky, A.E.; Soloshonok, V.A. Asymmetric synthesis of α-amino acids via homologation of Ni(II) complexes of glycine Schiff bases. Part 3: Michael addition reactions and miscellaneous transformations. Amino Acids 2014, 46, 2047–2073. [Google Scholar] [CrossRef] [PubMed]
- Sorochinsky, A.E.; Aceña, J.L.; Moriwaki, H.; Sato, T.; Soloshonok, V.A. Asymmetric synthesis of α-amino acids via homologation of Ni(II) complexes of glycine Schiff bases. Part 2: Aldol, Mannich addition reactions, deracemization and (S) to (R) interconversion of α-amino acids. Amino Acids 2013, 45, 1017–1033. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Song, X.; Wang, J.; Moriwaki, H.; Soloshonok, V.A.; Liu, H. Recent approaches for asymmetric synthesis of α-amino acids via homologation of Ni(II) complexes. Amino Acids 2017, 49, 1487–1520. [Google Scholar] [CrossRef] [PubMed]
- Ellis, T.K.; Ueki, H.; Yamada, T.; Ohfune, Y.; Soloshonok, V.A. The Design, Synthesis and Evaluation of a New Generation of Modular Nucleophilic Glycine Equivalents for the Efficient Synthesis of Sterically Constrained α-Amino Acids. J. Org. Chem. 2006, 71, 8572–8578. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Ueki, H.; Ellis, T.K.; Yamada, T.; Ohfune, Y. Application of Modular Nucleophilic Glycine Equivalents for Truly Practical Asymmetric Synthesis of β-Substituted Pyroglutamic Acids. Tetrahedron Lett. 2005, 46, 1107–1110. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Ellis, T.K.; Ueki, H.; Ono, T. Resolution/Deracemization of Chiral α-Amino Acids Using Resolving Reagents with Flexible Stereogenic Centers. J. Am. Chem. Soc. 2009, 131, 7208–7209. [Google Scholar] [CrossRef]
- Takeda, R.; Kawamura, A.; Kawashima, A.; Sato, T.; Moriwaki, H.; Izawa, K.; Akaji, K.; Wang, S.; Liu, H.; Aceña, J.L.; et al. Chemical Dynamic Kinetic Resolution and (S)/(R)-Interconversion of Unprotected α-Amino Acids. Angew. Chem. Int. Ed. 2014, 53, 12214–12217. [Google Scholar] [CrossRef]
- Bergagnini, M.; Fukushi, K.; Han, J.; Shibata, N.; Roussel, C.; Ellis, T.K.; Aceña, J.L.; Soloshonok, V.A. NH-type of chiral Ni(II) complexes of glycine Schiff base: Design, structural evaluation, reactivity and synthetic applications. Org. Biomol. Chem. 2014, 12, 1278–1291. [Google Scholar] [CrossRef]
- Nian, Y.; Wang, J.; Moriwaki, H.; Soloshonok, V.A.; Liu, H. Analysis of crystallographic structures of Ni(II) complexes of α-amino acid Schiff bases; Elucidation of the substituents effect on stereochemical preferences. Dalton Trans. 2017, 46, 4191–4198. [Google Scholar] [CrossRef]
- Tang, X.; Soloshonok, V.A.; Hruby, V.J. Convenient Asymmetric Synthesis of Enantiomerically Pure 2′,6′-Dimethyltyrosine (DMT) via Alkylation of Chiral Nucleophilic Glycine Equivalent. Tetrahedron Asymmetry 2000, 11, 2917–2925. [Google Scholar] [CrossRef]
- Nian, Y.; Wang, J.; Zhou, S.; Wang, S.; Moriwaki, H.; Kawashima, A.; Soloshonok, V.A.; Liu, H. Recyclable Ligands for the Non-Enzymatic Dynamic Kinetic Resolution of Challenging α-Amino Acids. Angew. Chem. Int. Ed. 2015, 54, 12918–12922. [Google Scholar] [CrossRef] [PubMed]
- Nian, Y.; Wang, J.; Zhou, S.; Dai, W.; Wang, S.; Moriwaki, H.; Kawashima, A.; Soloshonok, V.A.; Liu, H. Purely Chemical Approach for Preparation of D-alpha-amino Acids via (S)-to-(R)-interconversion of Unprotected Tailor-made alpha-amino Acids. J. Org. Chem. 2016, 81, 3501–3508. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Wang, J.; Chen, X.; Aceña, J.L.; Soloshonok, V.A.; Liu, H. Chemical Kinetic Resolution of Unprotected β-Substituted-β-Amino Acids Using Recyclable Chiral Ligands. Angew. Chem. Int. Ed. 2014, 53, 7883–7886. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Hiramatsu, T.; Takeda, R.; Moriwaki, H.; Abe, H.; Han, J.; Soloshonok, V.A. Expedient Asymmetric Synthesis of (S)-2-Amino-4,4,4-trifluorobutanoic Acid via Alkylation of Chiral Nucleophilic Glycine Equivalent. Org. Process. Res. Dev. 2019. [Google Scholar] [CrossRef]
