
Synchronization in Non-Mirror-Symmetrical Chirogenesis: Non-Helical π–Conjugated Polymers with Helical Polysilane Copolymers in Co-Colloids

 ,
,  ,
, 
Abstract
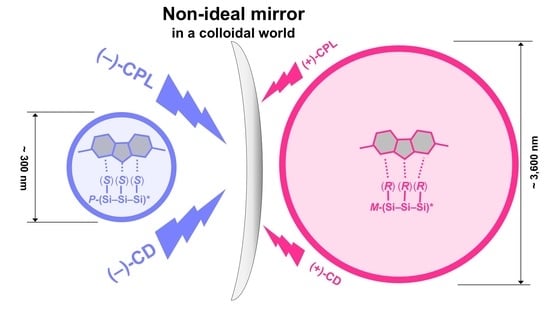
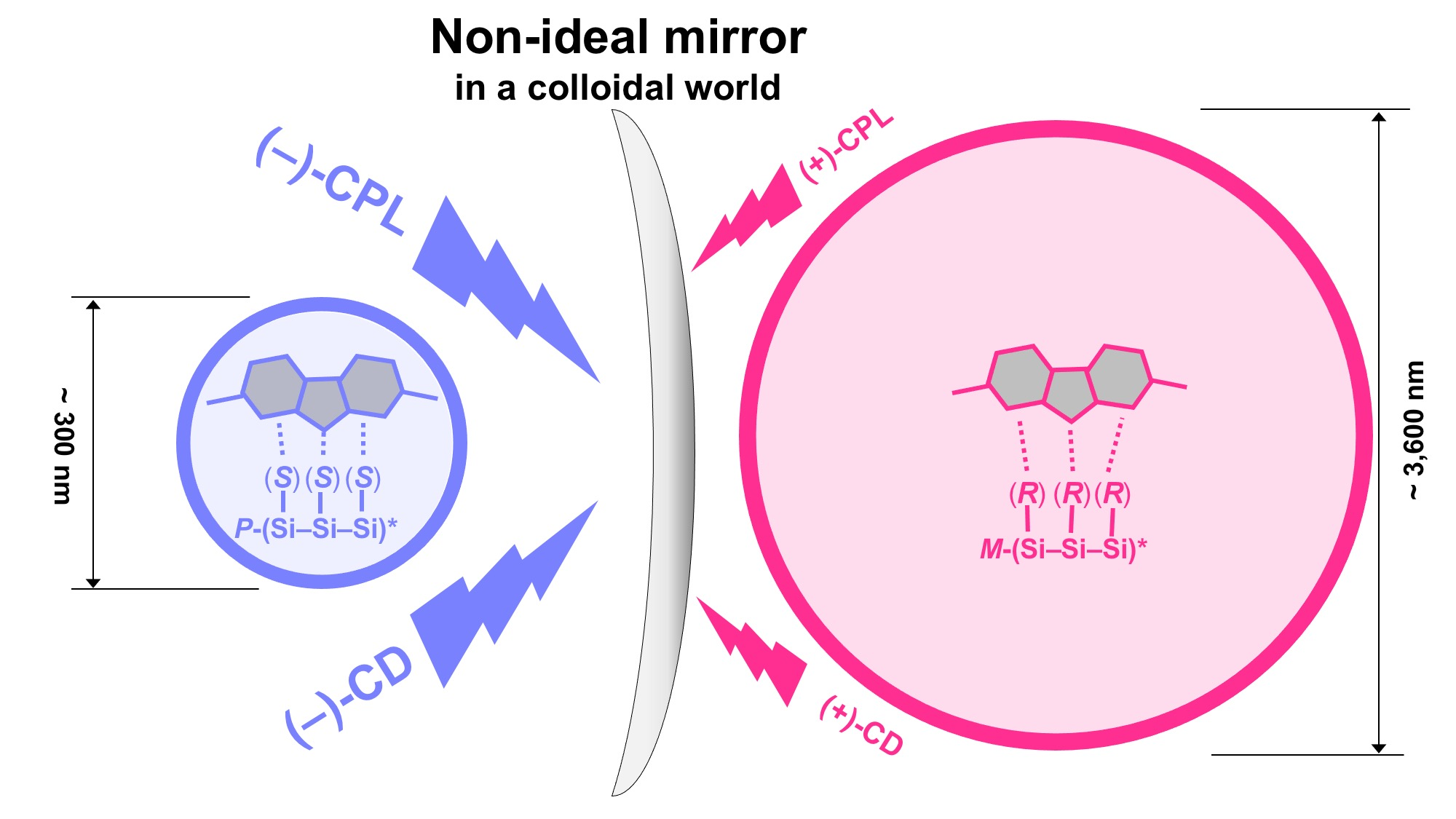
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Sergeants-and-Soldiers and Majority-Rule of Type I HCPS and Type II HCPS
2.1.1. Sergeants-and-Soldiers of Type I HCPS-S and -R in Isooctane and Homo-Colloids in CHCl3-MeOH Cosolvents
2.1.2. Majority-Rule of Type II HCPS in Isooctane and Homo-Colloids in CHCl3-MeOH Cosolvents
2.2. Sergeants-and-Soldiers in PFV8 Co-Colloids with Type I HCPS-S and -R
2.3. Majority-Rule in PFV8 Co-Colloids with Type II HCPS
2.4. Retention in CD and CPL Signals of PFV8 Co-Colloids with Type I HCPS and Type II HCPS by Si–Si Bond Selective Photolysis
2.5. Sergeants-and-Soldiers in PF8 Co-Colloids with Type I HCPS
2.6. Majority Rule in PF8 Co-Colloids with Type II HCPS
3. Discussion
3.1. Main-Chain Rigidity of HPS-S, HPS-R, HPS-IB, Type I HCPS, and Type II HCPS in Solution
3.2. Higher-Order Structures of Helical Polysilane Homo- and Copolymers in the Homo- and Co-Colloids
3.3. Possible Structures of Highly Emmisive PFV8 and PF8 in the Co-Colloids
3.4. Co-Colloidal Stucture of PFV8 and HPS-S by Atomic Force Microscopy
3.5. Possible Inter-Macromolecular Interactions of PFV8/PF8 with HPS/HCPS in the Co-Colloids
3.6. Film-State CD Spectra of PFV8 with Type I HCPS and Type II HCPS
4. Perspectives
5. Methods and Materials
5.1. Instrumentation
5.2. Preparation and Fractionation of Polysilanes and the Corresponding Dialkyldichlorosilanes
5.3. Fractionations of PFV8, PF8, and Polysilanes
5.4. Prepararing PFV8 and PF8 Co-Colloids with HPS, Type I HCPS, and Type II HCPS
5.5. Optimizing Optofluidic Effect of PFV8 Co-Colloids with HPS-S
5.6. Choosing Good-and-Poor Co-Solvent to Maximize Ser-Sol and Maj Effects of PFV8 and PF8 Co-Colloids with HPS and Type I HCPS
5.7. Calculating Mulliken Charges for Oigomeric Models of HPS-S, PFV8, and PF8
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schrodinger, E. What Is Life? With Mind and Matter and Autobiographical Sketches; Cambridge University Press: Cambridge, UK, 1944. [Google Scholar]
- Miller, S.L. A Production of Amino Acids under Possible Primitive Earth Conditions. Science 1953, 117, 528–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akabori, S.; Okawa, K.; Sato, M. Introduction of Side Chains into Polyglycine Dispersed on Solid Surface. I. Bull. Chem. Soc. Jpn 1956, 29, 608–611. [Google Scholar] [CrossRef] [Green Version]
- Hanafusa, H.; Akabori, S. Polymerization of Aminoacetonitrile. Bull. Chem. Soc. Jpn 1959, 32, 626–630. [Google Scholar] [CrossRef] [Green Version]
- Harada, K.; Fox, S.W. Thermal Synthesis of Natural Amino-Acids from a Postulated Primitive Terrestrial Atmosphere. Nature 1964, 201, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Rohlfing, D.L.; Oparin, A.I. Molecular Evolution: Prebiological and Biological; Rohlfing, D.L., Oparin, A.I., Eds.; Springer: New York, NY, USA, 1972. [Google Scholar]
- Gardner, M. The New Ambidextrous Universe–Symmetry and Asymmetry from Mirror Reflections to Superstrings, 3rd ed.; Freeman: New York, NY, USA, 1990; ISBN 9780486442440. [Google Scholar]
- Mason, S.F. Chemical Evolution: Origin of the Elements, Molecules, and Living Systems; Oxford University Press: New York, NY, USA, 1991; ISBN 900–19–8552726. [Google Scholar]
