
iRG-4mC: Neural Network Based Tool for Identification of DNA 4mC Sites in Rosaceae Genome




Abstract
:1. Introduction
2. Benchmark Dataset
3. Methodology
4. Figure of Merits
False Positive (FP) = Non 4mC incorrectly classified as 4mC
True Negative (TN) = Non 4mC correctly classified as Non 4mC
False Negative (FN) = 4mC incorrectly classified as Non 4mC
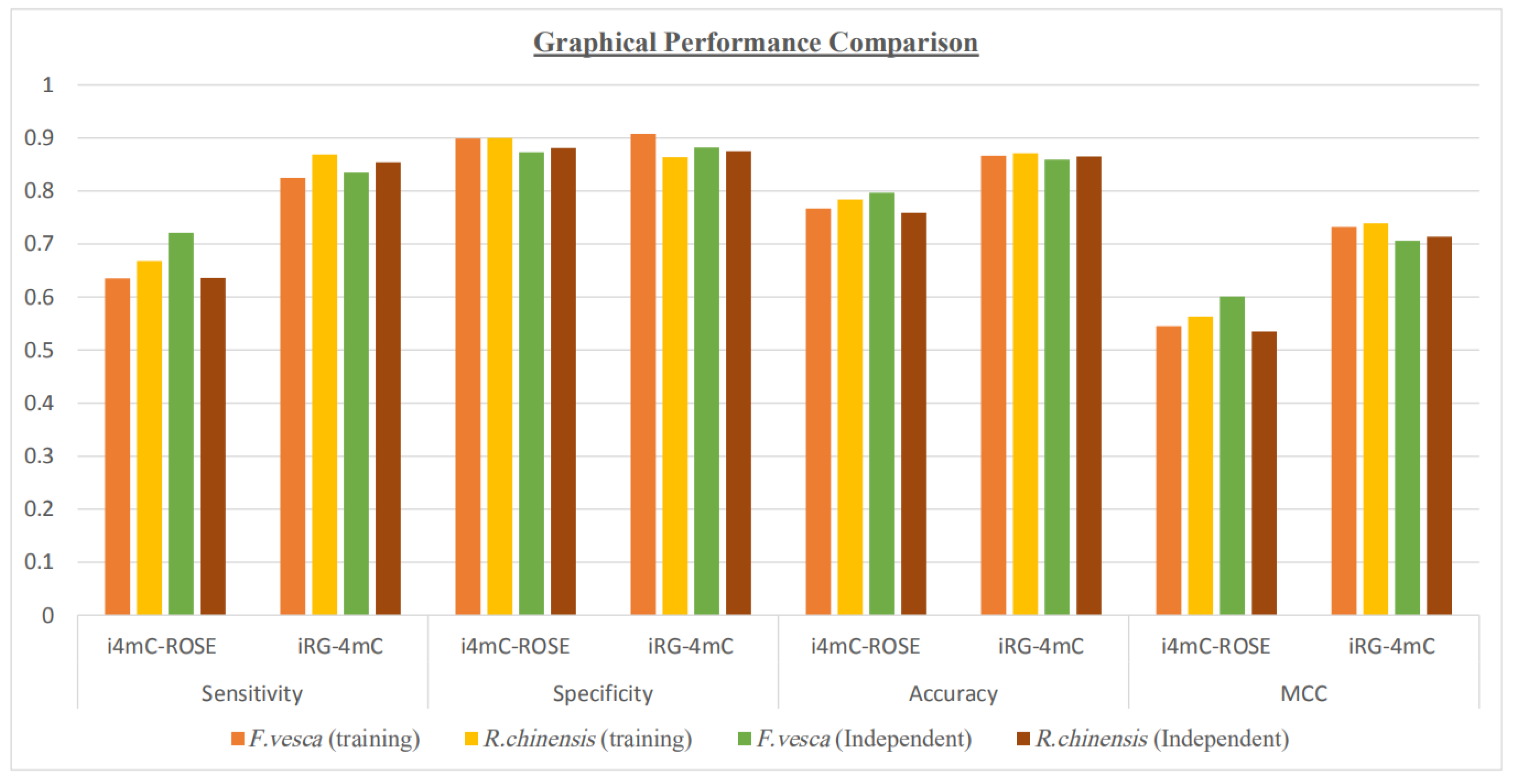
5. Results and Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Rathi, P.; Maurer, S.; Summerer, D. Selective recognition of N 4-methylcytosine in DNA by engineered transcription-activator-like effectors. Philos. Trans. R. Soc. Biol. Sci. 2018, 373, 20170078. [Google Scholar] [CrossRef] [Green Version]
- Jeltsch, A.; Jurkowska, R.Z. New concepts in DNA methylation. Trends Biochem. Sci. 2014, 39, 310–318. [Google Scholar] [CrossRef]
- Jin, Z.; Liu, Y. DNA methylation in human diseases. Genes Dis. 2018, 5, 1–8. [Google Scholar] [CrossRef]
- Zhang, H.; Lang, Z.; Zhu, J.-K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 8, 489–506. [Google Scholar]
- Liang, Z.; Shen, L.; Cui, X.; Bao, S.; Geng, Y.; Yu, G.; Liang, F.; Xie, S.; Lu, T.; Gu, X.; et al. DNA N6-adenine methylation in Arabidopsis thaliana. Dev. Cell 2018, 45, 406–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Eccles, M.R. DNA methylation and epigenomics: New technologies and emerging concepts. Genome Biol. 2015, 16. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Luo, G.Z.; Chen, K.; Deng, X.; Yu, M.; Han, D.; Hao, Z.; Liu, J.; Lu, X.; Doré, L.C.; et al. N6-methyldeoxyadenosine marks active transcription start sites in Chlamydomonas. Cell 2015, 161, 879–892. [Google Scholar] [CrossRef] [Green Version]
- Blow, M.J.; Clark, T.A.; Daum, C.G.; Deutschbauer, A.M.; Fomenkov, A.; Fries, R.; Froula, J.; Kang, D.D.; Malmstrom, R.R.; Morgan, R.D.; et al. The epigenomic landscape of prokaryotes. PLoS Genet. 2016, 12, e1005854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Yang, H.; Feng, P.; Ding, H.; Lin, H. iDNA4mC: Identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics 2017, 33, 3518–3523. [Google Scholar] [CrossRef]
- Heyn, H.; Esteller, M. An adenine code for DNA: A second life for N6-methyladenine. Cell 2015, 161, 710–713. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X. DNA modification by methyltransferases. Curr. Opin. Struct. Biol. 1995, 5, 4–10. [Google Scholar] [CrossRef]
