
Can an Intermediate Rate of Nitrogen Inversion Affect Drug Efficacy?

 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Prelude
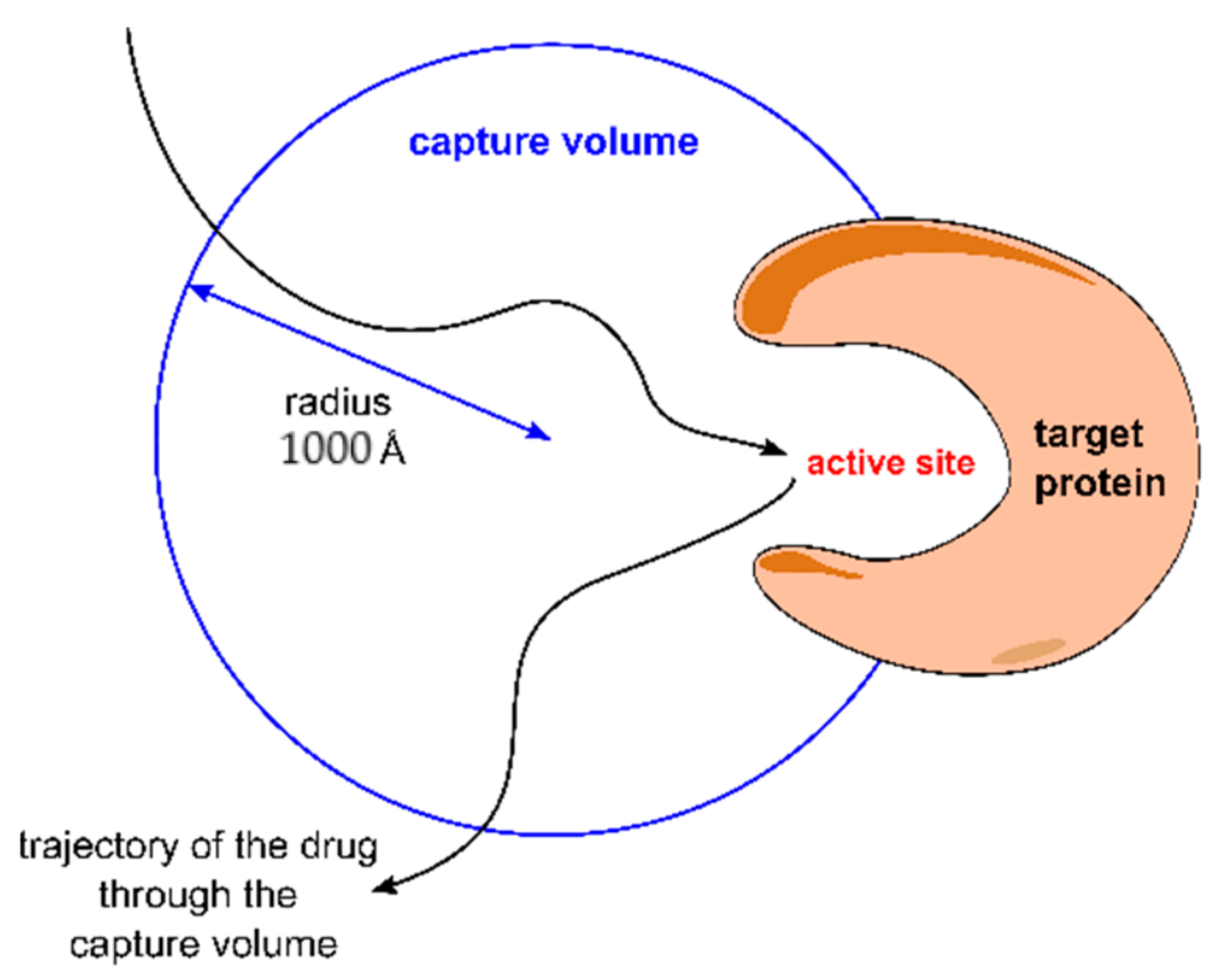
2.2. The Capture-Volume Concept
- The rate of the interconversion process is infinitely fast relative to the molecule’s motional diffusion so that binding of the drug to the active site in the protein occurs irrespective of the state of the drug molecule as the favored state is quickly adopted from the disfavored state before the drug molecule diffuses away from the active site. The result is that there is no effect at all on the action of the drug due to the rapid interconversion process.