- Seckbach, J.; Chela-Flores, J.; Owen, T.; Raulin, F. Life in the Universe: From the Miller Experiment to the Search for Life on Other Worlds; Seckbach, J., Chela-Flores, J., Owen, T., Raulin, F., Eds.; Kluwer: Dordrecht, Germany, 2004; ISBN 1–4020–2371–5. [Google Scholar]
- Fujiki, M. Possible Scenarios for Homochirality on Earth; Fujiki, M., Ed.; MDPI: Basel, Switzerland, 2019; ISBN 978–3-03921–722–9. [Google Scholar]
- Rauchfuss, H. Chemical Evolution and the Origin of Life; Springer: Berlin, Germany, 2008; ISBN 978–3-540–78822–5. [Google Scholar]
- Guijarro, A.; Yus, M. Origin of Chirality in the Molecules of Life: A Revision from Awareness to the Current Theories and Perspectives of this Unsolved Problem; RSC Publishing: Cambridge, UK, 2008; ISBN 978–0-85404–156–5. [Google Scholar]
- Breslow, R. A Likely Possible Origin of Homochirality in Amino Acids and Sugars on Prebiotic Earth. Tetrahedron Lett. 2011, 52, 2028–2032. [Google Scholar] [CrossRef]
- Wagnière, G.H. On Chirality and the Universal Asymmetry: Reflections on Image and Mirror Image; Wiley-VCH: Weinheim, Germany, 2007; ISBN 978–3-90639–038–3. [Google Scholar]
- Oparin., A.I. Life: Its Nature, Origin, and Development; Academic Press: New York, NY, USA, 1961. [Google Scholar]
- Haldane, J.B.S. An Exact Test for Randomness of Mating. New Biol. 1954, 16, 12–27. [Google Scholar]
- Terayama, H. Method of colloid titration (a new titration between polymer ions. J. Polym. Sci. 1952, 8, 243–253. [Google Scholar] [CrossRef]
- Kipping, F.S.; Pope, W.J. Enantiomorphism. J. Chem. Soc. Trans. 1898, 73, 606–617. [Google Scholar] [CrossRef] [Green Version]
- Perucca, E. Nuove Osservazioni e Misure su Cristalli Otticamente Attivi (NaClO3). II Nuovo Cim. 1919, 18, 112–154. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, P.; Quehl, K. Aktivierung von Komplexsalzen in Wäßriger Lösung. Chem. Ber. 1932, 65, 560–565. [Google Scholar] [CrossRef]
- Borovkov, V.V.; Lintuluoto, J.M.; Inoue, Y. Supramolecular Chirogenesis in Bis(zinc porphyrin): An Absolute Configuration Probe Highly Sensitive to Guest Structure. Org. Lett. 2000, 2, 1565–1568. [Google Scholar] [CrossRef]
- Borovkov, V.V.; Hembury, G.A.; Inoue, Y. Origin, Control, and Application of Supramolecular Chirogenesis in Bisporphyrin-Based Systems. Acc. Chem. Res. 2004, 37, 449–459. [Google Scholar] [CrossRef]
- Jalilah, A.J.; Asanoma, F.; Fujiki, M. Unveiling Controlled Breaking of the Mirror Symmetry of Eu(fod)3 with α-/β-Pinene and BINAP by Circularly Polarised Luminescence (CPL), CPL Excitation, and 19F-/31P {1H}-NMR Spectra and Mulliken Charges. Inorg. Chem. Front. 2018, 5, 2718–2733. [Google Scholar] [CrossRef]
- Fujiki, M.; Wang, L.; Ogata, N.; Asanoma, F.; Okubo, A.; Okazaki, S.; Kamite, H.; Jalilah, A.J. Chirogenesis and Pfeiffer Effect in Optically Inactive EuIII and TbIII Tris (β-diketonate) Upon Intermolecular Chirality Transfer from Poly-and Monosaccharide Alkyl Esters and α-Pinene: Emerging Circularly Polarized Luminescence (CPL) and Circular Dichroism (CD). Front. Chem. 2020, 8, 685. [Google Scholar]
- Fujiki, M. Resonance in Chirogenesis and Photochirogenesis: Colloidal Polymers Meet Chiral Optofluidics. Symmetry 2021, 13, 199. [Google Scholar] [CrossRef]
- Qian, S.X.; Snow, J.B.; Tzeng, H.M.; Chang, R.K. Lasing Droplets—Highlighting the Liquid-Air Interface by Laser-Emission. Science 1986, 23, 486–488. [Google Scholar] [CrossRef] [PubMed]
- McCall, S.L.; Levi, A.F.J.; Slusher, R.E.; Pearton, S.J.; Logan, R.A. Whispering-Gallery Mode Microdisk Lasers. App. Phys. Lett. 1992, 60, 289–291. [Google Scholar] [CrossRef]
