
Highly Diastereoselective Chelation-Controlled 1,3-anti-Allylation of (S)-3-(Methoxymethyl)hexanal Enabled by Hydrate of Scandium Triflate



Abstract
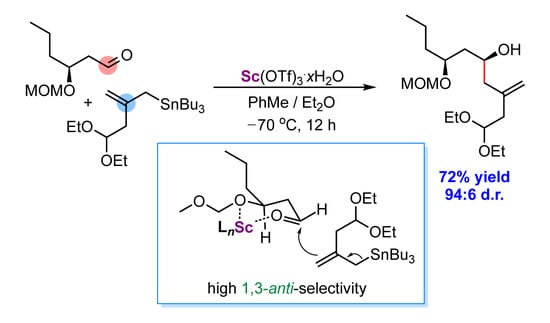
:
1. Introduction
2. Results and Discussion
2.1. Improved Protocol for the Preparation of Functionalized (Allyl)Tributylstannane 2
2.2. Diastereoselective Allylation of Aldehyde 3 with (Allyl)Tributylstannane 2
3. Experimental Section
3.1. General Experimental Methods
3.2. Preparation of 4,4-Diethoxy-2-methylenebutyl Bromide (1)
3.2.1. 1-(2,2-Diethoxyethyl)cyclopropan-1-ol (5)
3.2.2. 1-(2,2-Diethoxyethyl)cyclopropyl Methanesulfonate (6)
3.2.3. 4,4-Diethoxy-2-methylenebutyl Bromide (1)
3.3. Preparation of Tributyl(4,4-diethoxy-2-methylenebutyl)stannane (2)
3.4. Preparation of Hydrated Scandium Triflate Catalyst
3.5. (5S,7S)-1,1-Diethoxy-7-(methoxymethoxy)-3-methylenedecan-5-ol (anti-8)
3.6. Preparation of C7–C16 Building Block 15 for the Synthesis of (+)-Neopeltolide
3.6.1. (6S)-2-Ethoxy-6-((S)-2-(methoxymethoxy)pentyl)-4-methylenetetrahydro-2H-pyran (16)
3.6.2. (6S)-6-((S)-2-(Methoxymethoxy)pentyl)-4-methylenetetrahydro-2H-pyran-2-ol (9)
3.6.3. ((S)-6-((S)-2-(Methoxymethoxy)pentyl)-4-methylenetetrahydro-2H-pyran-2-one (17)
3.6.4. (S)-6-((S)-2-(Methoxymethoxy)pentyl)-4-methyl-5,6-dihydro-2H-pyran-2-one (10)
3.6.5. (4S,6S)-6-((S)-2-(Methoxymethoxy)pentyl)-4-methyltetrahydro-2H-pyran-2-one (11)
3.6.6. (3R,5S,7S)-7-(Methoxymethoxy)-3-methyldecane-1,5-diol (12)
3.6.7. (R)-4-((4S,6S)-2,2-Dimethyl-6-propyl-1,3-dioxan-4-yl)-3-methylbutan-1-ol (13)
3.6.8. (5S,7S,9R)-9,13,13,14,14-Pentamethyl-5-propyl-2,4,12-trioxa-13-silapentadecan-7-ol (18)
3.6.9. (5S,7S,9R)-7-Methoxy-9,13,13,14,14-pentamethyl-5-propyl-2,4,12-trioxa-13-silapentadecane (14)
3.6.10. (3R,5S,7S)-5-Methoxy-7-(methoxymethoxy)-3-methyldecan-1-ol (19)
3.6.11. (3S,5S,7S)-5-Methoxy-7-(methoxymethoxy)-3-methyldecanal (15) [55]
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yamamoto, Y.; Asao, N. Selective reactions using allylic metals. Chem. Rev. 1993, 93, 2207–2293. [Google Scholar] [CrossRef]
- Denmark, S.E.; Fu, J. Catalytic enantioselective addition of allylic organometallic reagents to aldehydes and ketones. Chem. Rev. 2003, 103, 2763–2794. [Google Scholar] [CrossRef] [PubMed]
- Yus, M.; González-Gómez, J.C.; Foubelo, F. Catalytic enantioselective allylation of carbonyl compounds and imines. Chem. Rev. 2011, 111, 7774–7854. [Google Scholar] [CrossRef]
- Yus, M.; González-Gómez, J.C.; Foubelo, F. Diastereoselective allylation of carbonyl compounds and imines: Application to the synthesis of natural products. Chem. Rev. 2013, 113, 5595–5698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fattah, T.A.; Saeed, A. Applications of Keck allylation in the synthesis of natural products. New J. Chem. 2017, 41, 14804–14821. [Google Scholar] [CrossRef]
- Evans, D.A.; Rajapakse, H.A.; Stenkamp, D. Asymmetric syntheses of Pectenotoxins-4 and -8, part I: Synthesis of the C1–C19 subunit. Angew. Chem. Int. Ed. 2002, 41, 4569–4573. [Google Scholar] [CrossRef]
