


Electronic and Optical Properties of Alkaline Earth Metal Fluoride Crystals with the Inclusion of Many-Body Effects: A Comparative Study on Rutile MgF2 and Cubic SrF2

Abstract
:1. Introduction
2. Computational Methods and Resulting Ground-State Properties
3. Electronic Excitations in r-MF and c-SF
3.1. Energy Gaps for r-MgF and c-SrF
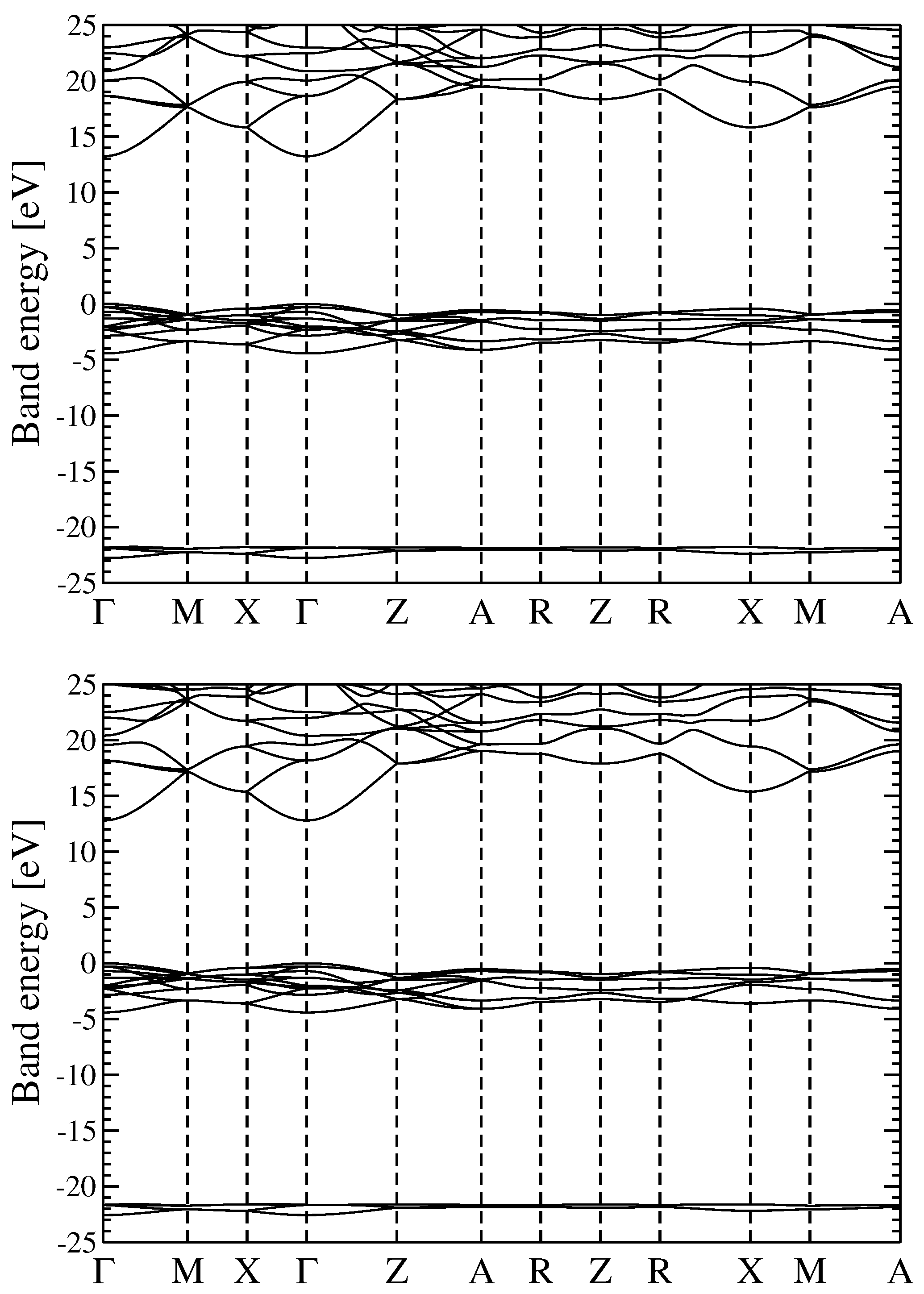
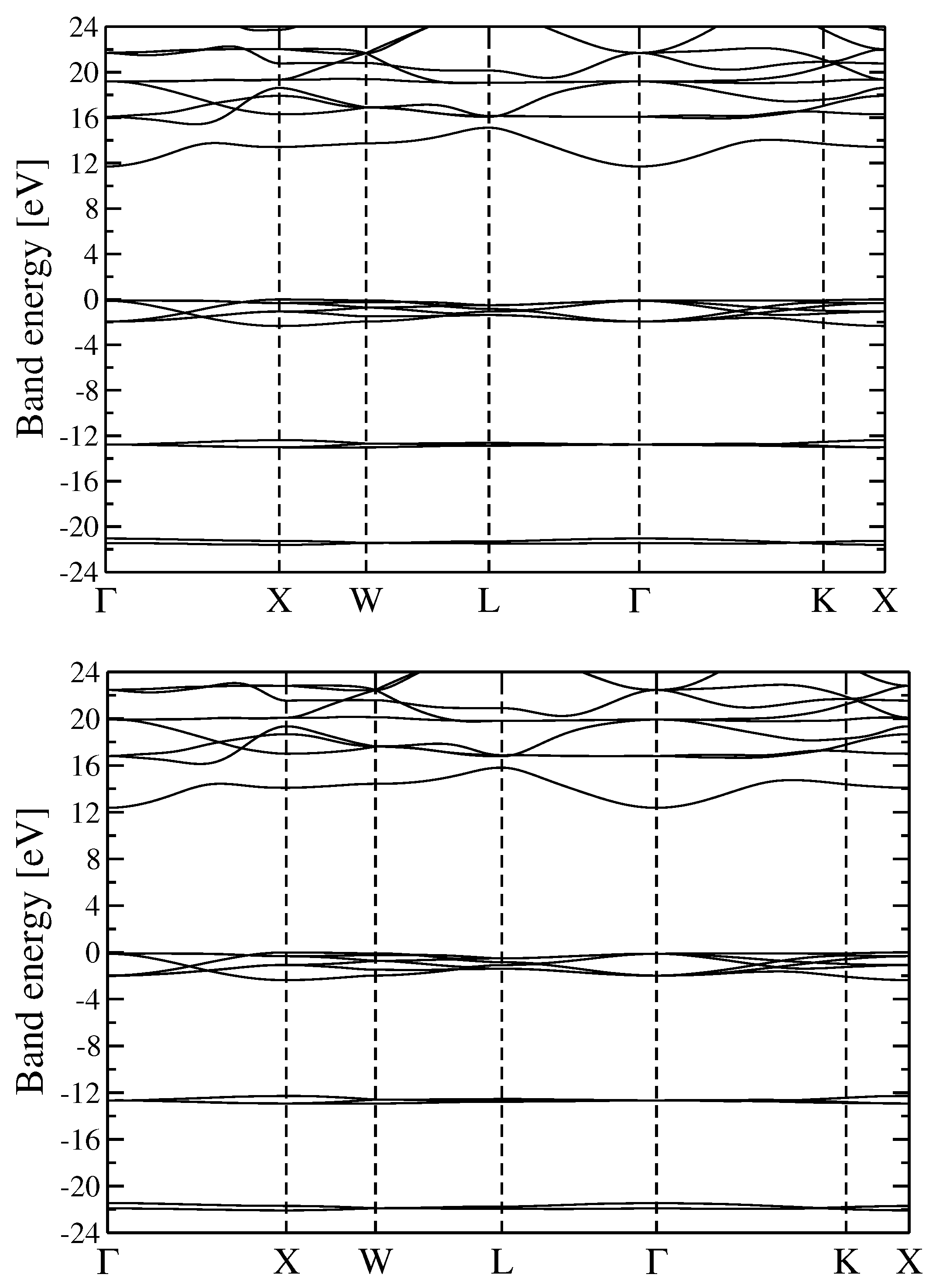
3.2. Quasiparticle Energy Bands for r-MgF and c-SrF
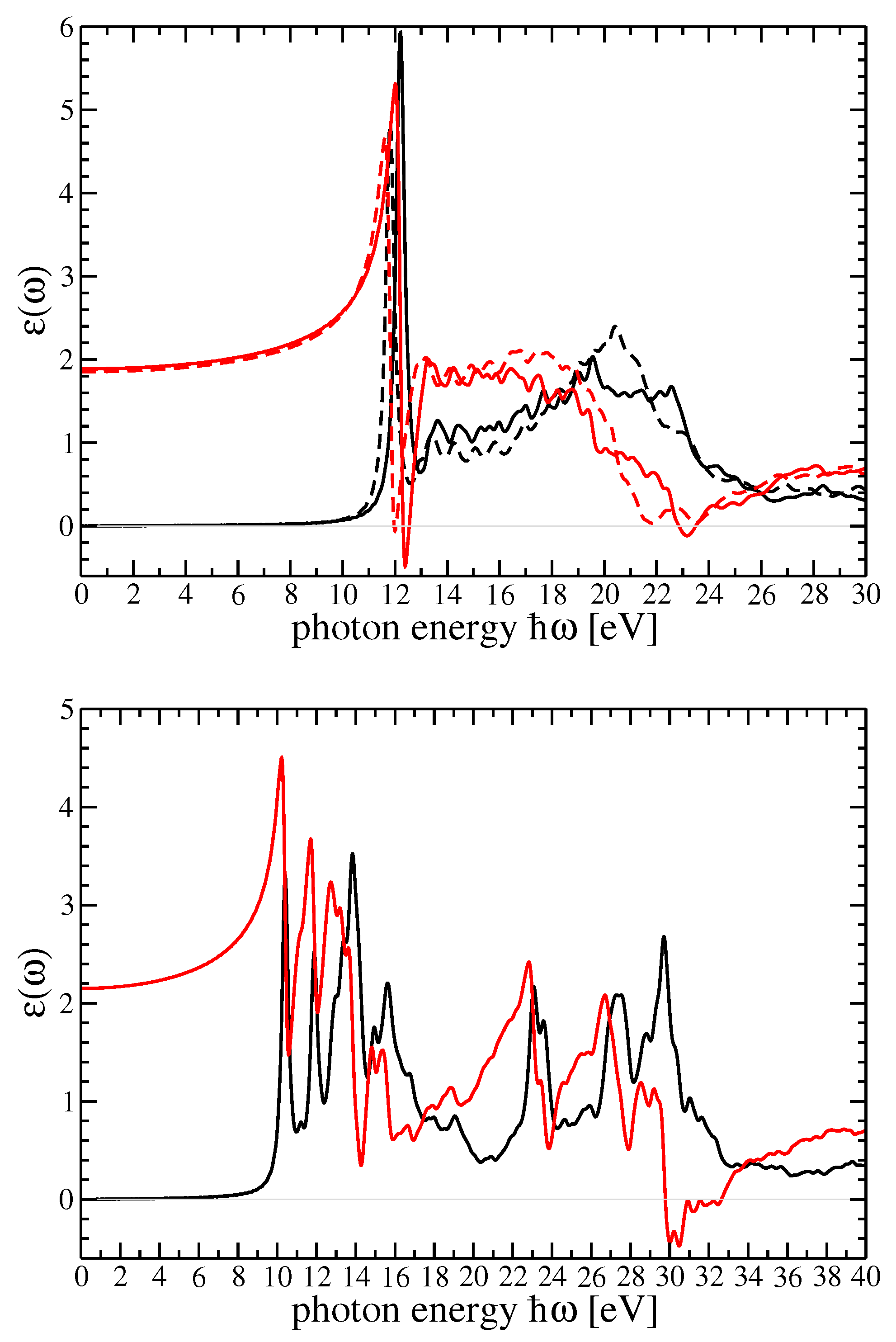
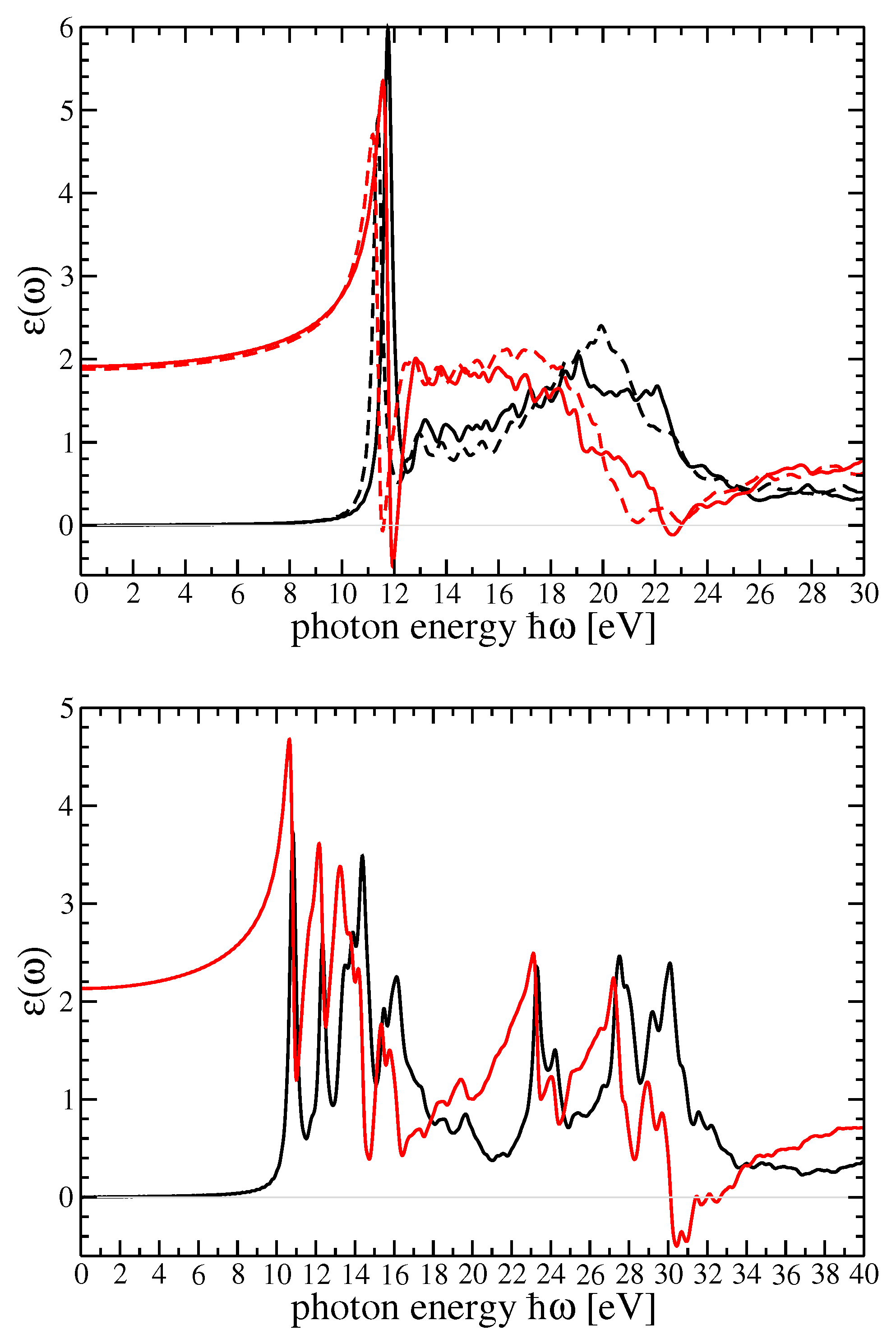
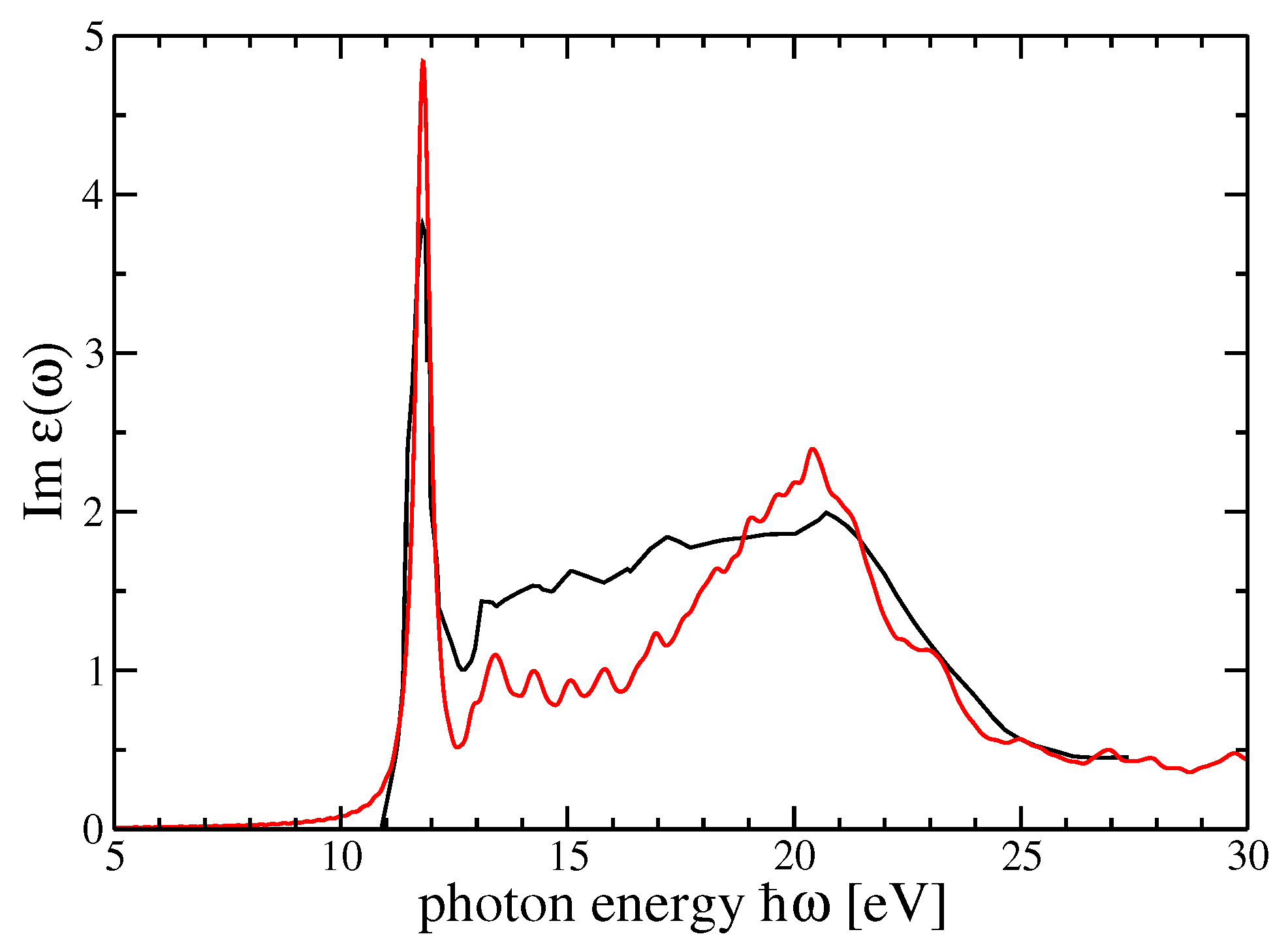
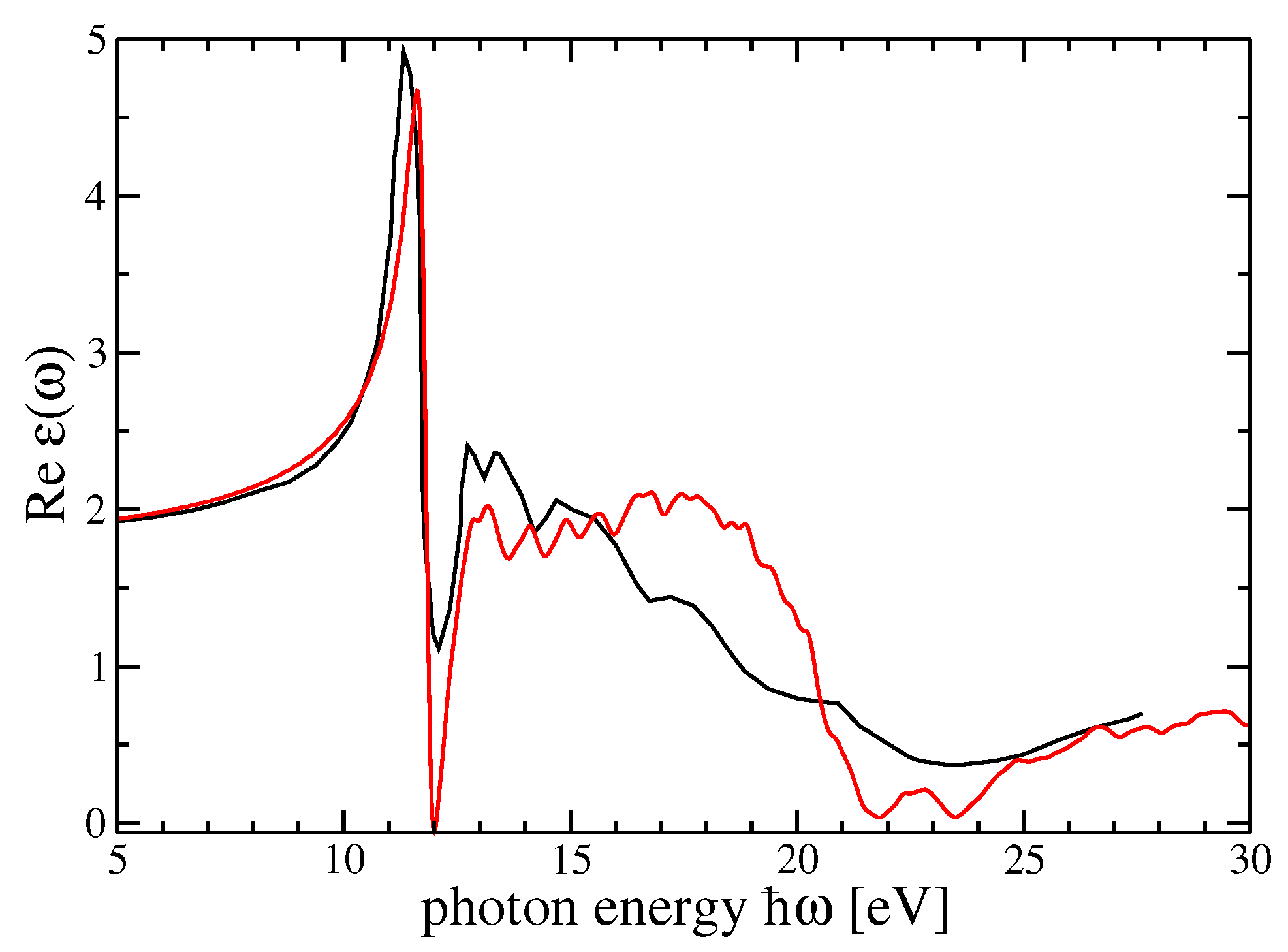
4. Dielectric Function and Optical Absorption Spectrum of MF and SF
5. Summary and Conclusions
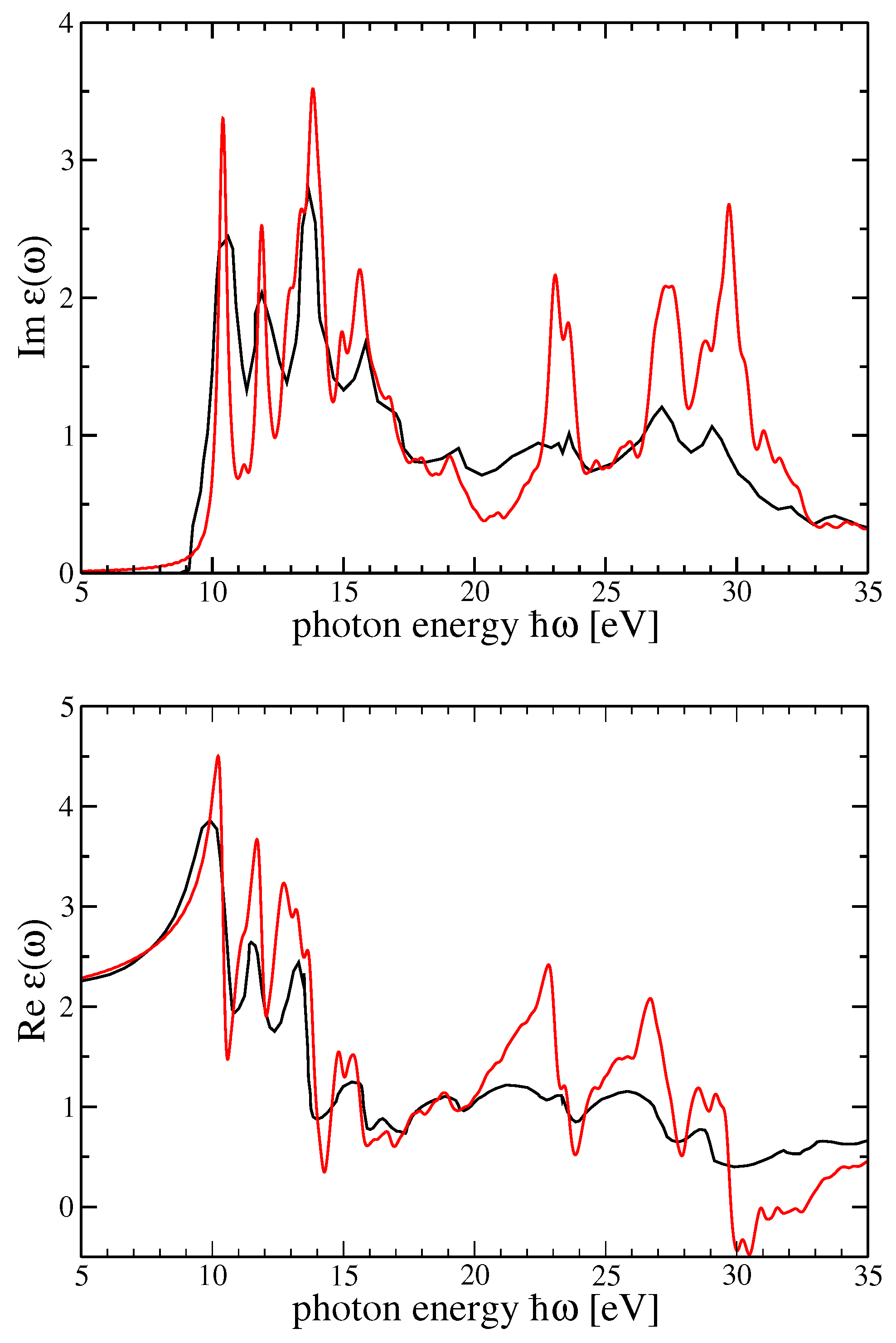
6. Additional Material: Bulk Systems versus Clusters