- Armani, D.K.; Kippenberg, T.J.; Spillane, S.M.; Vahala, K.J. Ultra-high-Q Toroid Microcavity on a Chip. Nature 2003, 421, 925–928. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.J.; Yan, C.; Yu, N.; Unterhinninghofen, J.; Wiersigb, J.; Pflügla, C.; Diehla, L.; Edamura, T.; Yamanishi, M.; Kan, H.; et al. Whispering-Gallery Mode Resonators for Highly Unidirectional Laser Action. Proc. Natl. Acad. Sci. USA 2010, 107, 22407–22412. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Pinar, M.; Roselló-Mechó, X.; Rivera-Pérez, E.; Díez, A.; Cruz, J.L.; Andrés, M.V. Whispering Gallery Modes for Accurate Characterization of Optical Fibers’ Parameters. In Applications of Optical Fibers for Sensing; IntechOpen: London, UK, 2019; pp. 1–19. [Google Scholar]
- Lord Rayleigh, O.M. The Problem of the Whispering Gallery. Lond. Edinb. Dubl. Phil. Mag. 1910, 20, 1001–1004. [Google Scholar] [CrossRef] [Green Version]
- Raman, C.V.; Sutherland, G.A. Whispering-Gallery Phenomena at St. Paul’s Cathedral. Nature 1921, 108, 42. [Google Scholar] [CrossRef]
- Budden, K.G.; Martin, H.G. The Ionosphere as a Whispering Gallery. Proc. Roy. Soc. Lond. A 1962, 265, 554–569. [Google Scholar]
- Garrett, C.G.B.; Kaiser, W.; Bond, W.L. Stimulated Emission into Optical Whispering Gallery Modes of Spheres. Phys. Rev. 1961, 124, 1807–1809. [Google Scholar] [CrossRef]
- Chiasera, A.; Dumeige, Y.; Feron, P.; Ferrari, M.; Jestin, Y.; Conti, G.N.; Pelli, S.; Soria, S.; Righini, G.C. Spherical Whispering-Gallery-Mode Microresonators. Laser Photon. Rev. 2010, 4, 457–482. [Google Scholar] [CrossRef]
- Ngara, Z.S.; Yamamoto, Y. Modulation of Whispering Gallery Modes from Fluorescent Copolymer Microsphere Resonators by Protonation/Deprotonation. Chem. Lett. 2019, 48, 607–610. [Google Scholar] [CrossRef]
- Nesvizhevsky, V.A.; Voronin, A.Y.; Cubitt, R.; Protasov, K.V. Neutron Whispering Gallery. Nat. Phys. 2010, 6, 114–117. [Google Scholar] [CrossRef] [Green Version]
- Psaltis, D.; Quack, S.R.; Yang, C. Developing Optofluidic Technology through the Fusion of Microfluidics and Optics. Nature 2006, 442, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Domachuk, P.; Littler, I.C.M.; Cronin-Golomb, M.; Eggletona, B.J. Compact Resonant Integrated Microfluidic Refractometer. Appl. Phys. Lett. 2006, 88, 093513. [Google Scholar] [CrossRef]
- Schäfer, J.; Mondia, J.P.; Sharma, R.; Lu, Z.H.; Susha, A.S.; Rogach, A.L.; Wang, L.J. Quantum Dot Microdrop Laser. Nanolett. 2008, 8, 1709–1712. [Google Scholar] [CrossRef]
- Fainman, Y.; Lee, L.P.; Psaltis, D.; Yang, C. Optofluidic; Fainman, Y., Lee, L.P., Psaltis, D., Yang, C., Eds.; McGraw-Hill: New York, NY, USA, 2010. [Google Scholar]
- Fan, X.; White, I.M. Optofluidic Microsystems for Chemical and Biological Analysis. Nat. Photon. 2011, 5, 591–597. [Google Scholar] [CrossRef]
- Schmidt, A.H.; Hawkins, A.R. The Photonic Integration of Non-Solid Media using Optofluidics. Nat. Photonics 2011, 5, 598–604. [Google Scholar] [CrossRef]
- Wolfe, D.B.; Conroy, R.S.; Garstecki, P.; Mayers, B.T.; Fischbach, M.A.; Paul, K.E.; Prentiss, M.; Whitesides, G.M. Dynamic Control of Liquid-Core/Liquid-Cladding Optical Waveguides. Proc. Natl. Acad. Sci. USA 2004, 101, 12434–12438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, S.K.Y.; Li, Z.; Abate, A.R.; Agresti, J.J.; Weitz, D.A.; Psaltis, D.; Whitesides, G.M. A Multi-Color Fast-Switching Microfluidic Droplet Dye Laser. Lab. Chip 2009, 9, 2767–2771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.M.; Reidy, M.P.; Johnson, R.D.; Darling, G.; O’Leary, D.J.; Willson, G. Macromolecular Stereochemistry: The Out-of-Proportion Influence of Optically Active Comonomers on the Conformational Characteristics of Polyisocyanates. The Sergeants and Soldiers Experiment. J. Am. Chem. Soc. 1989, 16, 6452–6454. [Google Scholar] [CrossRef]
