1. Introduction
Molecular aggregates are ubiquitous in nature. They may be found on many occasions, especially in the field of soft matter, where structures like micelles, liposomes, or vesicles are often encountered, and can be used as nanoscale probes for nonlinear optics alongside other nanoparticles [
1,
2]. The organization of the molecules at the microscopic level in these aggregates defines the properties or the function of these nanostructures. Hence, developing new tools and techniques to investigate how molecules arranges at the nanoscale nanostructures is always welcome, but is often hindered by the dimensions of the structures, often of sizes much smaller than the wavelength of light. Hence, one reverts to methods where the wavelength of the probe is of the order of the size of the aggregates, using X-ray or neutron scattering, for instance [
3,
4]. As a result, linear optical techniques are of limited use because of the diffraction limit rule. Nonlinear optical techniques, however, may offer new strategies in these studies, especially for even order techniques, like second harmonic generation (SHG), obeying the inversion symmetry cancellation rule. This rule states that SHG is forbidden in a medium possessing inversion symmetry within the electric dipole approximation [
5]. Hence, when the nonlinear optical sources are strongly correlated, the coherent response from the system is markedly different from that of an assembly of non-correlated sources. Furthermore, when going beyond the electric dipole approximation, a direct view is opened on the molecular organization in the system [
6,
7,
8]. To investigate this question more thoroughly, the spatial organization in small molecular aggregates has been probed by the frequency doubling conversion process known as second harmonic scattering (SHS). The latter terminology emphasizes that both an incoherent and a coherent response are observed as opposed to hyper-Rayleigh scattering (HRS), where only the incoherent response is observed. The first observation of this process has been reported by R.W. Terhune et al. and discussed by P.D. Maker, S.J. Cyvin et al. and R. Besohn et al. [
9,
10,
11,
12]. It has then been extensively used in the past to characterize the first hyper-polarizability of molecular compounds, supporting the extensive molecular engineering design of new organic compounds able to act as molecular probes [
13,
14,
15]. The investigation of elaborated structures, like micelles, liposomes, or even metallic nanoparticles, has also been performed [
16,
17,
18,
19]. In these systems, the question of the phase retardation in the SHS response must be raised. In particular, in the case of metallic gold nanoparticles, where the diameter of the system can be tuned without changing other properties, this question has turned into the determination of the role of retardation in the electromagnetic fields to correctly describe the polarization-resolved response at the harmonic frequency [