- Lee, E.; Jeong, E.J.; Kang, E.J.; Sung, L.T.; Hong, S.K. Total synthesis of Pamamycin-607. J. Am. Chem. Soc. 2001, 123, 10131–10132. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Kedei, N.; Michalowski, A.; Lewin, N.E.; Keck, G.E.; Blumberg, P.M. Deletion of the C26 methyl substituent from the Bryostatin analogue Merle 23 has negligible impact on its biological profile and potency. ChemBioChem 2018, 19, 1049–1059. [Google Scholar] [CrossRef]
- Timothy, N.; Trotter, T.N.; Albury, A.M.M.; Jennings, M.P. Total synthesis of 7-Deoxy-6-O-methylfusarentin featuring a chelation-controlled 1,3-Reetz–Keck-type allylation. J. Org. Chem. 2012, 77, 7688–7692. [Google Scholar]
- Sabitha, G.; Raoa, A.S.; Yadav, J.S. Synthesis of the C1–C25 southern domain of Spirastrellolides B and F. Org. Biomol. Chem. 2013, 11, 7218–7231. [Google Scholar] [CrossRef]
- Kim, W.H.; Hong, S.K.; Lim, S.M.; Ju, M.-A.; Jung, S.K.; Kim, Y.W.; Jung, J.H.; Kwon, M.S.; Lee, E. Total synthesis of IKD-8344. Angew. Chem. Int. Ed. 2006, 45, 7072–7075. [Google Scholar] [CrossRef] [PubMed]
- Keck, G.E.; Truong, A.P. Synthetic studies on the Bryostatins: preparation of a truncated BC-ring intermediate by pyran annulation. Org. Lett. 2005, 7, 2149–2152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joarder, D.D.; Jennings, M.P. Convergent enantioselective syntheses of two potential C25–C40 subunits of (−)-Caylobolide A. Tetrahedron Lett. 2011, 52, 5124–5127. [Google Scholar]
- Keck, G.E.; Yu, T.; McLaws, M.D. Enantio- and diastereoselective additions to aldehydes using the bifunctional reagent 2-(chloromethyl)-3-(tributylstannyl) propene: application to a synthesis of the C16−C27 segment of Bryostatin 1. J. Org. Chem. 2005, 70, 2543–2550. [Google Scholar] [CrossRef] [Green Version]
- Bartolo, N.D.; Read, J.A.; Valentín, E.M.; Woerpel, K.A. Reactions of allylmagnesium reagents with carbonyl compounds and compounds with C = N double bonds: Their diastereoselectivities generally cannot be analyzed using the Felkin-Anh and chelation-control models. Chem. Rev. 2020, 120, 1513–1619. [Google Scholar] [CrossRef]
- Mengel, A.; Reiser, O. Around and beyond Cram′s rule. Chem. Rev. 1999, 99, 1191–1224. [Google Scholar] [CrossRef]
- Reetz, M.T. Chelation or non-chelation control in addition reactions of chiral α- and β-alkoxy carbonyl compounds. Angew. Chem. Int. Ed. Engl. 1984, 23, 556–569. [Google Scholar] [CrossRef]
- Cherest, M.; Felkin, H.; Prudent, N. Torsional strain involving partial bonds. The stereochemistry of the lithium aluminium hydride reduction of some simple open-chain ketones. Tetrahedron Lett. 1968, 9, 2199–2204. [Google Scholar] [CrossRef]
- Cherest, M.; Felkin, H. Torsional strain involving partial bonds. The steric course of the reaction between allyl magnesium bromide and 4-t-butyl-cyclohexanone. Tetrahedron Lett. 1968, 9, 2205–2208. [Google Scholar] [CrossRef]
- Anh, N.T.; Eisenstein, O. Induction asymetrique 1–2: Comparaison ab initio des modeles de Cram, de Cornforth, de Karabatsos et de Felkin. Tetrahedron Lett. 1976, 17, 155–158. [Google Scholar] [CrossRef]
- Anh, N.T. Regio- and stereo-selectivities in some nucleophilic reactions. Top. Curr. Chem. 1980, 88, 145–162. [Google Scholar]