- Green, M.M.; Garetz, B.A.; Munoz, B.; Chang, H.; Hoke, S.; Cooks, R.G. Majority Rules in the Copolymerization of Mirror Image Isomers. J. Am. Chem. Soc. 1995, 117, 4181–4182. [Google Scholar] [CrossRef]
- Green, M.M.; Park, J.W.; Sato, T.; Teramoto, A.; Lifson, S.; Selinger, R.L.; Selinger, J.V. The Macromolecular Route to Chiral Amplification. Angew. Chem. Int. Ed. 1999, 38, 3138–3154. [Google Scholar] [CrossRef]
- Ohsawa, S.; Sakurai, S.-i.; Nagai, K.; Banno, M.; Maeda, K.; Yashima, E. Hierarchical Amplification of Macromolecular Helicity of Dynamic Helical Poly(phenylacetylene)s Composed of Chiral and Achiral Phenylacetylenes in Dilute solution, Liquid crystal, and Two-dimensional Crystal. J. Am. Chem. Soc. 2011, 133, 108–114. [Google Scholar] [CrossRef]
- Ito, H.; Ikeda, M.; Hasegawa, T.; Furusho, Y.; Yashima, E. Synthesis of Complementary Double-Stranded Helical Oligomers through Chiral and Achiral Amidinium-Carboxylate Salt Bridges and Chiral Amplification in Their Double-Helix Formation. J. Am. Chem. Soc. 2011, 133, 3419–3432. [Google Scholar] [CrossRef]
- Ohsawa, S.; Sakurai, S.-I.; Nagai1, K.; Maeda, K.; Kumaki, J.; Yashima, E. Amplification of Macromolecular Helicity of Dynamic helical Poly(phenylacetylene)s bearing Non-racemic Alanine Pendants in Dilute Solution, Liquid Crystal and Two-dimensional Crystal. Polym. J. 2012, 44, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Maeda, K.; Yashima, E. Helical Polyacetylenes Induced via Noncovalent Chiral Interactions and Their Applications as Chiral Materials. Top. Curr. Chem. 2017, 375, 72. [Google Scholar] [CrossRef] [Green Version]
- Ishidate, R.; Markvoort, A.J.; Maeda, K.; Yashima, E. Unexpectedly Strong Chiral Amplification of Chiral/Achiral and Chiral/Chiral Copolymers of Biphenylylacetylenes and Further Enhancement/ Inversion and Memory of the Macromolecular Helicity. J. Am. Chem. Soc. 2019, 141, 7605–7614. [Google Scholar] [CrossRef]
- Maeda, K.; Nozaki, M.; Hashimoto, K.; Shimomura, K.; Hirose, D.; Nishimura, T.; Watanabe, G.; Yashima, E. Helix-Sense-Selective Synthesis of Right-and Left-handed Helical Luminescent Poly (diphenylacetylene)s with Memory of the Macromolecular Helicity and Their Helical Structures. J. Am. Chem. Soc. 2020, 142, 7668–7682. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Yamada, T.; Adachi, T.; Akai, Y.; Yamamoto, T.; Suginome, M. Solvent-Dependent Switch of Helical Main-Chain Chirality in Sergeants-and-Soldiers-Type Poly(quinoxaline-2,3-diyl)s: Effect of the Position and Structures of the “Sergeant” Chiral Units on the Screw-Sense Induction. J. Am. Chem. Soc. 2013, 135, 10104–10113. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Nishikawa, T.; Suginome, M. Solvent Effect on the Sergeants-and-Soldiers Effect Leading to Bidirectional Induction of Single-Handed Helical Sense of Poly(quinoxaline-2,3-diyl)s Copolymers in Aromatic Solvents. ACS Macro Lett. 2016, 5, 519–522. [Google Scholar] [CrossRef]
- Ke, Y.-Z.; Nagata, Y.; Yamada, T.; Suginome, M. Majority-Rules-Type Helical Poly(quinoxaline-2,3-diyl)s as Highly Efficient Chirality-Amplification Systems for Asymmetric Catalysis. Angew. Chem. Int. Ed. 2015, 54, 9333–9337. [Google Scholar] [CrossRef] [PubMed]
- Langeveld-Voss, B.M.W.; Waterval, R.J.M.; Janssen, R.A.J.; Meijer, E.W. Principles of “Majority Rules” and “Sergeants and Soldiers” Applied to the Aggregation of Optically Active Polythiophenes: Evidence for a Multichain Phenomenon. Macromolecules 1999, 32, 227–230. [Google Scholar] [CrossRef]