20,
21].
In this work, we first present a theoretical foundation of the theory of second harmonic scattering of molecular aggregates. This is an extension of the standard theory developed for second harmonic scattering from molecules, also known as the theory of hyper-Rayleigh scattering, to encompass the case of molecular aggregates. In a second step, we illustrate this theory with two specific cases of molecular aggregates, the two cases differing by the internal molecular organization of the aggregate. In particular, we show unambiguously how the experimental data provide a quantitative insight into this aggregate molecular organization.
Hence, a general model for the SHS response of an assembly of a nonlinear dipole is first developed in order to investigate the impact of molecular organization of small molecular aggregates on the polarization-resolved SHS intensity. As a direct consequence, the weight of the dipolar and quadrupolar contributions is determined through a retardation parameter. This parameter describes the retardation effects, namely the spatial dependence of the electromagnetic field over the volume occupied by the aggregate, introduced in order to fully describe the SHS intensity response from aggregates. It is shown that this model encompasses the previous approaches restricted to the electric dipole approximation and used for the analysis of liquid solutions of well-dispersed, non-interacting molecular compounds, which now account for molecular correlations [
22,
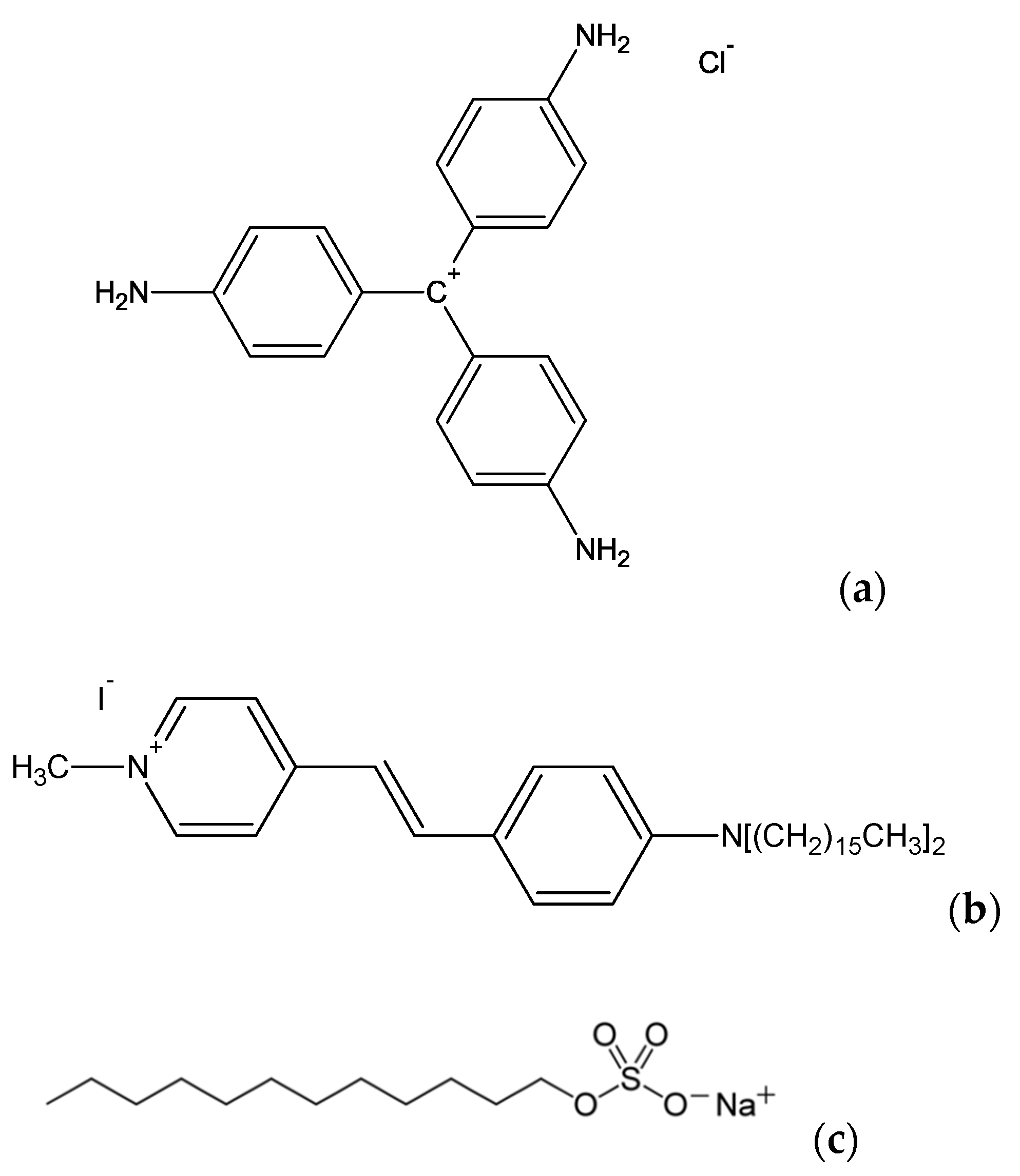
23]. Experimental data are provided to illustrate the model. These data are obtained from aqueous solutions of Crystal Violet (CV), a non-centrosymmetric octupolar dye, in the presence of sodium dodecyl sulfate (SDS) and a mixed water-methanol solution of 4-(4–dihexadecylaminostyryl)-N-methylpyridinium iodide (DiA), a cationic amphiphilic probe molecule with a strong first hyper-polarizability.
3. Theory
SHS intensity for molecular aggregates: The electric field amplitude of light scattered by a molecule
i located at position
at the harmonic frequency
, where
is the fundamental frequency in the direction
, is
, the expression of which is given by [
25]:
where
is the harmonic wave vector modulus, with
being the optical index of the medium at the harmonic frequency, and
being the unit vector in the direction of collection. The expression of the nonlinear dipole induced at the harmonic frequency is related to the molecular first hyper-polarizability
through:
where
is the fundamental wave electric field, with