- Cornforth, J.W.; Cornforth, R.H.; Mathew, K.K. A general stereoselective synthesis of olefins. J. Chem. Soc. 1959, 1, 112–127. [Google Scholar] [CrossRef]
- Evans, D.A.; Siska, S.J.; Cee, V.J. Resurrecting the Cornforth model for carbonyl addition: Studies on the origin of 1,2-asymmetric induction in enolate additions to heteroatom-substituted aldehydes. Angew. Chem. Int. Ed. 2003, 42, 1761–1765. [Google Scholar] [CrossRef]
- Cee, V.J.; Cramer, C.J.; Evans, D.A. Theoretical investigation of enolborane addition to α-heteroatom-substituted aldehydes. Relevance of the Cornforth and polar Felkin−Anh models for asymmetric induction. J. Am. Chem. Soc. 2006, 128, 2920–2930. [Google Scholar] [CrossRef]
- Cram, D.J.; Elhafez, F.A.A. Studies in stereochemistry. X. The rule of “steric control of asymmetric induction” in the syntheses of acyclic systems. J. Am. Chem. Soc. 1952, 74, 5828–5835. [Google Scholar] [CrossRef]
- Cram, D.J.; Kopecky, K.R. Studies in stereochemistry. XXX. Models for steric control of asymmetric induction. J. Am. Chem. Soc. 1959, 81, 2748–2755. [Google Scholar] [CrossRef]
- Leitereg, T.J.; Cram, D.J. Studies in stereochemistry. XXXVIII. Open-chain vs. cyclic models for 1,3-asymmetric induction in addition reactions. J. Am. Chem. Soc. 1968, 90, 4019–4026. [Google Scholar] [CrossRef]
- Reetz, M.T.; Jung, A. 1,3-Asymmetric induction in addition reactions of chiral β-alkoxy aldehydes: Efficient chelation control via Lewis acidic titanium reagents. J. Am. Chem. Soc. 1983, 105, 4833–4835. [Google Scholar] [CrossRef]
- Reetz, M.T.; Kesseler, K.; Jung, A. Concerning the role of Lewis acids in chelation controlled addition to chiral alkoxy aldehydes. Tetrahedron Lett. 1984, 25, 729–732. [Google Scholar] [CrossRef]
- Reetz, M.T. Structural, mechanistic, and theoretical aspects of chelation-controlled carbonyl addition reactions. Acc. Chem. Res. 1993, 26, 462–468. [Google Scholar] [CrossRef]
- Keck, G.E.; Castellino, S. On the origins of stereoselectivity in chelation controlled nucleophilic additions to β-alkoxy aldehydes: Solution structures of Lewis acid complexes via NMR spectroscopy. J. Am. Chem. Soc. 1986, 108, 3847–3849. [Google Scholar] [CrossRef]
- Keck, G.E.; Castellino, S.; Wiley, M.R. Dramatic effects of oxygen substituents on 1,3-asymmetric induction in additions of allyltriphenylstannane to β-alkoxy aldehydes: A chemical and spectroscopic investigation. J. Org. Chem. 1986, 51, 5478–5480. [Google Scholar] [CrossRef]
- Keck, G.E.; Boden, E.P.; Wiley, M.R. Total synthesis of (+)-Colletodiol: New methodology for the synthesis of macrolactones. J. Org. Chem. 1989, 54, 896–906. [Google Scholar] [CrossRef]
- Keck, G.E.; Abbott, D.E.; Wiley, M.R. New allylstannanes for the connective construction of monoprotected vicinal diols. Tetrahedron Lett. 1987, 28, 139–142. [Google Scholar] [CrossRef]
- Keck, G.E.; Murry, J.A. Total synthesis of (−)-Colletol. J. Org. Chem. 1991, 56, 6606–6611. [Google Scholar] [CrossRef]
- Keck, G.E.; Truong, A.P. Synthetic studies on the Bryostatins: synthetic routes to analogues containing the tricyclic macrolactone core. Org. Lett. 2005, 7, 2153–2156. [Google Scholar] [CrossRef] [Green Version]