- van Gestel, J.; Palmans, A.R.A.; Titulaer, B.; Vekemans, J.A.J.M.; Meijer, E.W. “Majority-Rules” Operative in Chiral Columnar Stacks of C3-Symmetrical Molecules. J. Am. Chem. Soc. 2005, 127, 5490–5494. [Google Scholar] [CrossRef]
- Smulders, M.M.J.S.; Schenning, A.P.H.J.; Meijer, E.W. Insight into the Mechanisms of Cooperative Self-Assembly: The “Sergeants-and-Soldiers” Principle of Chiral and Achiral C3-Symmetrical Discotic Triamides. J. Am. Chem. Soc. 2008, 130, 606–611. [Google Scholar] [CrossRef]
- Palmans, A.R.A.; Meijer, E.W. Amplification of Chirality in Dynamic Supramolecular Aggregates. Angew. Chem. Int. Ed. 2007, 46, 8948–8968. [Google Scholar] [CrossRef] [PubMed]
- Fujiki, M. Optically Active Polysilylenes: State-of-the-Art Chiroptical Polymers. Macromol. Rapid Commun. 2001, 22, 539–563. [Google Scholar] [CrossRef]
- Fujiki, M.; Koe, J.R.; Terao, K.; Sato, T.; Teramoto, A.; Watanabe, J. Optically Active Polysilanes. Ten Years of Progress and New Polymer Twist for Nanoscience and Nanotechnology. Polym. J. 2003, 35, 297–344. [Google Scholar] [CrossRef] [Green Version]
- Poddar, S.; Chakravarty, D.; Chakrabarti, P. Structural Changes in DNA-Binding Proteins on Complexation. Nucleic Acids Res. 2018, 46, 3298–3308. [Google Scholar] [CrossRef]
- Woo, H.; Beck, S.E.; Boczek, L.A.; Carlson, K.M.; Brinkman, N.E.; Linden, K.G.; Lawal, O.R.; Hayes, S.L.; Ryu, H. Efficacy of Inactivation of Human Enteroviruses by Dual-Wavelength Germicidal Ultraviolet (UV-C) Light Emitting Diodes(LEDs). Water 2019, 11, 1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.D.; Michl, J. Polysilane High Polymers. Chem. Rev. 1989, 89, 1359–1410. [Google Scholar] [CrossRef]
- Rahim, N.A.A.; Fujiki, M. Aggregation-induced Scaffolding: Photoscissable Helical Polysilane Generates Circularly Polarized Luminescent Polyfluorene. Polym. Chem. 2016, 7, 4618–4629. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, N.; Ito, R.; Fujiki, M.; Geerts, Y.; Nomura, K. Synthesis of All-Trans High Molecular Weight Poly(N-alkyl-carbazole-2,7-vinylene)s and Poly(9,9-dialkylfluorene-2,7-vinylene)s by Ayclic Diene Metathesis (ADMET) Polymerization Using Ruthenium-Carbene Complex Catalysts. Macromolecules 2009, 42, 5104–5111. [Google Scholar] [CrossRef]
- Takamizu, K.; Inagaki, A.; Nomura, K. Precise Synthesis of Poly(fluorene vinylene)s Capped with Chromophores: Efficient Fluorescent Polymers Modified by Conjugation Length and End-Groups. Acs Macro Lett. 2013, 2, 980–984. [Google Scholar] [CrossRef]
- Fujiki, M.; Jalilah, A.J.; Suzuki, N.; Taguchi, M.; Zhang, W.; Abdellatif, M.M.; Nomura, K. Chiral Optofluidics: Gigantic Circularly Polarized Light Enhancement of all-trans-Poly(9,9-di-n-octylfluorene-2,7-vinylene) during Mirror-Symmetry-Breaking Aggregation by Optically Tuning Fluidic Media. RSC Adv. 2012, 2, 6663–6671. [Google Scholar] [CrossRef]
- Fujiki, M. Effect of Main Chain Length in the Exciton Spectra of Helical-Rod Polysilanes as a Model of a 5 Å Wide Quantum Wire. Appl. Phys. Lett. 1994, 65, 3251–3253. [Google Scholar] [CrossRef]
- Nakano, Y.; Fujiki, M. Circularly Polarized Light Enhancement by Helical Polysilane Aggregates Suspension in Organic Optofluids. Macromolecules 2011, 44, 7511–7919. [Google Scholar] [CrossRef]
- Hauser, E.A. The History of Colloid Science: In Memory of Wolfgang Ostwald. J. Chem. Educ. 1955, 32, 2–9. [Google Scholar] [CrossRef]
- Ostwald Ripening. Available online: https://en.wikipedia.org/wiki/Ostwald_ripening (accessed on 14 January 2021).