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rubloff, G.W. Far-Ultraviolet Reflectance Spectra and the Electronic Structure of Ionic Crystals. Phys. Rev. B 1972, 5, 662–684. [Google Scholar] [CrossRef]
- Zemann, J. Crystal structures, 2nd edition. Vol. 1, by R. W. G. Wyckoff. Acta Crystallogr. 1965, 18, 139. [Google Scholar] [CrossRef] [Green Version]
- Bechstedt, F. Many-Body Approach to Electronic Excitations; Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar] [CrossRef]
- Bordonaro, G.J. DUV Photolithography and Materials. In Encyclopedia of Nanotechnology; Bhushan, B., Ed.; Springer: Dordrecht, The Netherlands, 2012; pp. 590–604. [Google Scholar] [CrossRef]
- Bertoni, C.M.; Cappellini, G.; Finocchi, F.; Monachesi, P. 7.3.4 CaF2 and other fluorides surfaces. In Physics of Solid Surfaces; Springer: Berlin/Heidelberg, Germany, 2015; pp. 387–391. [Google Scholar] [CrossRef]
- Mattila, T.; Pöykkö, S.; Nieminen, R.M. Ab initio study of point defects in CdF2. Phys. Rev. B 1997, 56, 15665–15671. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Eglitis, R.I.; Borstel, G. Ab initio calculations of the BaF2 bulk and surface F centers. J. Phys. Condens. Matter 2006, 18, 8367–8381. [Google Scholar] [CrossRef] [Green Version]
- Jia, R.; Shi, H.; Borstel, G. The atomic and electronic structure of CaF2 and BaF2 crystals with H centers: A hybrid DFT calculation study. J. Phys. Condens. Matter 2010, 22, 055501. [Google Scholar] [CrossRef]
- Tousey, R. Optical Constants of Fluorite in the Extreme Ultraviolet. Phys. Rev. 1936, 50, 1057–1066. [Google Scholar] [CrossRef]
- Samara, G.A. Temperature and pressure dependences of the dielectric properties of PbF2and the alkaline-earth fluorides. Phys. Rev. B 1976, 13, 4529–4544. [Google Scholar] [CrossRef]