- Keck, G.E.; Giles, R.L.; Cee, V.J.; Wager, C.A.; Yu, T.; Kraft, M.B. Total synthesis of Epothilones B and D: Stannane equivalents for β-keto ester dianions. J. Org. Chem. 2008, 73, 9675–9691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keck, G.E.; Welch, D.S.; Vivian, P.K. Synthetic studies toward the Bryostatins: a substrate-controlled approach to the A-ring. Org. Lett. 2006, 8, 3667–3670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keck, G.E.; Welch, D.S.; Poudel, Y.B. Synthetic studies toward Bryostatin 1: Preparation of a C1–C16 fragment by pyran annulation. Tetrahedron Lett. 2006, 47, 8267–8270. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Murai, Y.; Kishi, T.; Takamura, H.; Kadota, I. Convergent total synthesis of (−)-Dactylolide. Tetrahedron Lett. 2018, 59, 763–766. [Google Scholar] [CrossRef]
- Williams, D.R.; Plummer, S.V.; Patnaik, S. Formal synthesis of leucascandrolide A. Angew. Chem. Int. Ed. 2003, 42, 3934–3938. [Google Scholar] [CrossRef]
- Williams, D.R.; Patnaik, S.; Plummer, S.V. Leucascandrolide A: a second generation formal synthesis. Org. Lett. 2003, 5, 5035–5038. [Google Scholar] [CrossRef] [PubMed]
- Skepper, C.K.; Quach, T.; Molinski, T.F. Total synthesis of Enigmazole A from cinachyrella enigmatica. Bidirectional bond constructions with an ambident 2,4-disubstituted oxazole synthon. J. Am. Chem. Soc. 2010, 132, 10286–10292. [Google Scholar] [CrossRef] [PubMed]
- Ahlers, A.; Haro, T.; Gabor, B.; Fürstner, A. Concise total synthesis of Enigmazole A. Angew. Chem. Int. Ed. 2016, 55, 1406–1411. [Google Scholar] [CrossRef] [Green Version]
- El-Gaber, M.K.A.; Yasuda, S.; Iida, E.; Mukai, C. Enantioselective total synthesis of (+)-Sieboldine A. Org. Lett. 2017, 19, 320–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.B.; Zhu, W.; Shirakami, S.; Sfouggatakis, C.; Doughty, V.A.; Bennett, C.S.; Sakamoto, Y. Total synthesis of (+)-Spongistatin 1. An effective second-generation construction of an advanced EF wittig salt, fragment union, and final elaboration. Org. Lett. 2003, 5, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Woo, S.K.; Krische, M.J. Total synthesis of Bryostatin 7 via C–C bond-forming hydrogenation. J. Am. Chem. Soc. 2011, 133, 13876–13879. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, C.C.; Keck, G.E. Total synthesis of (+)-Dactylolide. Org. Lett. 2005, 7, 3053–3056. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Liang, W.; Dai, M. Total syntheses via cyclopropanols. Tetrahedron 2019, 75, 193–208. [Google Scholar] [CrossRef]
- Haym, I.; Brimble, M.A. The Kulinkovich hydroxycyclopropanation reaction in natural product synthesis. Org. Biomol. Chem. 2012, 10, 7649–7665. [Google Scholar] [CrossRef] [PubMed]
- Elek, G.Z.; Koppel, K.; Zubrytski, D.M.; Konrad, N.; Järving, I.; Lopp, M.; Kananovich, D.G. Divergent access to histone deacetylase inhibitory cyclopeptides via a late-stage cyclopropane ring cleavage strategy. Short synthesis of Chlamydocin. Org. Lett. 2019, 21, 8473–8478. [Google Scholar] [CrossRef]