- Wei, L.; Luan, S.; Nagai, L.A.E.; Su, R.; Zou, Q. Exploring sequence-based features for the improved prediction of DNA N4-methylcytosine sites in multiple species. Bioinformatics 2019, 35, 1326–1333. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, H.P. Bacterial genetics: Past achievements, present state of the field, and future challenges. Biotechniques 2008, 44, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005, 6, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Yao, B.; Jin, P. Cytosine modifications in neurodevelopment and diseases. Cell. Mol. Life Sci. 2014, 71, 405–418. [Google Scholar] [CrossRef] [Green Version]
- Ling, C.; Groop, L. Epigenetics: A molecular link between environmental factors and type 2 diabetes. Diabetes 2009, 58, 2718–2725. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Zhao, B.S.; He, C. Nucleic acid modifications in regulation of gene expression. Cell Chem. Biol. 2016, 23, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Doherty, R.; Couldrey, C. Exploring genome wide bisulfite sequencing for DNA methylation analysis in livestock: A technical assessment. Front. Genet. 2014, 5, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buryanov, Y.I.; Shevchuk, T. DNA methyltransferases and structural-functional specificity of eukaryotic DNA modification. Biochemistry 2005, 70, 730–742. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, J.; Wang, Y.; Li, S.; Jia, C.; Song, J.; Li, F. DeepTorrent: A deep learning-based approach for predicting DNA N4-methylcytosine sites. Brief. Bioinform. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Khanal, J.; Nazari, I.; Tayara, H.; Chong, K.T. 4mCCNN: Identification of N4-methylcytosine sites in prokaryotes using convolutional neural network. IEEE Access 2019, 7, 145455–145461. [Google Scholar] [CrossRef]
- Hasan, M.M.; Manavalan, B.; Khatun, M.S.; Kurata, H. i4mC-ROSE, a bioinformatics tool for the identification of DNA N4-methylcytosine sites in the Rosaceae genome. Int. J. Biol. Macromol. 2020, 157, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Raymond, O.; Gouzy, J.; Just, J.; Badouin, H.; Verdenaud, M.; Lemainque, A.; Vergne, P.; Moja, S.; Choisne, N.; Pont, C.; et al. The Rosa genome provides new insights into the domestication of modern roses. Nat. Genet. 2018, 50, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Edger, P.P.; VanBuren, R.; Colle, M.; Poorten, T.J.; Wai, C.M.; Niederhuth, C.E.; Alger, E.I.; Ou, S.; Acharya, C.B.; Wang, J.; et al. Single-molecule sequencing and optical mapping yields an improved genome of woodland strawberry (Fragaria vesca) with chromosome-scale contiguity. Gigascience 2018, 7, gix124. [Google Scholar] [CrossRef] [Green Version]