- Sugimoto, T. Reversed Ostwald Ripening. J. Soc. Photogr. Sci. Technol. Jpn. 1983, 46, 306–312. [Google Scholar]
- Burlakov, V.M.; Goriely, A. Reverse Coarsening and the Control of Particle Size Distribution through Surfactant. Appl. Sci. 2020, 10, 5359. [Google Scholar] [CrossRef]
- Rosowski, K.A.; Vidal-Henriquez, E.; Zwicker, D.; Style, R.W.; Dufresne, E.R. Elastic Stresses Reverse Ostwald Ripening. Soft Matter. 2020, 16, 5892–5897. [Google Scholar] [CrossRef] [PubMed]
- Grell, M.; Bradley, D.D.C.; Long, X.; Chamberlain, T.; Inbasekaran, M.; Woo, E.P.; Soliman, M. Chain Geometry, Solution Aggregation and Enhanced Dichroism in the Liquid-Crystalline Conjugated Polymer Poly(9,9-dioctylfluorene). Acta Polym. 1998, 49, 439–444. [Google Scholar] [CrossRef]
- Scherf, U.; List, E.J.W. Semiconducting Polyfluorenes–Towards Reliable Structure–Property Relationships. Adv. Mater. 2002, 14, 477–487. [Google Scholar] [CrossRef]
- Knaapila, M.; Monkman, A.P. Methods for Controlling Structure and Photophysical Properties in Polyfluorene Solutions and Gels. Adv. Mater. 2013, 25, 1090–1108. [Google Scholar] [CrossRef]
- Mason, S.F.; Tranter, G.E. The Electroweak Origin of Biomolecular Handedness. Proc. R. Soc. Lond. A 1985, 397, 45–65. [Google Scholar]
- Quack, M. How Important is Parity Violation for Molecular and Biomolecular Chirality? Angew. Chem. Int. Ed. 2002, 41, 4618–4630. [Google Scholar] [CrossRef]
- Berger, R. Molecular Parity Violation in Electronically Excited States. Phys. Chem. Chem. Phys. 2003, 5, 12–17. [Google Scholar] [CrossRef]
- Schwerdtfeger, P.; Gierlich, J.; Bollwein, T. Large Parity-Violation Effects in Heavy-Metal-Containing Chiral Compounds. Angew. Chem. Int. Ed. 2003, 42, 1293–1296. [Google Scholar] [CrossRef]
- Faglioni, F.; Passalacqua, A.; Lazzeretti, P. Parity Violation Energy of Biomolecules–I: Polypeptides. Orig. Life Evol. Biosph. 2005, 35, 461–475. [Google Scholar] [CrossRef]
- Quack, M.; Stohner, J.; Willeke, M. High-Resolution Spectroscopic Studies and Theory of Parity Violation in Chiral Molecules. Annu. Rev. Phys. Chem. 2008, 59, 741–769. [Google Scholar] [CrossRef]
- Schwerdtfeger, P. The Search for Parity Violation in Chiral Molecules. In Computational Spectroscopy: Methods, Experiments and Applications, Chapter 6; Grunenberg, J., Ed.; Wiley: Hoboken, NJ, USA, 2010; pp. 201–221. [Google Scholar]
- Daussy, C.; Marrel, T.; Amy-Klein, A.A.-K.; Nguyen, V.T.; Bordé, C.J.; Chardonnet, C. Limit on the Parity Nonconserving Energy Difference between the Enantiomers of a Chiral Molecule by Laser Spectroscopy. Phys. Rev. Lett. 1999, 83, 1554–1557. [Google Scholar] [CrossRef]
- Darquié, B.; Stoeffler, C.; Shelkovnikov, A.; Daussy, C.; Amy-Klein, A.; Chardonnet, C.; Zrig, S.; Guy, L.; Crassous, J.; Soulard, P.; et al. Progress Toward the First Observation of Parity Violation in Chiral Molecules by High-Resolution Laser Spectroscopy. Chirality 2010, 22, 870–884. [Google Scholar] [CrossRef] [Green Version]
- Szabo-Nagy, A.; Keszthelyi, L. Demonstration of the Parity-Violating Energy Difference Between Enantiomers. Proc. Natl. Acad. Sci. USA 1999, 96, 4252–4255. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.Q.; Yi, F.; Ni, Y.M.; Zhao, Z.X.; Jin, X.; Tang, Y. Parity Violations of Electroweak Force in Phase Transitions of Single Crystals of D- and L-Alanine and Valine. J. Biol. Phys. 2000, 26, 51–65. [Google Scholar] [CrossRef]
- Fujiki, M. Experimental Tests of Parity Violation at Helical Polysilylene Level. Macromol. Rapid Commun. 2001, 22, 669–674. [Google Scholar] [CrossRef]
- Shinitzky, M.; Nudelman, F.; Barda, Y.; Haimovitz, R.; Chen, E.; Deamer, D.W. Unexpected Differences Between D-and L-Tyrosine Lead to Chiral Enhancement in Racemic Mixtures. Orig. Life Evol. Biosph. 2002, 32, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.; Pyda, M.; Pak, J.; Wunderlich, B.; Thompson, J.R.; Pagni, R.; Barnes, C.; Schwerdtfeger, P.; Compton, R. Search for Electroweak Interactions in Amino Acid Crystals II—The Salam Hypothesis. J. Phys. Chem. A 2003, 107, 6674–6680. [Google Scholar] [CrossRef]
- Lahav, M.; Weissbuch, I.; Shavit, E.; Reiner, C.; Nicholson, G.J.; Schurig, V. Parity Violating Energetic Difference and Enantiomorphous Crystals–Caveats; Reinvestigation of Tyrosine Crystallization. Orig. Life Evol. Biosph. 2006, 36, 151–170. [Google Scholar] [CrossRef]
- Scolnik, Y.; Portnaya, I.; Cogan, U.; Tal, S.; Haimovitz, R.; Fridkin, M.; Elitzur, A.C.; Deamer, D.W.; Shinitzky, M. Subtle Differences in Structural Transitions between Poly-L-and Poly-D-Amino Acids of Equal Length in Water. Phys. Chem. Chem. Phys. 2006, 8, 333–339. [Google Scholar] [CrossRef]