- The rate of the interconversion process is infinitely slow, or even does not occur at all prior to clearance, and the result is that the dosage of the drug is effectively half that dispensed in the case, say, of rapid enantiomeric interconversion. An obvious example would be a chiral drug that is configurationally stable and is dispensed as a racemate.
- 3.
- The rate of the interconversion process is intermediate. Thus, while a drug molecule in the disfavored state will not bind to the protein when it encounters the active site and will diffuse away, later, after it has converted to the favored state, it is then able to bind to the protein when it again encounters it. The result is that while all of the drug is, in principle, available to bind to the protein, the availability of the drug is restricted as it is present at an effectively lower concentration even though it might be interconverting at a reasonable rate. The question is, what constitutes a reasonable rate?
2.3. Detailed Examination of 2 by 1H NMR
2.4. Hydroxamic Acids 1 and 3–14
2.5. Compound 15, the Epimeric Case
3. Conclusions and Summary
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- US Food and Drug Administration. FDA’s Policy Statement for the Development of New Stereoisomeric Drugs. Chirality 1992, 4, 338–340. [Google Scholar] [CrossRef] [PubMed]
- Committee for Proprietary Medicinal Products. Note for Guidance: Investigation of Chiral Active Substances; Document III/3501/91-EN, draft 13. 1993. Available online: https://docplayer.net/57670332-Investigation-of-chiral-active-substances.html (accessed on 11 June 2021).
- Ballard, A.; Narduolo, S.; Ahmad, H.O.; Cosgrove, D.A.; Leach, A.G.; Buurma, N.J. The problem of racemization in drug discovery and tools to predict it. Expert Opin. Drug Discov. 2019, 14, 527–539. [Google Scholar] [CrossRef]
- Landoni, M.F.; Soraci, A.L.; Delatour, P.; Lees, P. Enantioselective behaviour of drugs used in domestic animals: A review. J. Vet. Pharmacol. Therap. 1997, 20, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Cleeton, C.E.; Williams, N.H. Electromagnetic Waves of 1.1 cm Wave-Length and the Absorption Spectrum of Ammonia. Phys. Rev. 1934, 45, 234–237. [Google Scholar] [CrossRef]
- Rauk, A.; Allen, L.C.; Mislow, K. Pyramidal Inversion. Angew. Chem. Int. Ed. 1970, 9, 400–414. [Google Scholar] [CrossRef]
- Zawatzky, K.; Kamuf, M.; Trapp, O. Chiral 1,2-Dialkenyl Diaziridines: Synthesis, Enantioselective Separation, and Nitrogen Inversion Barriers. Chirality 2015, 27, 156–162. [Google Scholar] [CrossRef]
- Rodríguez-Franco, M.I.; Dorronsoro, I.; Castro, A.; Martínez, A. Hindered Inversion/Rotation in Diheteroaryl Alkyl Amines with a N-(1-Pyrazolyl) Group: Dynamic NMR and Molecular Modelling Studies. Tetrahedron 2000, 56, 1739–1743. [Google Scholar] [CrossRef]