- Scrocco, M. Satellites in X-ray photoelectron spectroscopy of insulators. I. Multielectron excitations in CaF2, SrF2, and BaF2. Phys. Rev. B 1985, 32, 1301–1305. [Google Scholar] [CrossRef]
- Weesner, F.J.; Wright, J.C.; Fontanella, J.J. Laser spectroscopy of ion-size effects on point-defect equilibria in PbF2:Eu3+. Phys. Rev. B 1986, 33, 1372–1380. [Google Scholar] [CrossRef]
- Kosacki, I.; Langer, J.M. Fundamental absorption edge of PbF2 and Cd1-xPbxF2 crystals. Phys. Rev. B 1986, 33, 5972–5973. [Google Scholar] [CrossRef]
- Hull, S.; Keen, D.A. Effect of hydrostatic pressure on the crystal structure and superionic behavior of lead (II) fluoride. Phys. Rev. B 1998, 58, 14837–14844. [Google Scholar] [CrossRef]
- Fujita, M.; Itoh, M.; Bokumoto, Y.; Nakagawa, H.; Alov, D.L.; Kitaura, M. Optical spectra and electronic structures of lead halides. Phys. Rev. B 2000, 61, 15731–15737. [Google Scholar] [CrossRef]
- Burnett, J.; Levine, Z.; Shirley, E. Intrinsic birefringence in calcium fluoride and barium fluoride. Phys. Rev. B 2001, 64, 241102–1–241102–4. [Google Scholar] [CrossRef]
- Inagaki, H.; Saito, A.; Sugiyama, H.; Okabayashi, T.; Fujimoto, S. Rapid inactivation of SARS-CoV-2 with deep-UV LED irradiation. Emerg. Microbes Infect. 2020, 9, 1744–1747. [Google Scholar] [CrossRef]
- Hessling, M.; Hoenes, K.; Vatter, P.; Lingenfelder, C. Ultraviolet irradiation doses for coronavirus inactivation - review and analysis of coronavirus photoinactivation studies. GMS Hyg. Infect. Control. 2020, 15, Doc08. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, W. UVGI Disinfection Theory. In Ultraviolet Germicidal Irradiation Handbook; Springer: Berlin/Heidelberg, Germany, 2009; pp. 17–50. [Google Scholar] [CrossRef]
