
Crystal Structure of Mixed Np(V)-Ammonium Carbonate
 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of (NH4)[NpO2CO3] Crystals
2.2. X-ray Structural Analysis
2.3. X-ray Photoelectron Spectroscopy
2.4. Vibrational Spectroscopy
3. Results
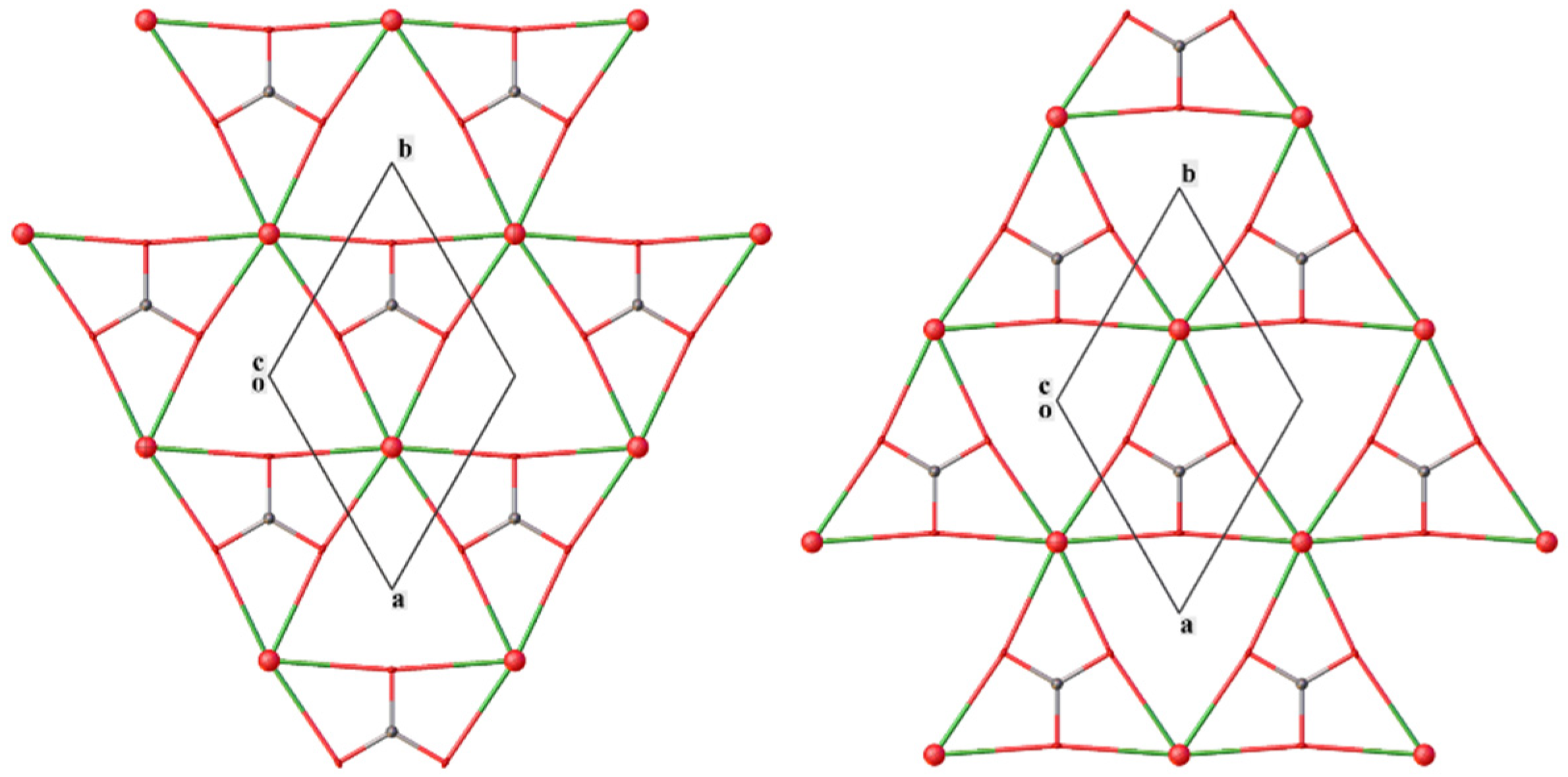
3.1. X-ray Structure Analysis
3.2. X-ray Photoelectron Spectroscopy
3.3. Vibrational Spectroscopy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McKinley, I.G.; Russell Alexander, W.; Blaser, P.C. Development of Geological Disposal Concepts. In Radioactivity in the Environment; Elsevier: Amsterdam, The Netherlands, 2007; pp. 41–76. [Google Scholar]
- Davis, J.A.; Kent, D.B. Surface Complexation Modeling in Aqueous Geochemistry. Miner. Interface Geochem. 1990, 23, 177–260. [Google Scholar]
- Fernandes, M.M.; Scheinost, A.C.; Baeyens, B. Sorption of Trivalent Lanthanides and Actinides onto Montmorillonite: Macroscopic, Thermodynamic and Structural Evidence for Ternary Hydroxo and Carbonato Surface Complexes on Multiple Sorption Sites. Water Res. 2016, 99, 74–82. [Google Scholar] [CrossRef]
- Romanchuk, A.Y.; Vlasova, I.E.; Kalmykov, S.N. Speciation of Uranium and Plutonium From Nuclear Legacy Sites to the Environment: A Mini Review. Front. Chem. 2020, 8, 630. [Google Scholar] [CrossRef] [PubMed]
- Geckeis, H.; Lützenkirchen, J.; Polly, R.; Rabung, T.; Schmidt, M. Mineral-Water Interface Reactions of Actinides. Chem. Rev. 2013, 113, 1016–1062. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.; Deschanels, X.; Bart, J.M.; Boubals, J.M.; Den Auwer, C.; Simoni, E. Solubility of Actinide Surrogates in Nuclear Glasses. J. Nucl. Mater. 2003, 312, 76–80. [Google Scholar] [CrossRef]
- Sweet, L.E.; Corbey, J.F.; Gendron, F.; Autschbach, J.; McNamara, B.K.; Ziegelgruber, K.L.; Arrigo, L.M.; Peper, S.M.; Schwantes, J.M. Structure and Bonding Investigation of Plutonium Peroxocarbonate Complexes Using Cerium Surrogates and Electronic Structure Modeling. Inorg. Chem. 2017, 56, 791–801. [Google Scholar] [CrossRef]
- Clark, D.L.; Hobart, D.E.; Neu, M.P. Actinide Carbonte Complexes and Their Importance in Actinide Environmental Chemistry. Chem. Rev. 1995, 95, 25–48. [Google Scholar] [CrossRef]
- Pastoor, K.J.; Kemp, R.S.; Jensen, M.P.; Shafer, J.C. Progress in Uranium Chemistry: Driving Advances in Front-End Nuclear Fuel Cycle Forensics. Inorg. Chem. 2021, 60, 8347–8367. [Google Scholar] [CrossRef]
- Keenan, T.K.; Kruse, F.H. Potassium Double Carbonates of Pentavalent Neptunium, Plutonium, and Americium. Inorg. Chem. 1964, 3, 1231–1232. [Google Scholar] [CrossRef]