- Bushweller, C.H.; Brown, J.H.; Harpp, K.S.; Hirth, B.H.; Barden, T.C.; D’Albis, J.N.; Gribble, G.W. Restricted Nitrogen Inversion in 9,10-Diazatetracyclo[6.3.0.0.4,110.5,9]undecanes. Dynamic NMR Studies. J. Org. Chem. 1998, 63, 3775–3777. [Google Scholar] [CrossRef]
- Jaźwiński, J. Interaction of amines with rhodium(II) tetracarboxylates in solution: Formation of nitrogenous stereogenic center. Tetrahedron Asymmetry 2006, 17, 2358–2365. [Google Scholar] [CrossRef]
- Tähtinen, P.; Sinkkonen, J.; Klika, K.D.; Nieminen, V.; Stájer, G.; Szakonyi, Z.; Fülöp, F.; Pihlaja, K. 1H, 13C, and 15N NMR Stereochemical Study of cis-Fused 7a(8a)-Methyl and 6-Phenyl Octa(hexa)hydrocyclopenta[d][1,3]oxazines and [3,1]Benzoxazines. Chirality 2002, 14, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Tähtinen, P.; Bagno, A.; Klika, K.D.; Pihlaja, K. Modeling NMR Parameters by DFT Methods as an Aid to the Conformational Analysis of cis-Fused 7a(8a)-Methyl Octa(hexa)hydrocyclopenta[d][1,3]oxazines and [3,1]benzoxazines. J. Am. Chem. Soc. 2003, 125, 4609–4618. [Google Scholar] [CrossRef] [PubMed]
- Rosling, A.; Klika, K.D.; Fülöp, F.; Sillanpää, R.; Mattinen, J. An NMR Conformational Study of Ring- and N-Inversion, and Prototropic Tautomerism in Stereoisomeric 2-[Arylamino(imino)]-4a,5,6,7,8,8a-hexahydro-(4H)-1,3,4-benzoxadiazines. Acta Chem. Scand. 1999, 53, 103–113. [Google Scholar] [CrossRef]
- Rosling, A.; Hotokka, M.; Klika, K.D.; Fülöp, F.; Sillanpää, R.; Mattinen, J. The Conformational Behaviour of 4,4a,5,6,7,8-Hexahydropyrido[1,2-d][1,3,4]oxadiazine Derivatives Studied by NMR Spectroscopy and Molecular Mechanics. Acta Chem. Scand. 1999, 53, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Rosling, A.; Klika, K.; Fülöp, F.; Sillanpää, R.; Mattinen, J. The Conformational Preference of Some Tetrahydropyrrolo[1,2-d][1,3,4]oxadiazine Derivatives as Studied by NMR Spectroscopy and X-ray Analysis. Heterocycles 1999, 51, 2575–2588. [Google Scholar]
- Viljanen, T.; Klika, K.D.; Fülöp, F.; Pihlaja, K. Coupling constants 1J(15N,31P) as a probe for the conformational equilibria of 2-amino-substituted 1,3,2λ5-oxazaphosphinan-2-ones. J. Chem. Soc. Perkin Trans. 1998, 2, 1479–1481. [Google Scholar] [CrossRef]
- Ertl, P.; Altmann, E.; McKenna, J.M. The Most Common Functional Groups in Bioactive Molecules and How Their Popularity Has Evolved over Time. J. Med. Chem. 2020, 63, 8408–8418. [Google Scholar] [CrossRef]
- James, L.C.; Roversi, P.; Tawfik, D.S. Antibody Multispecificity Mediated by Conformational Diversity. Science 2003, 299, 1362–1367. [Google Scholar] [CrossRef] [Green Version]
- Abe, I.; Kashiwagi, Y.; Noguchi, H.; Tanaka, T.; Ikeshiro, Y.; Kashiwada, Y. Ellagitannins and Hexahydroxydiphenoyl Esters as Inhibitors of Vertebrate Squalene Epoxidase. J. Nat. Prod. 2001, 64, 1010–1014. [Google Scholar] [CrossRef]