- Budowsky, E.I.; Bresler, S.E.; Friedman, E.A.; Zheleznova, N.V. Principles of selective inactivation of viral genome. Arch. Virol. 1981, 68, 239–247. [Google Scholar] [CrossRef]
- Molteni, E.; Fratesi, G.; Cappellini, G.; Onida, G. Optical Properties of Free and Si(001)-Adsorbed Pyrimidinic Nucleobases. Phys. Status Solidi (b) 2017, 255, 1700497. [Google Scholar] [CrossRef]
- Molteni, E.; Cappellini, G.; Onida, G.; Fratesi, G. Optical properties of organically functionalized silicon surfaces: Uracil-like nucleobases on Si(001). Phys. Rev. B 2017, 95, 05437–1–05437–8. [Google Scholar] [CrossRef] [Green Version]
- Cadelano, E.; Cappellini, G. Electronic structure of fluorides: General trends for ground and excited state properties. Eur. Phys. J. B 2011, 81, 115–120. [Google Scholar] [CrossRef]
- Cadelano, E.; Furthmüller, J.; Cappellini, G.; Bechstedt, F. One- and two-particle effects in the electronic and optical spectra of barium fluoride. J. Phys. Condens. Matter 2014, 26, 125501. [Google Scholar] [CrossRef]