being its polarization vector defined with the polarization angle
in the plane perpendicular to the propagation OZ direction. The frame transformation tensor
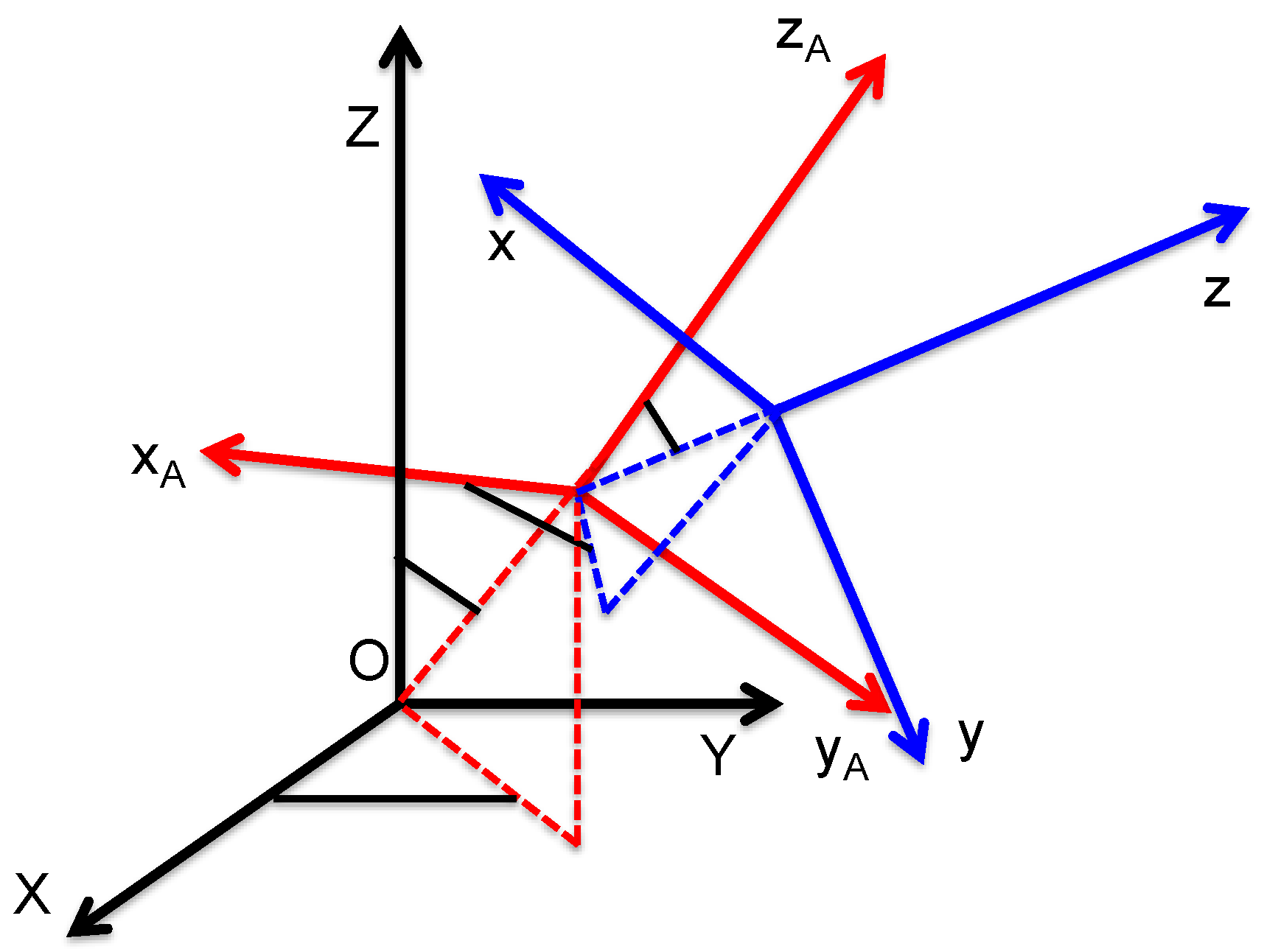
, accounting for the transformation of the molecular first hyper-polarizability tensor from the molecular frame to the laboratory frame, is defined with the standard Euler angles according to
Figure 1 below.
For an assembly of correlated molecules, the total field amplitude
is the sum of the individual amplitudes given in Equation (1). The
m molecules of the aggregate are assumed to be fixed relative to each other in space, hence, in position and orientation. Here, it is therefore simply assumed that the time scale of molecular motion in the aggregate is much longer than the time scale of the nonlinear interaction with the electromagnetic wave. With multiple scattering at both frequencies, and local field factors and molecular interactions neglected, then:
This superposition is built as a coherent superposition of the amplitudes scattered by single molecules. For a large number of molecules present in the aggregate, that is, if is large, the discrete sum may be replaced by a continuous integration. In a liquid suspension, however, the molecules are distributed into many aggregates, and the latter are randomly distributed in the sampled volume in position and orientation. Hence, for molecules distributed within aggregates containing molecules, one has , and the total intensity scattered at the harmonic frequency is the incoherent superposition of the intensities scattered by each molecular aggregate.
This intensity is therefore:
where the brackets stand for the spatial averaging of the orientation of the aggregate, since it is dispersed in a liquid suspension. This description is the standard theoretical foundation for hyper-Rayleigh scattering.
In the developments of the above foundation for molecular aggregates, it is now important to introduce three distinguished reference frames: the laboratory frame (OXYZ), the molecular frame (Oxyz), and the aggregate frame (Ox
Ay
Az
A) (see
Figure 1). Indeed, the molecules possess a fixed position and orientation within the aggregate, but the aggregate is randomly distributed in the liquid suspension in position and orientation. Hence, in Equation (4), the averaging procedure is performed on the orientation of the aggregate. Owing to the intermediate frame, the frame transformation from the molecular to the laboratory frame can be performed into two successive steps for a single molecule (see
Supplementary File Part A), namely:
with the introduction of the frame transformation tensor from the aggregate to the laboratory frame
and the molecular first hyper-polarizability tensor
of molecule
i expressed in the aggregate reference frame.