- Fraga, C.G.; Oteiza, P.I. Dietary flavonoids: Role of (−)-epicatechin and related procyanidins in cell signaling. Free Radic. Biol. Med. 2011, 51, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Imberty, A.; Bourne, Y.; Cambillau, C.; Rougé, P.; Pérez, S. Oligosaccharide conformation in protein/carbohydrate complexes. Adv. Biophys. Chem. 1993, 3, 71–118. [Google Scholar]
- Imberty, A. Oligosaccharide structures: Theory versus experiment. Curr. Opin. Struct. Biol. 1997, 7, 617–623. [Google Scholar] [CrossRef]
- Diaz-Mauriño, T.; Solis, D.; Jeminéz-Barbero, J.J.; Martín-Lomas, M.; Siebert, H.-C.; Vliegenthart, J.F.G. Lectin meets ligand. Carbohydr. Eur. 1998, 23, 36–41. [Google Scholar]
- Gilleron, M.; Siebert, H.-C.; Kaltner, H.; von der Lieth, C.-W.; Kozár, T.; Halkes, K.M.; Korchagina, E.Y.; Bovin, N.Y.; Gabius, H.-J.; Vliegenthart, J.F.G. Conformer selection and differential restriction of ligand mobility by plant lectin. Conformational behaviour of Galβ1-3GlcNAcβ1-R, Galβ1-3GalNAcβ1-R and Galβ1-2Galβ1-R′ in the free state and complexed with galactoside-specific mistletoe lectin as revealed by random-walk and conformational-clustering molecular-mechanics calculations, molecular-dynamics simulations and nuclear Overhauser experiments. Eur. J. Biochem. 1998, 252, 416–427. [Google Scholar]
- Von der Lieth, C.-W.; Kozár, T. Towards a better semiquantitative estimation of binding constants: Molecular dynamics exploration of the conformational behavior of isolated sialyllactose and sialyllactose complexed with influenza A hemagglutinin. J. Mol. Struct. 1996, 368, 213–222. [Google Scholar] [CrossRef]
- Von der Lieth, C.-W.; Kozár, T.; Hull, W.E. A (critical) survey of modelling protocols used to explore the conformational space of oligosaccharides. J. Mol. Struct. 1997, 395–396, 225–244. [Google Scholar] [CrossRef]
- Géraldy, M.; Morgen, M.; Sehr, P.; Steimbach, R.R.; Moi, D.; Ridinger, J.; Oehme, I.; Witt, O.; Malz, M.; Nogueira, M.S.; et al. Selective Inhibition of Histone Deacetylase 10: Hydrogen Bonding to the Gatekeeper Residue is Implicated. J. Med. Chem. 2019, 62, 4426–4443. [Google Scholar] [CrossRef] [PubMed]
- Steimbach, R.R.; Tihanyi, G.; Géraldy, M.N.E.; Herbst-Gervasoni, C.J.; Christianson, D.W.; Oehme, I.; Gunkel, N.; Miller, A.K. Rational Design Yields Selective Histone Deacetylase 10 Inhibitor. J. Med. Chem. 2021, submitted. [Google Scholar]
- Zheng, G.; Stait-Gardner, T.; Anil Kumar, P.G.; Torres, A.M.; Price, W.S. PGSTE-WATERGATE: An STE-based PGSE NMR sequence with excellent solvent suppression. J. Magn. Reson. 2008, 191, 159–163. [Google Scholar] [CrossRef]
- Price, W.S.; Elwinger, F.; Vigouroux, C.; Stilbs, P. PGSTE-WATERGATE, new tool for NMR diffusion-based studies of ligand–macromolecule binding. Magn. Reson. Chem. 2002, 40, 391–395. [Google Scholar] [CrossRef]