- Zubrytski, D.M.; Kananovich, D.G.; Matiushenkov, E.A. Preparation of stereochemically pure E- and Z-alkenoic acids and their methyl esters from bicyclo[n.1.0]alkan-1-ols. Application in the synthesis of insect pheromones. Russ. J. Org. Chem. 2017, 53, 813–823. [Google Scholar] [CrossRef]
- Mineeva, I.V.; Kulinkovich, O.G. Preparation of 3-bromomethyl-3-butenal diethylacetal and its conversion into isoprenoid aldehydes derivatives. Russ. J. Org. Chem. 2009, 45, 1623–1632. [Google Scholar] [CrossRef]
- Mineyeva, I.V.; Kulinkovich, O.G. Methyl 3-bromomethyl-3-butenoate as an isopentane building block for the stereoselective preparation of (S)-4-methyl-3,6-dihydro-2H-pyran-2-carbaldehyde and (+)-faranal. Tetrahedron Lett. 2010, 51, 1836–1839. [Google Scholar] [CrossRef]
- Mineeva, I.V. New approach to the synthesis of macrocyclic core of cytotoxic lactone (+)-Neopeltolide. Synthesis of C7–C14 segment basing on cyclopropanol intermediates. Russ. J. Org. Chem. 2015, 51, 1061–1070. [Google Scholar] [CrossRef]
- Masyuk, V.S.; Mineeva, I.V. Synthesis of phenyl analog of retinoic acid methyl ester proceeding from 3-(bromomethyl)but-3-enal diethylacetal. Russ. J. Org. Chem. 2017, 53, 1642–1650. [Google Scholar] [CrossRef]
- Masyuk, V.S.; Mineeva, I.V. Synthesis of β-(2,2-diethoxyethyl)-substituted (allyl)tributylstannane and its application to asymmetric allylation. Russ. J. Org. Chem. 2016, 52, 178–185. [Google Scholar] [CrossRef]
- Kulinkovich, O.G. The chemistry of cyclopropanols. Chem. Rev. 2003, 103, 2597–2632. [Google Scholar] [CrossRef] [PubMed]
- McDonald, T.R.; Mills, L.R.; West, M.S.; Rousseaux, S.A.L. Selective carbon–carbon bond cleavage of cyclopropanols. Chem. Rev. 2021, 121, 3–79. [Google Scholar] [CrossRef]
- Cha, J.K.; Kulinkovich, O.G. The Kulinkovich cyclopropanation of carboxylic acid derivatives. Org. React. 2012, 77, 1–160. [Google Scholar]
- Kozyrkov, Y.Y.; Kulinkovich, O.G. A simple and efficient conversion of tertiary cyclopropanols to 2-substituted allyl halides. Synlett 2002, 3, 443–446. [Google Scholar] [CrossRef]
- Kulinkovich, O.G.; Kozyrkov, Y.Y.; Bekish, A.V.; Matiushenkov, E.A.; Lysenko, I.L. A convenient way for the conversion of carboxylic esters into 2-substituted allyl halides. Synthesis 2005, 10, 1713–1717. [Google Scholar] [CrossRef]
- Kananovich, D.G.; Hurski, A.L.; Kulinkovich, O.G. A highly stereoselective transformation of carboxylic esters to trisubstituted olefins via cationic cyclopropyl–allyl rearrangement of sulfonates of cis-1,2-disubstituted cyclopropanols. Tetrahedron Lett. 2007, 48, 8424–8429. [Google Scholar] [CrossRef]
- Wright, A.E.; Botelho, J.C.; Guzman, E.; Harmody, D.; Linley, P.; McCarthy, P.J.; Pitts, T.P.; Pomponi, S.A.; Reed, J.K. Neopeltolide, a macrolide from a lithistid sponge of the family neopeltidae. J. Nat. Prod. 2007, 70, 412–416. [Google Scholar] [CrossRef]
- Bai, Y.; Dai, M. Strategies and methods for the synthesis of anticancer natural product Neopeltolide and its analogs. Curr. Org. Chem. 2015, 19, 871–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuwa, H. Contemporary strategies for the synthesis of tetrahydropyran derivatives: Application to total synthesis of Neopeltolide, a marine macrolide natural product. Mar. Drugs 2016, 14, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keck, G.E.; Tarbet, K.H.; Geraci, L.S. Catalytic asymmetric allylation of aldehydes. J. Am. Chem. Soc. 1993, 115, 8467–8468. [Google Scholar] [CrossRef]