In order to get a better insight into the different contributions to the scattered intensity, Equation (3) is rewritten using the first order approximation:
for simplicity and later discussion. Hence, keeping only the term in
, Equation (3) yields:
with the introduction of the molecular volume density
within the aggregate and the wave vector mismatch
, the expression of which is simply:
with the introduction of the fundamental wavelength
. If the direction of collection is in the forward direction, namely
, then the usual phase matching condition of second harmonic generation (SHG) is recovered. Because the condition
is also assumed, i.e., the aggregates are small before the fundamental wavelength, the expansion of the exponential factor of the integral in Equation (7) yields:
It is therefore convenient to split the harmonic field amplitude into two terms, namely:
The first term
in Equation (10) is the equivalent of Equation (3) at the level of the aggregate instead of the molecule. Hence, the integrated induced dipole of the aggregate
is simply:
where the dipolar first hyper-polarizability of the aggregate is given by:
i.e., the superposition of the first hyper-polarizabilities of the molecules present in the aggregate. This superposition takes into account both the position and the orientation of the molecules. The second term
of Equation (10) is the first correction to the non-vanishing spatial extension of the aggregate. Therefore,
is the integrated induced quadrupole of the aggregate, and is given by:
with the quadrupolar first hyper-polarizability
of the aggregate defined through:
Note that the vector has to be also transformed into the aggregate reference frame. In the molecular frame, it is simply given by . The quadrupolar first hyper-polarizability is a fourth rank tensor. Owing to the definition of the wave vector mismatch in Equation (8) and the geometrical configuration of the experiment, where the direction of incidence of the fundamental wave is along the OZ direction and the collection of the harmonic wave is performed in the OY direction in the laboratory, only the elements of the general form and are involved in Equation (13), but the calculation of these elements entails the transformation from the aggregate frame to the laboratory frame with the definition . Finally, it is observed