- Romoff, T.T.; Palmer, A.B.; Mansour, N.; Creighton, C.J.; Miwa, T.; Ejima, Y.; Moriwaki, H.; Soloshonok, V.A. Scale-up Synthesis of (R)- and (S)-N-(2-benzoyl-4-chlorophenyl)-1-(3,4-dichlorobenzyl)pyrrolidine-2-carboxamide hydrochloride, a Versatile Reagent for Preparation of Tailor-made α- and β-Amino Acids in Enantiomerically Pure Form. Org. Process. Res. Dev. 2017, 21, 732–739. [Google Scholar] [CrossRef]
- Ueki, H.; Ellis, T.K.; Martin, C.H.; Soloshonok, V.A. Efficient Large-Scale Synthesis of Picolinic Acid Derived Ni(II)-Complexes of Glycine. Eur. J. Org. Chem. 2003, 2003, 1954–1957. [Google Scholar] [CrossRef]
- Ellis, T.K.; Hochla, V.M.; Soloshonok, V.A. Efficient Synthesis of 2-Aminoindane-2-Carboxylic Acid via Dialkylation of Nucleophilic Glycine Equivalent. J. Org. Chem. 2003, 68, 4973–4976. [Google Scholar] [CrossRef] [PubMed]
- Houck, D.; Aceña, J.L.; Soloshonok, V.A. Alkylations of Chiral Nickel(II) Complexes of Glycine under Phase-Transfer Conditions. Helv. Chim. Acta 2012, 95, 2672–2679. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Ueki, H. Design, Synthesis and Characterization of Binuclear Ni(II) Complexes with Inherent Helical Chirality. J. Am. Chem. Soc. 2007, 129, 2426–2427. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Ono, T.; Ueki, H.; Vanthuyne, N.; Balaban, T.S.; Bürck, J.; Fliegl, H.; Klopper, W.; Naubron, J.V.; Tam, T.T.; et al. Ridge-tile-like chiral topology: Synthesis, resolution and complete chiroptical characterization of enantiomers of edge-sharing binuclear square planar complexes of Ni(II) bearing achiral ligands. J. Am. Chem. Soc. 2010, 132, 10477–10483. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Byproducts | ||||||
|---|---|---|---|---|---|---|---|
| Entry | Time (h) | 4 (%) | 6 (%) | 7 (%) | Dr (6:7) | 5 (%) | 8 + uk [b] (%) |
| 1 | 1.5 | 63.9 | 21.7 | 4.0 | 84:16 | 0.7 | 3.0 |
| 2 | 3.0 | 54.5 | 25.7 | 4.7 | 85:15 | 0.4 | 5.1 |
| 3 | 4.5 | 43.6 | 27.8 | 5.3 | 84:16 | 0.6 | 8.9 |
| 4 | 24.0 | 0.7 | 37.9 | 2.1 | 95:5 | 5.1 | 42.5 |
| Compounds | Byproducts | |||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Time (h) | NaOMe (Concentration) | 4 (%) | 6 (%) | 7 (%) | Dr (6:7) | 5 (%) | 8 + uk [b] (%) |
| 1 | 0.5 | 28% | 0.9 | 81.3 | 9.0 | 90:10 | 1.15 | 0.2 |
| 2 | 2.0 | 28% | <0.1 | 82.1 | 9.1 | 90:10 | 1.8 | 0.35 |
| 3 | 0.5 | 10% | 0.2 | 89.05 | 3.2 | 97:3 | 0.3 | 2.5 |
| 4 | 2.0 | 10% | 0.2 | 89.3 | 3.25 | 96.5:3.5 | 0.35 | 3.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mei, H.; Yin, Z.; Miwa, T.; Moriwaki, H.; Abe, H.; Han, J.; Soloshonok, V.A. Convenient Asymmetric Synthesis of Fmoc-(S)-6,6,6-Trifluoro-Norleucine. Symmetry 2019, 11, 578. https://doi.org/10.3390/sym11040578
Mei H, Yin Z, Miwa T, Moriwaki H, Abe H, Han J, Soloshonok VA. Convenient Asymmetric Synthesis of Fmoc-(S)-6,6,6-Trifluoro-Norleucine. Symmetry. 2019; 11(4):578. https://doi.org/10.3390/sym11040578
Chicago/Turabian StyleMei, Haibo, Zizhen Yin, Toshio Miwa, Hiroki Moriwaki, Hidenori Abe, Jianlin Han, and Vadim A. Soloshonok. 2019. "Convenient Asymmetric Synthesis of Fmoc-(S)-6,6,6-Trifluoro-Norleucine" Symmetry 11, no. 4: 578. https://doi.org/10.3390/sym11040578
APA StyleMei, H., Yin, Z., Miwa, T., Moriwaki, H., Abe, H., Han, J., & Soloshonok, V. A. (2019). Convenient Asymmetric Synthesis of Fmoc-(S)-6,6,6-Trifluoro-Norleucine. Symmetry, 11(4), 578. https://doi.org/10.3390/sym11040578