- Keck, G.E.; Geraci, L.S. Catalytic asymmetric allylation (CAA) reactions. II. A new enantioselective allylation procedure. Tetrahedron Lett. 1993, 34, 7827–7828. [Google Scholar] [CrossRef]
- Keck, G.E.; Krishnamurthy, D.; Grier, M.C. Catalytic asymmetric allylation reactions. 3. Extension to methallylstannane, comparison of procedures, and observation of a nonlinear effect. J. Org. Chem. 1993, 58, 6543–6544. [Google Scholar] [CrossRef]
- Wender, P.A.; Verma, V.A. The design, synthesis, and evaluation of C7 diversified Bryostatin analogs reveals a hot spot for PKC affinity. Org. Lett. 2008, 10, 3331–3334. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.-M.; Choi, H.-S.; Yoon, S.-K.; Jung, W.-H. Effects of subjoin Lewis Acid on the catalytic asymmetric allylic transfer reactions of aldehydes promoted by BINOL-Ti (IV) complex. Synlett 1997, 8, 889–890. [Google Scholar] [CrossRef]
- Banerjee, B. Sc(OTf)3 catalyzed carbon-carbon and carbon-heteroatom bond forming reactions: A review. Arkivoc 2017, 2017, 1–25. [Google Scholar] [CrossRef]
- Pellissier, H. Recent developments in enantioselective scandium-catalyzed transformations. Coord. Chem. Rev. 2016, 313, 1–37. [Google Scholar] [CrossRef]
- Kobayashi, S.; Sugiura, M.; Kitagawa, H.; Lam, W.W.-L. Rare-earth metal triflates in organic synthesis. Chem. Rev. 2002, 102, 2227–2302. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S. Scandium triflate in organic synthesis. Eur. J. Org. Chem. 1999, 1999, 15–27. [Google Scholar] [CrossRef]
- Rossini, A.J.; Hildebrand, M.P.; Hazendonk, P.A.; Schurko, R.W. Multinuclear solid-state NMR studies of polymer-supported scandium triflate catalysts. J. Phys. Chem. C 2014, 118, 22649–22662. [Google Scholar] [CrossRef]
- Kimbrough, T.J.; Roethle, P.A.; Mayer, P.; Trauner, D. Total synthesis of Coralloidolides A, B, C, and E. Angew. Chem. Int. Ed. 2010, 49, 2619–2621. [Google Scholar] [CrossRef]
- Bai, S.; Liao, Y.; Lin, L.; Luo, W.; Liu, X.; Feng, X. N, N’-Dioxide-scandium (III)-catalyzed asymmetric aza-Friedel-Crafts reaction of sesamol with aldimines. J. Org. Chem. 2014, 79, 10662–10668. [Google Scholar] [CrossRef]
- Kobayashi, S.; Tsuchiya, T.; Komoto, I.; Matsuo, J. Scandium perfluoroalkanesulfonate-catalyzed Diels–Alder reactions in an organic solvent. J. Organomet. Chem. 2001, 624, 392–394. [Google Scholar] [CrossRef]
- Rychnovsky, S.D.; Skalitzky, D.J. Stereochemistry of alternating polyol chains: 13C NMR analysis of 1,3-diol acetonides. Tetrahedron Lett. 1990, 31, 945–948. [Google Scholar] [CrossRef]
- Evans, D.A.; Rieger, D.L.; Gage, J.R. 13C NMR chemical shift correlations in 1,3-diol acetonides. Implications for the stereochemical assignment of propionate-derived polyols. Tetrahedron Lett. 1990, 31, 7099–7100. [Google Scholar] [CrossRef]
- Cambridge Structural Database. Available online: https://www.ccdc.cam.ac.uk/structures/ (accessed on 18 February 2021).