that, from Equation (10), the second contribution scales with the parameter . Since it has been set beforehand that this parameter is much smaller than unity, this contribution is usually neglected for molecular aggregates, since their average size is much smaller than the fundamental wavelength. Typical aggregates have diameters of the order of a few nanometers, whereas the fundamental wavelength is often in the range of hundreds of nanometers. However, depending on the molecular organization of the aggregate, the induced dipole may be very small or even vanish. In this case, the quadrupolar contribution may not be negligible any longer, and must be accounted for.
SHS scattered intensity for non-centrosymmetrical aggregates: In the particular case of a non-centrosymmetrical spatial orientational distribution of the molecules in the aggregates,
does not vanish, and possesses three components, namely
,
, and
. In most cases, this
term will dominate, and this case reverts to the rather well-known case of a single molecule, albeit at the level of the aggregate. The scattered intensity at the harmonic frequency vertically and horizontally polarized, respectively
and
, is now given by:
with the conditions
and
. The different coefficients are simply:
with the constant:
and
for
and
for
. In particular, the depolarization ratio
is now an indication of the symmetry of the aggregate, and no longer of the molecule. Furthermore, the total intensity scales linearly with the number of aggregates present in the liquid suspension. Note that the resulting apparent first hyper-polarizability of the aggregates depends now on the orientation of all molecules constituting the aggregate, and can be very large, since it is the superposition of the first hyper-polarizabilities of the
molecules contained in the aggregate. For instance, in a perfect alignment of all molecules, the aggregate first hyperpolarizability is indeed as large as
times that of a single molecule, and hence, the intensity scattered at the harmonic intensity scales with the product
.
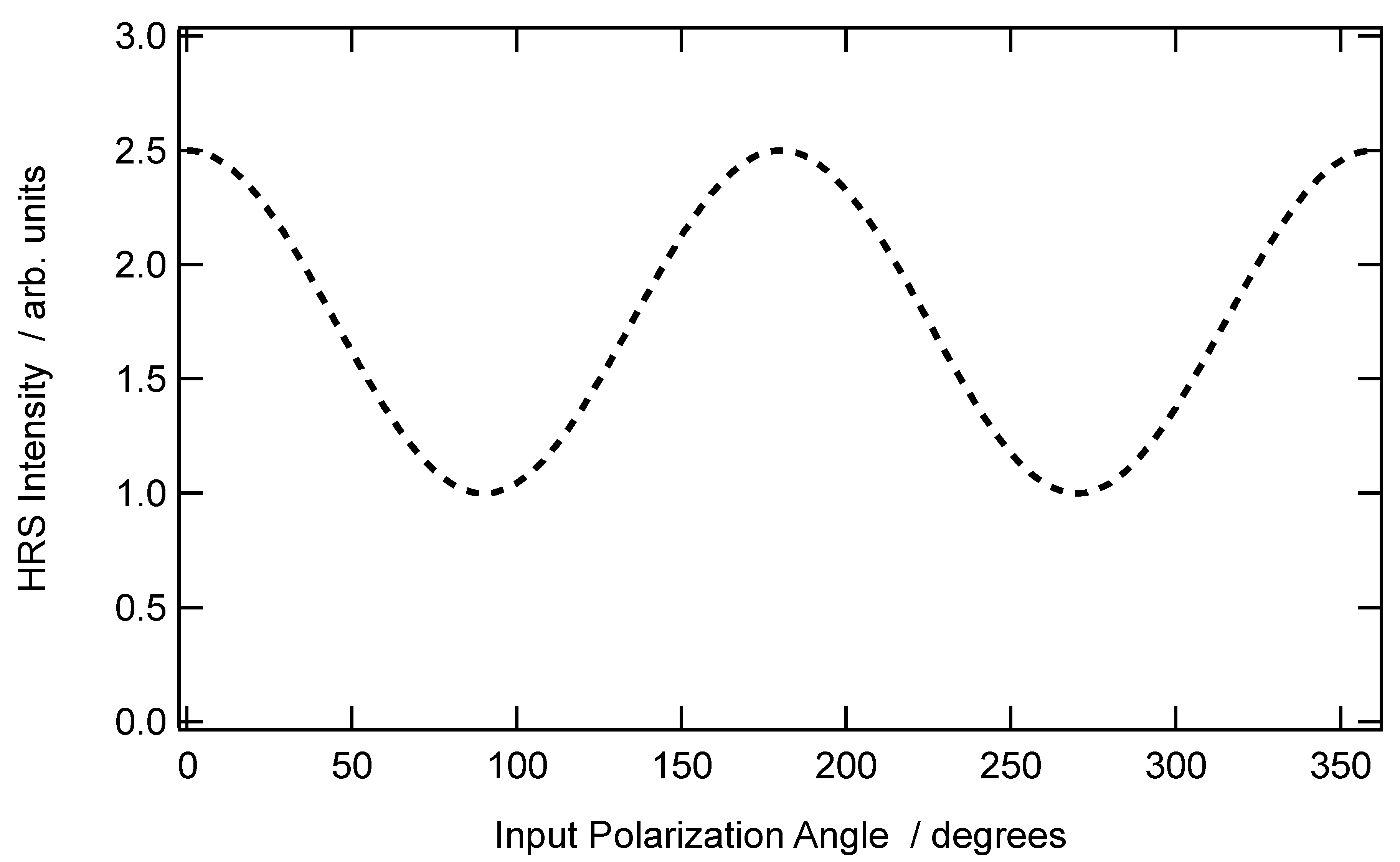
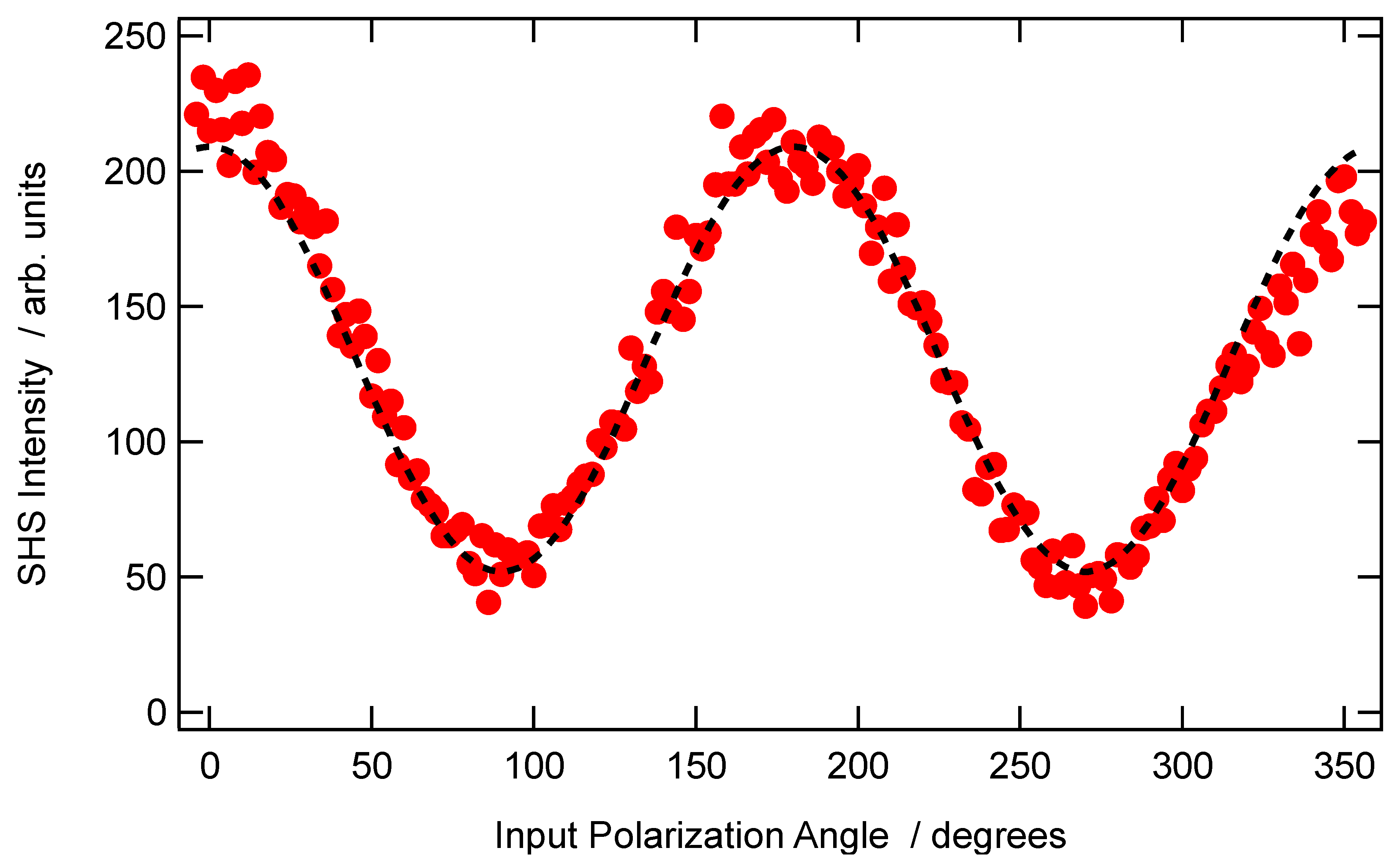
A typical graph of the vertically polarized SHS intensity collected in the case of a non-centrosymmetric aggregate with an arbitrary depolarization ratio of
is provided in
Figure 2 below.