- Viedma, C. Selective Chiral Symmetry Breaking during Crystallization: Parity Violation or Cryptochiral Environment in Control? Cryst. Growth Des. 2007, 7, 553–556. [Google Scholar] [CrossRef]
- Kodona, E.K.; Alexopoulos, C.; Panou-Pomonis, E.; Pomonis, P.J. Chirality and Helix Stability of Polyglutamic Acid Enantiomers. J. Colloid Interface Sci. 2008, 319, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Fujiki, M. Mirror Symmetry Breaking in Helical Polysilanes: Preference between Left and Right of Chemical and Physical Origin. Symmetry 2010, 2, 1625–1652. [Google Scholar] [CrossRef] [Green Version]
- Kamide, K.; Kataoka, A. Theoretical Relationships Between Parameters in the Kuhn-Mark-Houwink-Sakurada Equation. Macromol. Chem. Phys. 2003, 128, 217–228. [Google Scholar] [CrossRef]
- Fujiki, M. A Correlation between Global Conformation of Polysilane and UV Absorption Characteristics. J. Am. Chem. Soc. 1996, 118, 7424–7425. [Google Scholar] [CrossRef]
- Terao, K.; Terao, Y.; Teramoto, A.; Nakamura, N.; Terakawa, I.; Sato, T.; Fujiki, M. Stiffness of Polysilylenes Depending Remarkably on a Subtle Difference in Chiral Side Chain Structure: Poly{n-hexyl-[(S)-2-methylbutyl]silylene)} and poly{n-hexyl-[(S)-3-methylpentyl]silylene}. Macromolecules 2001, 34, 2682–2685. [Google Scholar] [CrossRef]
- Sato, T.; Terao, K.; Teramoto, A.; Fujiki, M. Molecular Properties of Helical Polysilylenes in Solution. Polymer 2003, 44, 5477–5495. [Google Scholar] [CrossRef] [Green Version]
- Okoshi, K.; Kamee, H.; Suzaki, G.; Tokita, M.; Fujiki, M.; Watanabe, J. Well-Defined Phase Sequence Including Cholesteric, Smectic A, and Columnar Phases Observed in a Thermotropic LC System of Simple Rigid-Rod Helical Polysilane. Macromolecules 2002, 35, 4556–4559. [Google Scholar] [CrossRef]
- Okoshi, K.; Saxena, A.; Naito, M.; Suzaki, G.; Tokita, M.; Watanabe, J.; Fujiki, M. First Observation of a Smectic A–Cholesteric Phase Transition in a Thermotropic Liquid Crystal Consisting of a Rigid-Rod Helical Polysilane. Liq. Cryst. 2004, 31, 279–283. [Google Scholar] [CrossRef]
- Peng, W.; Motonaga, M.; Koe, J.R. Chirality Control in Optically Active Polysilane Aggregates. J. Am. Chem. Soc. 2004, 126, 13822–13826. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Fujiki, M.; Kimpinde-Kalunga, R.; Koe, J.R. Chiroptical Inversion in Helical Si–Si Bond Polymer Aggregates. J. Am. Chem. Soc. 2013, 135, 13073–13079. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Nishikawa, T.; Suginome, M. Abnormal Sergeants-and-Soldiers effects of Poly-(quinoxaline-2,3-diyl)s Enabling Discrimination of One-Carbon Homologous n-Alkanes Through a Highly Sensitive Solvent-Dependent Helix Inversion. Chem. Commun. 2018, 54, 6867–6870. [Google Scholar] [CrossRef]
- Nagata, Y.; Hasegawa, H.; Terao, K.; Suginome, M. Main-Chain Stiffness and Helical Conformation of a Poly(quinoxaline-2,3-diyl) in Solution. Macromolecules 2015, 48, 7983–7989. [Google Scholar] [CrossRef]
- Kueia, B.; Gomez, E.D. Chain conformations and phase behavior of conjugated polymers. Soft Matter. 2017, 13, 49–67. [Google Scholar] [CrossRef]
- Fytas, G.; Nothofer, H.G.; Scherf, U.; Vlassopoulos, D.; Meier, G. Structure and Dynamics of Nondilute Polyfluorene Solutions. Macromolecules 2002, 35, 481–488. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, M.; Suzuki, N.; Fujiki, M. Intramolecular CH/π interaction of Poly(9,9-dialkylfluorene)s in Solutions: Interplay of the Fluorene Ring and Alkyl Side Chains Revealed by 2D 1H–1H NOESY NMR and 1D 1H-NMR experiments. Polym. J. 2013, 45, 1047–1057. [Google Scholar] [CrossRef]
- Suzuki, N.; Matsuda, T.; Nagai, T.; Yamazaki, K.; Fujiki, M. Investigation of the Intra-CH/π Interaction in Dibromo-9,9’-dialkylfluorenes. Cryst. Growth Des. 2016, 16, 6593–6599. [Google Scholar] [CrossRef]
- Nishio, M. The CH/π hydrogen bond in chemistry. Conformation, supramolecules, optical resolution and interactions involving carbohydrates. Phys. Chem. Chem. Phys. 2011, 13, 13873–13900. [Google Scholar] [CrossRef]
- Rajagopal, S.K.; Philip, A.M.; Nagarajan, K.; Hariharan, M. Progressive Acylation of Pyrene Engineers Solid State Packing and Colour via C–H⋯H–C, C–H⋯O and π–π Interactions. Chem. Commun. 2014, 50, 8644–8647. [Google Scholar] [CrossRef]
- Ribozyme. Available online: https://en.wikipedia.org/wiki/Ribozyme (accessed on 12 February 2021).