- Gruenbaum, Y.; Naveh-Many, T.; Cedar, H.; Razin, A. Sequence specificity of methylation in higher plant DNA. Nature 1981, 292, 860–862. [Google Scholar] [CrossRef]
- Rehman, M.U.; Cho, S.; Kim, J.H.; Chong, K.T. BU-Net: Brain Tumor Segmentation Using Modified U-Net Architecture. Electronics 2020, 9, 2203. [Google Scholar] [CrossRef]
- Rehman, M.U.; Cho, S.; Kim, J.; Chong, K.T. BrainSeg-Net: Brain Tumor MR Image Segmentation via Enhanced Encoder-Decoder Network. Diagnostics 2021, 11, 169. [Google Scholar] [CrossRef]
- Rehman, M.U.; Abbas, Z.; Khan, S.H.; Ghani, S.H. Diabetic retinopathy fundus image classification using discrete wavelet transform. In Proceedings of the 2018 IEEE 2nd International Conference on Engineering Innovation (ICEI), Bangkok, Thailand, 5–6 July 2018; pp. 75–80. [Google Scholar]
- Ilyas, T.; Khan, A.; Umraiz, M.; Kim, H. Seek: A framework of superpixel learning with cnn features for unsupervised segmentation. Electronics 2020, 9, 383. [Google Scholar] [CrossRef] [Green Version]
- Ilyas, T.; Umraiz, M.; Khan, A.; Kim, H. DAM: Hierarchical Adaptive Feature Selection Using Convolution Encoder Decoder Network for Strawberry Segmentation. Front. Plant Sci. 2021, 12, 189. [Google Scholar] [CrossRef]
- Okinda, C.; Nyalala, I.; Korohou, T.; Okinda, C.; Wang, J.; Achieng, T.; Wamalwa, P.; Mang, T.; Shen, M. A review on computer vision systems in monitoring of poultry: A welfare perspective. Artif. Intell. Agric. 2020, 4, 184–208. [Google Scholar] [CrossRef]
- Heinrich, F.; Wutke, M.; Das, P.P.; Kamp, M.; Gültas, M.; Link, W.; Schmitt, A.O. Identification of regulatory SNPs associated with vicine and convicine content of Vicia faba based on genotyping by sequencing data using deep learning. Genes 2020, 11, 614. [Google Scholar] [CrossRef] [PubMed]
- Yik, S.; Benjamin, M.; Lavagnino, M.; Morris, D. DIAT (Depth-Infrared Image Annotation Transfer) for Training a Depth-Based Pig-Pose Detector. In Proceedings of the 2020 IEEE/RSJ International Conference on Intelligent Robots and Systems (IROS), Las Vegas, NV, USA, 25–29 October 2020. [Google Scholar] [CrossRef]