SHS scattered intensity for centrosymmetrical aggregates: The second case occurs when the organization of the molecules within the aggregate possesses a center of inversion. This is the organization found in spherical micelles, for instance, or corresponding to a random orientation. As expected for this centrosymmetrical structure, the first contribution vanishes, namely
. The scattered amplitude at the harmonic frequency is therefore reduced to the second contribution only, which may be recast as:
Finally, the intensity again takes the expressions given by Equations (15a) and (15b), but this time:
with the constant:
and the effective parameters:
with
for
and
for
.
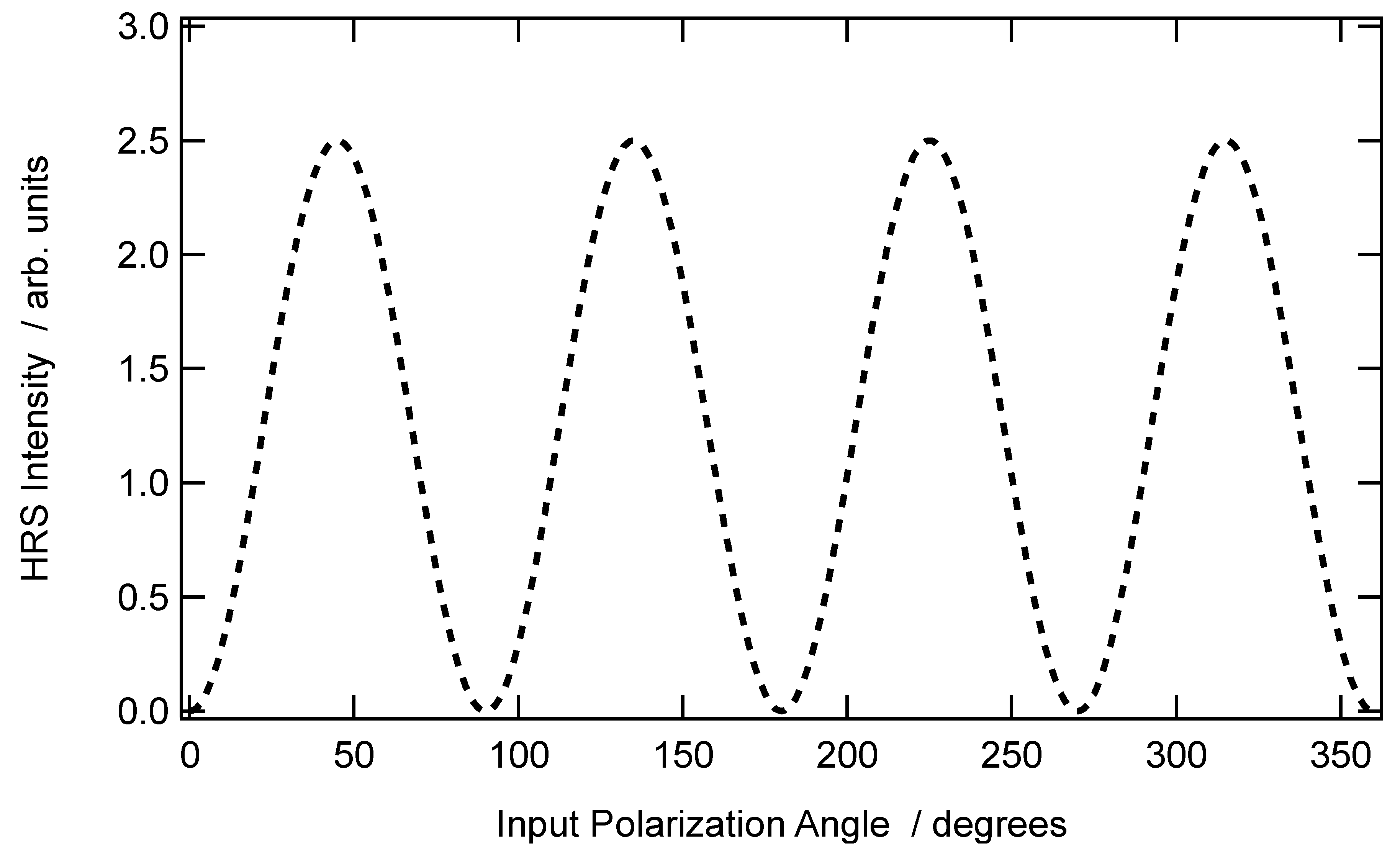
The case of spherical micelles of radius
a leads to simple expressions for the intensity coefficients of Equations (19a–c). With a uniform distribution of the molecular dye at the aggregate surface, the molecular density is simply:
if one assumes that the nonlinear optical chromophore is point-like. The volume integration for the effective quadrupolar first hyper-polarizability elements leads to a dependence of these elements with the third power of the radius of the sphere. Furthermore, the coefficients
and
for the vertical polarization vanish, whereas
does not, and is proportional to
(see
Figure 3). For the horizontal polarization, one gets
. Furthermore, for an experimental geometry where the collection is performed along the opposite direction of the OY axis of the laboratory, Equation (21) will involve the difference of
and
, rather than their sum.
The perfect centrosymmetry rule is not expected to be strictly fulfilled by aggregates. However, if this symmetry is close to perfect, contributions from the first contribution
will also contribute simultaneously to the total intensity scattered at the harmonic frequency without overwhelming the second one. Therefore, the resulting polarization-resolved plot may take a profile somewhere in between the two contributions presented in
Figure 2 and
Figure 3. Note also that other orientational distributions may be found, like the azimuthal one, instead of the radial one.
SHS of Molecular Aggregates: To illustrate the problem, we now discuss the case of aggregates constituted by Crystal Violet and DiA, two compound-forming aggregates with different properties depending on the conditions, see below.
Experiments are initially performed for a simple rod-like molecule, 4-(4–dihexadecylaminostyryl)-N-methylpyridinium (DiA) (see
Figure 4), the first hyper-polarizability tensor of which is dominated by a single element. This latter dominating element is