- Herbert, A.; Rich, A. The Biology of Left-handed Z-DNA. J. Biol. Chem. 1996, 271, 11595–11598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eschenmoser, A. Chemical Etiology of Nucleic Acid Structure. Science 1999, 284, 2118–2124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urata, H.; Shinohara, K.; Ogura, E.; Ueda, Y.; Akagi, M. Mirror-Image DNA. J. Am. Chem. Soc. 1991, 113, 8174–8175. [Google Scholar] [CrossRef]
- Zawadzke, L.E.; Berg, J.M. A Racemic Protein. J. Am. Chem. Soc. 1992, 114, 4002–84003. [Google Scholar] [CrossRef]
- Kobayashi, K.; Asakawa, Y.; Kikuchi, Y.; Toi, H.; Aoyama, Y. CH–π Interaction as an Important Driving Force of Host-Guest Complexation in Apolar Organic Media. Binding of Monools and Acetylated Compounds to Resorcinol Cyclic Tetramer as Studied by 1H NMR and Circular Dichroism Spectroscopy. J. Am. Chem. Soc. 1993, 115, 2648–2654. [Google Scholar] [CrossRef]
- Watanabe, K.; Osaka, I.; Yorozuya, S.; Akagi, K. Helically π-Stacked Thiophene-Based Copolymers with Circularly Polarized Fluorescence: High Dissymmetry Factors Enhanced by Self- Ordering in Chiral Nematic Liquid Crystal Phase. Chem. Mater. 2012, 24, 1011–1024. [Google Scholar] [CrossRef]
- San Jose, B.A.; Matsushita, S.; Akagi, K. Lyotropic Chiral Nematic Liquid Crystalline Aliphatic Conjugated Polymers Based on Disubstituted Polyacetylene Derivatives That Exhibit High Dissymmetry Factors in Circularly Polarized Luminescence. J. Am. Chem. Soc. 2012, 134, 19795–19807. [Google Scholar] [CrossRef]
- Takaishi, K.; Hinoide, S.; Matsumoto, T.; Ema, T. Axially Chiral peri-Xanthenoxanthenes as a Circularly Polarized Luminophore. J. Am. Chem. Soc. 2019, 141, 11852–11857. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, K.; Iwachido, K.; Takehana, R.; Uchiyama, M.; Ema, T. Evolving Fluorophores into Circularly Polarized Luminophores with a Chiral Naphthalene Tetramer: Proposal of Excimer Chirality Rule for Circularly Polarized Luminescence. J. Am. Chem. Soc. 2019, 141, 6185–6190. [Google Scholar] [CrossRef]
- Maeda, K.; Hirose, D.; Okoshi, N.; Shimomura, K.; Wada, Y.; Ikai, T.; Kanoh, S.; Yashima., E. Direct Detection of Hardly Detectable Hidden Chirality of Hydrocarbons and Deuterated Isotopomers by a Helical Polyacetylene through Chiral Amplification and Memory. J. Am. Chem. Soc. 2018, 140, 3270–3276. [Google Scholar] [CrossRef] [PubMed]
- Ribó, J.M.; Crusats, J.; Sagués, F.; Claret, J.; Rubires, R. Chiral Sign Induction by Vortices During the Formation of Mesophases in Stirred Solutions. Science 2001, 292, 2063–2066. [Google Scholar] [CrossRef] [PubMed]
- Kozlova, S.G.; Gabuda, S. Thermal Properties of Zn2(C8H4O4)2•C6H12N2 Metal-Organic Framework Compound and Mirror Symmetry Violation of Dabco Molecules. Sci. Rep. 2017, 7, 11505. [Google Scholar] [CrossRef]
- Fujiki, M.; Koe, J.R.; Mori, T.; Kimura, Y. Questions of Mirror Symmetry at the Photoexcited and Ground States of Non-Rigid Luminophores Raised by Circularly Polarized Luminescence and Circular Dichroism Spectroscopy: Part 1. Oligofluorenes, Oligophenylenes, Binaphthyls and Fused Aromatics. Molecules 2018, 23, 2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiki, M.; Koe, J.R.; Amazumi, S. Questions of Mirror Symmetry at the Photoexcited and Ground States of Non-Rigid Luminophores Raised by Circularly Polarized Luminescence and Circular Dichroism Spectroscopy, Part 2: Perylenes, BODIPYs, Molecular Scintillators, Coumarins, Rhodamine B, and DCM. Symmetry 2019, 11, 363. [Google Scholar]
- Puneet, P.; Singh, S.; Fujiki, M.; Nandan, B. Handed Mirror Symmetry Breaking at the Photo-Excited State of π-Conjugated Rotamers in Solutions. Symmetry 2021, 13, 272. [Google Scholar] [CrossRef]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fujiki, M.; Okazaki, S.; Rahim, N.A.A.; Yamada, T.; Nomura, K. Synchronization in Non-Mirror-Symmetrical Chirogenesis: Non-Helical π–Conjugated Polymers with Helical Polysilane Copolymers in Co-Colloids. Symmetry 2021, 13, 594. https://doi.org/10.3390/sym13040594
Fujiki M, Okazaki S, Rahim NAA, Yamada T, Nomura K. Synchronization in Non-Mirror-Symmetrical Chirogenesis: Non-Helical π–Conjugated Polymers with Helical Polysilane Copolymers in Co-Colloids. Symmetry. 2021; 13(4):594. https://doi.org/10.3390/sym13040594
Chicago/Turabian StyleFujiki, Michiya, Shun Okazaki, Nor Azura Abdul Rahim, Takumi Yamada, and Kotohiro Nomura. 2021. "Synchronization in Non-Mirror-Symmetrical Chirogenesis: Non-Helical π–Conjugated Polymers with Helical Polysilane Copolymers in Co-Colloids" Symmetry 13, no. 4: 594. https://doi.org/10.3390/sym13040594
APA StyleFujiki, M., Okazaki, S., Rahim, N. A. A., Yamada, T., & Nomura, K. (2021). Synchronization in Non-Mirror-Symmetrical Chirogenesis: Non-Helical π–Conjugated Polymers with Helical Polysilane Copolymers in Co-Colloids. Symmetry, 13(4), 594. https://doi.org/10.3390/sym13040594