- Keenan, T.K. Lattice Constants of Some Alkali Metal Actinyl(V) Compounds. Inorg. Chem. 1965, 4, 1500–1501. [Google Scholar] [CrossRef]
- Ellinger, F.H.; Zachariasen, W.H. The Crystal Structure of KPuO2CO3, NH4PuO2CO3 and RbAmO2CO3. J. Phys. Chem. 1954, 58, 405–408. [Google Scholar] [CrossRef]
- Burney, G.A. Separation of Americium from Curium by Precipitation of K3AmO2(CO3)2. Nucl. Appl. 1968, 4, 217–221. [Google Scholar] [CrossRef]
- Choppin, G.R. Actinide Speciation in the Environment. J. Radioanal. Nucl. Chem. 2007, 273, 695–703. [Google Scholar] [CrossRef]
- Kaszuba, J.P.; Runde, W.H. The Aqueous Geochemistry of Neptunium: Dynamic Control of Soluble Concentrations with Applications to Nuclear Waste Disposal. Environ. Sci. Technol. 1999, 33, 4427–4433. [Google Scholar] [CrossRef]
- Ikeda-Ohno, A.; Tsushima, S.; Takao, K.; Rossberg, A.; Funke, H.; Scheinost, A.C.; Bernhard, G.; Yaita, T.; Hennig, C. Neptunium Carbonato Complexes in Aqueous Solution: An Electrochemical, Spectroscopic, and Quantum Chemical Study. Inorg. Chem. 2009, 48, 11779–11787. [Google Scholar] [CrossRef]
- Ikeda, A.; Hennig, C.; Tsushima, S.; Takao, K.; Ikeda, Y.; Scheinost, A.C.; Bernhard, G. Comparative Study of Uranyl(VI) and -(V) Carbonato Complexes in an Aqueous Solution. Inorg. Chem. 2007, 46, 4212–4219. [Google Scholar] [CrossRef]
- Vitova, T.; Pidchenko, I.; Schild, D.; Prüßmann, T.; Montoya, V.; Fellhauer, D.; Gaona, X.; Bohnert, E.; Rothe, J.; Baker, R.J.; et al. Competitive Reaction of Neptunium(V) and Uranium(VI) in Potassium–Sodium Carbonate-Rich Aqueous Media: Speciation Study with a Focus on High-Resolution X-Ray Spectroscopy. Inorg. Chem. 2020, 59, 8–22. [Google Scholar] [CrossRef]
- Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D. Comparison of Silver and Molybdenum Microfocus X-Ray Sources for Single-Crystal Structure Determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Le Bail, A. Whole Powder Pattern Decomposition Methods and Applications: A Retrospection. Powder Diffr. 2005, 20, 316–326. [Google Scholar] [CrossRef]
- Petříček, V.; Dušek, M.; Palatinus, L. Crystallographic Computing System JANA2006: General Features. Zeitschrift Für Krist.-Cryst. Mater. 2014, 229, 345–352. [Google Scholar] [CrossRef]
- Vlasova, I.E.; Yapaskurt, V.O.; Averin, A.A.; Melnik, O.E.; Zolotov, D.A.; Senin, R.A.; Poliakova, T.R.; Nevolin, I.M.; Kalmykov, S.N.; Shiryaev, A.A. Nuclear Melt Glass from Experimental Field, Semipalatinsk Test Site. Energies 2022, 15, 9121. [Google Scholar] [CrossRef]
- Finch, R.J.; Cooper, M.A.; Hawthorne, F.C.; Ewing, R.C. The Crystal Structure of Schoepite, [(UO2)8O2(OH)12](H2O)12. Can. Mineral. 1996, 34, 1071–1088. [Google Scholar]