where the molecular Oz axis is taken along the axis of the charge transfer. Since DiA is rod-like, this molecular axis is nearly oriented along the axis direction of DiA. Since DiA is also amphiphilic due to the two long alkyl chains, it is expected to form micelle-like aggregates, with the Oz axis oriented towards the center of the aggregate. The orientation distribution within the molecular aggregate is therefore expected to closely resemble that of spherical micelles. DiA dissolves in methanol but not in water, and therefore aggregation was induced by varying the volume fraction of water into methanol at a fixed DiA concentration of 12.5 μM [
25]. A typical vertically polarized resolved SHS intensity for DiA dispersed in pure methanol is given in
Figure 5. This graph allows to immediately identify
as
,
as
, and
as
, although an adjustment procedure with Equation (15a) is usually performed, and the condition
is obeyed.
In order to have a quantified characterization of the aggregate, the following
parameter is defined:
also found as [
6]:
Note in particular the sign inversion between and . As examples, it is immediate to see that the pure dipolar response of a non-centrosymmetric aggregate leads to , whereas the purely retarded response of a centrosymmetric aggregate in the shape of a spherical micelle, i.e., with a radial distribution, leads to . Interestingly, an azimuthal distribution of the dyes forming a centrosymmetric aggregate leads to a negative value for the retarded parameter .
As an illustration of a quantitative analysis of the aggregate structure, one may discuss the results reported in Ref. [
25], comparing the vertically polarized SHS intensity obtained for DiA dispersed in pure methanol and in a mixed 5:1
v/v water-methanol solution. The depolarization ratio
is found to be 0.25, along with a vanishing retardation parameter
for DiA dispersed in pure methanol (see
Figure 5). Oppositely, in a mixed 5:1
v/v water-methanol solvent, the depolarization ratio
is found to be 0.28, along with a retardation parameter
[
25].
The depolarization ratio obtained for the aggregate is different from the value of +0.25 measured for DiA dissolved in pure methanol and closely associated with a single first hyper-polarizability tensor element compound [
25]. It is concluded that some distortion in the electronic structure of DiA occurs in the aggregate, leading to the appearance of contributions from other tensor elements. Furthermore, the retardation parameter, vanishing in the pure methanol solution where DiA is ideally dispersed, now increases, indicating an organization tending to the spherical micellar-like structure, as expected for a compound with two hydrophobic long alkyl chains.