- Wutke, M.; Schmitt, A.O.; Traulsen, I.; Gültas, M. Investigation of Pig Activity Based on Video Data and Semi-Supervised Neural Networks. AgriEngineering 2020, 2, 39. [Google Scholar] [CrossRef]
- Rehman, M.U.; Chong, K.T. DNA6mA-MINT: DNA-6mA modification identification neural tool. Genes 2020, 11, 898. [Google Scholar] [CrossRef] [PubMed]
- Abbas, Z.; Tayara, H.; to Chong, K. SpineNet-6mA: A Novel Deep Learning Tool for Predicting DNA N6-Methyladenine Sites in Genomes. IEEE Access 2020, 8, 201450–201457. [Google Scholar] [CrossRef]
- Rehman, M.U.; Hong, K.J.; Tayara, H.; to Chong, K. m6A-NeuralTool: Convolution Neural Tool for RNA N6-Methyladenosine Site Identification in Different Species. IEEE Access 2021, 9, 17779–17786. [Google Scholar] [CrossRef]
- Alam, W.; Ali, S.D.; Tayara, H.; to Chong, K. A CNN-based RNA n6-methyladenosine site predictor for multiple species using heterogeneous features representation. IEEE Access 2020, 8, 138203–138209. [Google Scholar] [CrossRef]
- Abbas, Z.; Tayara, H.; Chong, K.T. 4mCPred-CNN—Prediction of DNA N4-Methylcytosine in the Mouse Genome Using a Convolutional Neural Network. Genes 2021, 12, 296. [Google Scholar] [CrossRef]
- Ali, S.D.; Alam, W.; Tayara, H.; Chong, K. Identification of functional piRNAs using a convolutional neural network. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020. [Google Scholar] [CrossRef] [PubMed]
- Alam, W.; Tayara, H.; Chong, K.T. XG-ac4C: Identification of N4-acetylcytidine (ac4C) in mRNA using eXtreme gradient boosting with electron-ion interaction pseudopotentials. Sci. Rep. 2020, 10, 20942. [Google Scholar] [CrossRef] [PubMed]
- Shujaat, M.; Wahab, A.; Tayara, H.; Chong, K.T. pcPromoter-CNN: A CNN-Based Prediction and Classification of Promoters. Genes 2020, 11, 1529. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Y.; Xing, J.F.; Chen, W.; Luan, M.W.; Xie, R.; Huang, J.; Xie, S.Q.; Xiao, C.L. MDR: An integrative DNA N6-methyladenine and N4-methylcytosine modification database for Rosaceae. Hortic. Res. 2019, 6, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Jeong, B.S.; Bari, A.G.; Reaz, M.R.; Jeon, S.; Lim, C.G.; Choi, H.J. Codon-based encoding for DNA sequence analysis. Methods 2014, 67, 373–379. [Google Scholar] [CrossRef] [PubMed]




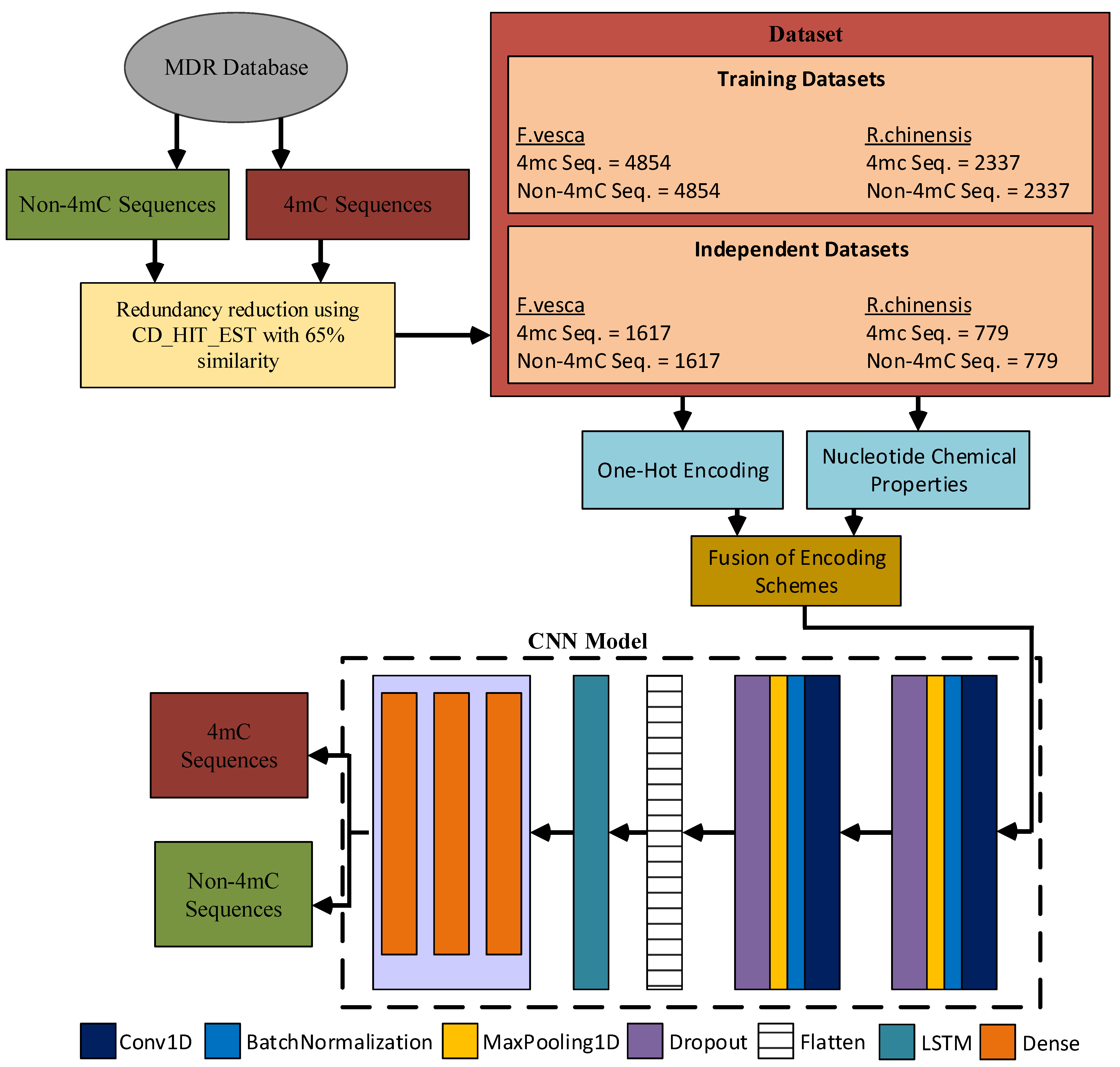{kind=link}
{kind=link}
{kind=link}
| Specie | Sequence Class | Number of Sequences of Seq | Sequence Length |