- Veenstra, D.L.; Gerig, J.T. Fluorine NMR studies of the human carbonic anhydrase–3,5-difluorobenzenesulfonamide complex. Magn. Reson. Chem. 1998, 36, S169–S178. [Google Scholar] [CrossRef]
- Kwiatkowska, M.; Wzorek, A.; Kolbus, A.; Urbaniak, M.; Han, J.; Soloshonok, V.A.; Klika, K.D. Flurbiprofen: A Study of the Behavior of the Scalemate by Chromatography, Sublimation, and NMR. Symmetry 2021, 13, 543. [Google Scholar] [CrossRef]
- Baumann, A.; Wzorek, A.; Soloshonok, V.A.; Klika, K.D.; Miller, A.K. Potentially Mistaking Enantiomers for Different Compounds Due to the Self-Induced Diastereomeric Anisochronism (SIDA) Phenomenon. Symmetry 2020, 12, 1106. [Google Scholar] [CrossRef]
- Wu, D.H.; Chen, A.D.; Johnson, C.S., Jr. An Improved Diffusion-Ordered Spectroscopy Experiment Incorporating Bipolar-Gradient Pulses. J. Magn. Reson. Ser. A 1995, 115, 260–264. [Google Scholar] [CrossRef]
- McDonald, G.G.; Leigh, J.S., Jr. A New Method for Measuring Longitudinal Relaxation Times. J. Magn. Reson. 1973, 9, 358–362. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compd | Conc., mm | pH | Solvent | Temp., °C | Inv. Rate, s−1 | D × 10−10, m2s−1 |
|---|---|---|---|---|---|---|---|
| 1 | 2·HCl | 19.8 | – | CD3OD | 25 | 2.33, 0.25 a (Tc = 90 °C) | 8.53 |
| 2 | 2·HCl | 19.8 | 6 | D2O | 25 | – | 5.12 |
| 3 | 1·TFA | 64.4 | 8 | D2O | 25 | 35.47 | 3.51 |
| 4 | 1·TFA | unknown | 3 | D2O | 25 | 3.60 | 3.51 |
| 5 | 3·TFA | unknown, sat. soln. | 3 | D2O | 25 | 6.86, 8.9 a (Tc = 45 °C) | 3.20 |
| 6 | 4·TFA | 25.7 | 3 | D2O | 25 | 1.43 | 3.26 |
| 7 | 14·HCl | 69.9 | 3.5 | D2O | 25 | 1.24 | 3.81 |
| 8 | 11 | 156.2 | 4 | D2O | 25 | 1.50 | 3.23 |
| 9 | 14·HCl | 69.9 | 3.5 | D2O | 37 | 5.32 | 5.40 |
| 10 | 14·HCl | 69.9 | 6 | D2O | 25 | 30–70 b | 3.79 |
| 11 | 10 | 133.7 | 3 | D2O | 25 | 5.46 | 3.45 |
| 12 | 12 | 107.8 | 3 | D2O | 25 | 1.35 | 3.70 |
| 13 | 10 | 133.7 | 3 | D2O | 37 | 13.96 | 4.91 |
| 14 | 15·HCl | 20.0 | 7.5 | D2O | 25 | 1.19 | 4.47 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steimbach, R.R.; Tihanyi, G.; Géraldy, M.N.E.; Wzorek, A.; Miller, A.K.; Klika, K.D. Can an Intermediate Rate of Nitrogen Inversion Affect Drug Efficacy? Symmetry 2021, 13, 1753. https://doi.org/10.3390/sym13091753
Steimbach RR, Tihanyi G, Géraldy MNE, Wzorek A, Miller AK, Klika KD. Can an Intermediate Rate of Nitrogen Inversion Affect Drug Efficacy? Symmetry. 2021; 13(9):1753. https://doi.org/10.3390/sym13091753
Chicago/Turabian StyleSteimbach, Raphael R., Gergely Tihanyi, Magalie N. E. Géraldy, Alicja Wzorek, Aubry K. Miller, and Karel D. Klika. 2021. "Can an Intermediate Rate of Nitrogen Inversion Affect Drug Efficacy?" Symmetry 13, no. 9: 1753. https://doi.org/10.3390/sym13091753
APA StyleSteimbach, R. R., Tihanyi, G., Géraldy, M. N. E., Wzorek, A., Miller, A. K., & Klika, K. D. (2021). Can an Intermediate Rate of Nitrogen Inversion Affect Drug Efficacy? Symmetry, 13(9), 1753. https://doi.org/10.3390/sym13091753