In the case of aqueous solutions of Crystal Violet, the same experiments were performed, this time as a function of SDS concentration, as this anionic surfactant is known to form micelles beyond the critical micelle concentration (CMC) of 7 mM [
24]. It is expected that these molecular structures favor adsorption [
26].
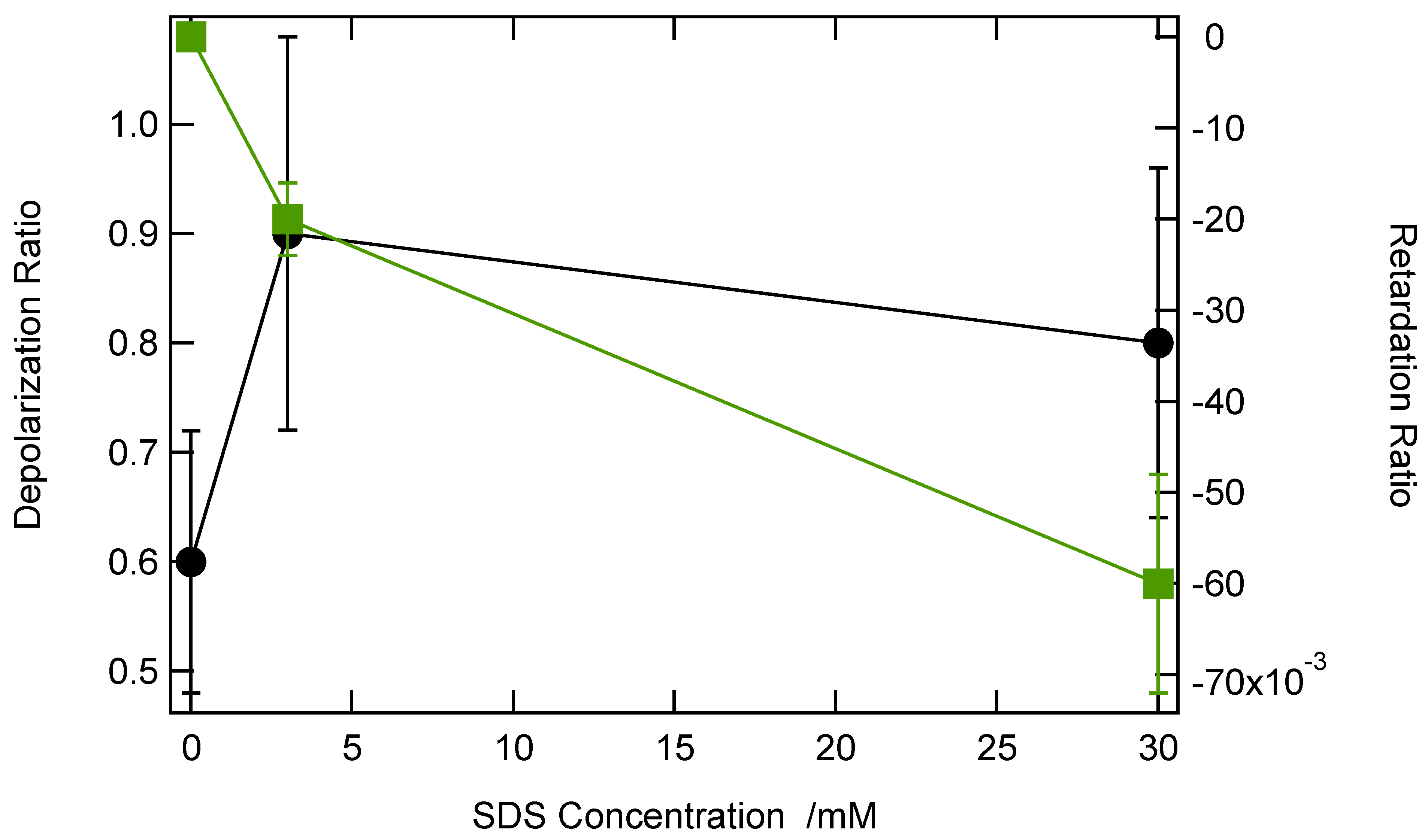
Figure 6 shows the depolarization ratio
along with the retardation parameter
as a function of the SDS concentration, below and above the SDS CMC.
The depolarization ratio is 0.6 in the absence of SDS, as expected for the close to planar octupolar symmetry of CV [
27]. It increases when SDS is present, but does not change much, indicating a change in symmetry of the whole aggregate compared to the CV monomer. Oppositely, the retardation parameter
is initially zero, as expected for a compound ideally dissolved in its solvent, but then decreases as SDS is introduced into the solution. The negative value of this parameter is not in agreement with a spherical micellar-like organization, but rather with an azimuthal distribution, i.e., adsorption onto the SDS aggregates. Interestingly, this organization already starts at 3 mM, a value below the SDS CMC, suggesting the appearance of aggregates below the CMC.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