- Cordfunke, E.H.P. Composition and Structure of Ammonium Uranates. J. Inorg. Nucl. Chem. 1970, 32, 3129–3131. [Google Scholar] [CrossRef]
- Li, Y.; Cahill, C.L.; Burns, P.C. Synthesis, Structural Characterization, and Topological Rearrangement of a Novel Open Framework U−O Material: (NH4)3(H2O)2{[(UO2)10O10(OH)][(UO4)(H2O)2]}. Chem. Mater. 2001, 13, 4026–4031. [Google Scholar] [CrossRef]
- Teterin, Y.A.; Teterin, A.Y. The Structure of X-Ray Photoelectron Spectra of Light Actinide Compounds. Russ. Chem. Rev. 2004, 73, 541–580. [Google Scholar] [CrossRef]
- Shchukarev, A.; Korolkov, D. XPS Study of Group IA Carbonates. Open Chem. 2004, 2, 347–362. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhou, J.; Zhang, J.; Su, J.; Zhang, S.; Chen, N.; Jia, Y.; Li, J.; Wang, Y.; Wang, J.-Q. Extraction of Local Coordination Structure in a Low-Concentration Uranyl System by XANES. J. Synchrotron Radiat. 2016, 23, 758–768. [Google Scholar] [CrossRef]
- Basile, L.J.; Sullivan, J.C.; Ferraro, J.R.; LaBonville, P. The Raman Scattering of Uranyl and Transuranium V, VI, and VII Ions. Appl. Spectrosc. 1974, 28, 142–145. [Google Scholar] [CrossRef]
- Fujii, T.; Uehara, A.; Kitatsuji, Y.; Yamana, H. Raman Spectroscopic Study on NpO2+–Ca2+ Interaction in Highly Concentrated Calcium Chloride. J. Radioanal. Nucl. Chem. 2014, 301, 293–296. [Google Scholar] [CrossRef] [Green Version]
- Krot, A.; Vlasova, I.; Trigub, A.; Averin, A.; Yapaskurt, V.; Kalmykov, S. From EXAFS of Reference Compounds to U(VI) Speciation in Contaminated Environments. J. Synchrotron Radiat. 2022, 29, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Tlili, M.M.; Amor, M.B.; Gabrielli, C.; Joiret, S.; Maurin, G.; Rousseau, P. Characterization of CaCO3 Hydrates by Micro-Raman Spectroscopy. J. Raman Spectrosc. 2002, 33, 10–16. [Google Scholar] [CrossRef]
- Graziani, R.; Bombieri, G.; Forsellini, E. Crystal Structure of Tetra-Ammonium Uranyl Tricarbonate. J. Chem. Soc. Dalt. Trans. 1972, 19, 2059–2061. [Google Scholar] [CrossRef]
- Frost, R.L.; Carmody, O.; Erickson, K.L.; Weier, M.L.; Čejka, J. Molecular Structure of the Uranyl Mineral Andersonite—A Raman Spectroscopic Study. J. Mol. Struct. 2004, 703, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Morgan, P.A.; Staats, H.W.; Goldstein, J.H. Infrared Spectra of N15H3 and N15H4+. J. Chem. Phys. 1957, 27, 4–6. [Google Scholar] [CrossRef]
- Faulques, E.; Perry, D.L.; Kalashnyk, N. Vibrational Spectroscopy of a Crystallographically Unsettled Uranyl Carbonate: Structural Impact and Model. Vib. Spectrosc. 2018, 99, 184–189. [Google Scholar] [CrossRef]