|---|---|---|---|
| F. vesca (training) | 4mC | 4854 | 41 bp |
| non-4mC | 4854 | ||
| R. chinensis (training) | 4mC | 2337 | 41 bp |
| non-4mC | 2337 | ||
| F. vesca (Independent) | 4mC | 1617 | 41 bp |
| non-4mC | 1617 | ||
| R. chinensis (Independent) | 4mC | 779 | 41 bp |
| non-4mC | 779 |
| Nucleotide | One-Hot | NCP | Fusion |
|---|---|---|---|
| A | 1,0,0,0 | 1,1,1 | 1,0,0,0,1,1,1 |
| C | 0,1,0,0 | 0,0,1 | 0,1,0,0,0,0,1 |
| G | 0,0,1,0 | 1,0,0 | 0,0,1,0,1,0,0 |
| T | 0,0,0,1 | 0,1,0 | 0,0,0,1,0,1,0 |
| Layer | Output Shape | Number of Parameters |
|---|---|---|
| Input | (41,7) | - |
| Conv1D (32,9,1) | (33,32) | 2048 |
| Batch Normalization | (33,32) | 128 |
| Max Pooling (4,2) | (15,32) | 0 |
| Dropout (0.1) | (15,32) | 0 |
| Conv1D (8,5,1) | (11,8) | 1288 |
| Batch Normalization | (11,8) | 32 |
| Max Pooling (4,2) | (4,8) | 0 |
| Dropout (0.25) | (4,8) | 0 |
| LSTM (16) | (4,16) | 0 |
| Flatten | 64 | 0 |
| Dense (64) | 64 | 4160 |
| Dense (32) | 32 | 2080 |
| Dense (1) | 1 | 33 |
| Dataset | Tool | Sensitivity | Specificity | Accuracy | MCC |
|---|---|---|---|---|---|
| F. vesca (training) | i4mC-ROSE | 0.635 | 0.899 | 0.767 | 0.545 |
| iRG-4mC | 0.825 | 0.908 | 0.8665 | 0.732 | |
| R. chinensis (training) | i4mC-ROSE | 0.668 | 0.9 | 0.784 | 0.563 |
| iRG-4mC | 0.869 | 0.864 | 0.871 | 0.739 | |
| F. vesca (independent) | i4mC-ROSE | 0.721 | 0.873 | 0.797 | 0.601 |
| iRG-4mC | 0.835 | 0.882 | 0.859 | 0.706 | |
| R. chinensis (independent) | i4mC-ROSE | 0.636 | 0.881 | 0.759 | 0.535 |
| iRG-4mC | 0.854 | 0.875 | 0.865 | 0.714 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, D.Y.; Rehman, M.U.; Chong, K.T. iRG-4mC: Neural Network Based Tool for Identification of DNA 4mC Sites in Rosaceae Genome. Symmetry 2021, 13, 899. https://doi.org/10.3390/sym13050899
Lim DY, Rehman MU, Chong KT. iRG-4mC: Neural Network Based Tool for Identification of DNA 4mC Sites in Rosaceae Genome. Symmetry. 2021; 13(5):899. https://doi.org/10.3390/sym13050899
Chicago/Turabian StyleLim, Dae Yeong, Mobeen Ur Rehman, and Kil To Chong. 2021. "iRG-4mC: Neural Network Based Tool for Identification of DNA 4mC Sites in Rosaceae Genome" Symmetry 13, no. 5: 899. https://doi.org/10.3390/sym13050899
APA StyleLim, D. Y., Rehman, M. U., & Chong, K. T. (2021). iRG-4mC: Neural Network Based Tool for Identification of DNA 4mC Sites in Rosaceae Genome. Symmetry, 13(5), 899. https://doi.org/10.3390/sym13050899