- Limbach, M.; Dalai, S.; de Meijere, A. Cyclopropyl building blocks for organic synthesis, Part 100. Advanced syntheses of cyclopropylideneacetates—versatile multifunctional building blocks for organic synthesis. Adv. Synth. Catal. 2004, 346, 760–766. [Google Scholar] [CrossRef]
- Mineeva, I.V.; Kulinkovich, O.G. Synthesis of methyl 3-bromomethylbut-3-enoate and its reactions with aldehydes and tributylchlorostannane in the presence of zinc. Russ. J. Org. Chem. 2008, 44, 1261–1266. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Lewis Acid | Solvent | Time, h | T, °C | Conv. % b | d.r. anti/syn b |
|---|---|---|---|---|---|---|
| 1 | Ti((S)-BINOL)2 | CH2Cl2 | 120 | −20 | trace | – |
| 2 | TiCl4 | CH2Cl2 | 1 | −78 | – c | – |
| 3 | SnCl4 | CH2Cl2 | 1 | −78 | – c | – |
| 4 | TiCl(Oi-Pr)3 | CH2Cl2 | 15 | −20 | no reaction | |
| 5 | Cp2TiCl2 | CH2Cl2 | 15 | −20 | no reaction | |
| 6 | MgBr2∙Et2O d | CH2Cl2 | 5 | −78 | 72 | 76:24 |
| 7 | ZnCl2∙Et2O d | CH2Cl2 | 15 | −20 | 100 | 60:40 |
| 8 | ZrCl4 d | CH2Cl2 | 1 | −60 | 78 | 73:27 |
| 9 | Sc(OTf)3, old batch | CH2Cl2 | 4 | −25 | 65 | 84:16 |
| 10 | Sc(OTf)3, old batch | toluene | 4 | −25 | 86 | 88:12 |
| 11 | In(OTf)3 | toluene | 5 | −70 | 48 | 75:25 |
| 12 | Y(OTf)3 | toluene | 2 | −20 | 45 | 55:45 |
| 13 | Hf(OTf)4 | toluene | 4 | +20 | 28 | 65:35 |
| 14 | Sc(OTf)3, fresh batch | toluene | 5 | −70 | 60 | 76:24 e |
| 15 | Sc(OTf)3 + H2O | toluene | 2 | −25 | 65 | 83:14 |
| 16 | Sc(OTf)3 + 2H2O | toluene | 4 | −25 | 72 | 88:12 |
| 17 | Sc(OTf)3 + 2H2O | toluene/Et2O | 4 | −70 | 78 | 91:9 |
| 18 | Sc(OTf)3 + 2H2O | toluene/Et2O | 12 | −70 | 92 (72) f | 94:6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masiuk, U.S.; Mineyeva, I.V.; Kananovich, D.G. Highly Diastereoselective Chelation-Controlled 1,3-anti-Allylation of (S)-3-(Methoxymethyl)hexanal Enabled by Hydrate of Scandium Triflate. Symmetry 2021, 13, 470. https://doi.org/10.3390/sym13030470
Masiuk US, Mineyeva IV, Kananovich DG. Highly Diastereoselective Chelation-Controlled 1,3-anti-Allylation of (S)-3-(Methoxymethyl)hexanal Enabled by Hydrate of Scandium Triflate. Symmetry. 2021; 13(3):470. https://doi.org/10.3390/sym13030470
Chicago/Turabian StyleMasiuk, Uladzimir S., Iryna V. Mineyeva, and Dzmitry G. Kananovich. 2021. "Highly Diastereoselective Chelation-Controlled 1,3-anti-Allylation of (S)-3-(Methoxymethyl)hexanal Enabled by Hydrate of Scandium Triflate" Symmetry 13, no. 3: 470. https://doi.org/10.3390/sym13030470
APA StyleMasiuk, U. S., Mineyeva, I. V., & Kananovich, D. G. (2021). Highly Diastereoselective Chelation-Controlled 1,3-anti-Allylation of (S)-3-(Methoxymethyl)hexanal Enabled by Hydrate of Scandium Triflate. Symmetry, 13(3), 470. https://doi.org/10.3390/sym13030470