- Bessonov, A.A.; Afonasieva, T.V.; Krot, N.N. On Np (V) and (VI) formation at thermal decomposition of Np (IV) compounds. Sov. Radiochem. 1989, 31, 9–13. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Empirical Formula | CH4NNpO5 | Dcalc, (g cm−3) | 4.714 |
| Crystal size | 0.04 × 0.04 × 0.03 mm | μ, mm−1 | 21.18 |
| Fw | 374.05 | Reflections collected | 4630 |
| T (K) | 150 (2) | Independent reflections | 165 |
| Crystal system | Hexagonal | Observed reflections [I > 2σ(I)] | 158 |
| Space group | P63/mmc | Rint | 0.0559 |
| a, (Å) | 5.0788 (6) | Number of parameters | 19 |
| c, (Å) | 10.9456 (19) | R1, wR2 [I > 2σ(I)] | 0.0102, 0.0234 |
| V, (Å3) | 244.51 (7) | R1, wR2 [all data] | 0.0114, 0.0237 |
| Z | 2 | GOOF | 1.203 |
| Dcalc, (g cm−3) | 4.714 | Δρmax, Δρmin, (e·Å−3) | 0.687, −0.541 |
| Bond | D |
|---|---|
| Np1-O1 | 2 × 1.815 (5) |
| Np1-O2 | 6 × 2.5464 (4) |
| C1-O2 | 3 × 1.278 (3) |
| N1-H1A | 0.86 (8) |
| N1-H1B | 3 × 0.86 (8) |
| Compound | a, Å | c, Å | Ref. |
|---|---|---|---|
| K[NpO2CO3] | 5.120 (1) | 9.971 (3) | [10] |
| K[PuO2CO3] | 5.093 (1) | 9.815 (2) | [10] |
| K[PuO2CO3] | 5.09 (1) | 9.83 (2) | [12] |
| K[AmO2CO3] | 5.112 (1) | 9.740 (2) | [10] |
| (NH4)[NpO2CO3] | 5.09858 (4) | 10.89394 (14) | This work |
| (NH4)[PuO2CO3] | 5.09 (1) | 10.39 (2) | [12] |
| Rb[AmO2CO3] | 5.12 (1) | 10.46 (4) | [12] |
| Cs[AmO2CO3] | 5.123 (1) | 11.538 (7) | [11] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nevolin, I.M.; Petrov, V.G.; Grigoriev, M.S.; Averin, A.A.; Shiryaev, A.A.; Krot, A.D.; Maslakov, K.I.; Teterin, Y.A.; Fedoseev, A.M. Crystal Structure of Mixed Np(V)-Ammonium Carbonate. Symmetry 2022, 14, 2634. https://doi.org/10.3390/sym14122634
Nevolin IM, Petrov VG, Grigoriev MS, Averin AA, Shiryaev AA, Krot AD, Maslakov KI, Teterin YA, Fedoseev AM. Crystal Structure of Mixed Np(V)-Ammonium Carbonate. Symmetry. 2022; 14(12):2634. https://doi.org/10.3390/sym14122634
Chicago/Turabian StyleNevolin, Iurii M., Vladimir G. Petrov, Mikhail S. Grigoriev, Alexei A. Averin, Andrey A. Shiryaev, Anna D. Krot, Konstantin I. Maslakov, Yury A. Teterin, and Alexander M. Fedoseev. 2022. "Crystal Structure of Mixed Np(V)-Ammonium Carbonate" Symmetry 14, no. 12: 2634. https://doi.org/10.3390/sym14122634
APA StyleNevolin, I. M., Petrov, V. G., Grigoriev, M. S., Averin, A. A., Shiryaev, A. A., Krot, A. D., Maslakov, K. I., Teterin, Y. A., & Fedoseev, A. M. (2022). Crystal Structure of Mixed Np(V)-Ammonium Carbonate. Symmetry, 14(12), 2634. https://doi.org/10.3390/sym14122634