- Cappellini, G.; Furthmüller, J.; Cadelano, E.; Bechstedt, F. Electronic and optical properties of cadmium fluoride: The role of many-body effects. Phys. Rev. B 2013, 87, 075203–1–075203–9. [Google Scholar] [CrossRef] [Green Version]
- Filippetti, A.; Fiorentini, V. A practical first-principles band-theory approach to the study of correlated materials. Eur. Phys. J. B 2009, 71, 139–183. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Pelá, R.R.; Teles, L.K.; Marques, M., Jr.; Furthmülller, J. The LDA-1/2 technique: Recent Developments. In Proceedings of the 31st International Conference on the Physics of Semiconductors (ICPS), Zurich, Switzerland, 29 July–3 August 2012; AIP: Long Island, NY, USA, 2013; Volume 1566, pp. 27–28. [Google Scholar] [CrossRef]
- Matusalem, F.; Marques, M.; Teles, L.K.; Filippetti, A.; Cappellini, G. Electronic properties of fluorides by approximated quasiparticle DFT-1/2 and PSIC methods: BaF2, CaF2 and CdF2 as test cases. J. Phys. Condens. Matter 2018, 30, 365501. [Google Scholar] [CrossRef] [PubMed]
- Frandon, J.; Lahaye, B.; Pradal, F. Spectra of Electronic Excitations in CaF2, SrF2, and BaF2 in the 8 to 150 eV Range. Phys. Status Solidi (b) 1972, 53, 565–575. [Google Scholar] [CrossRef]
- Raisin, C.; Berger, J.M.; Robin-Kandare, S. UPS and XPS spectra of CdF2 and SrF2 and interpretation of optical properties of these compounds. J. Phys. C Solid State Phys. 1980, 13, 1835–1844. [Google Scholar] [CrossRef]
- Kudrnovský, J.; Christensen, N.E.; Maek, J. Electronic structure of fluorite-type compounds and mixed crystals. Phys. Rev. B 1991, 43, 12597–12606. [Google Scholar] [CrossRef]
- Khenata, R.; Daoudi, B.; Sahnoun, M.; Baltache, H.; Rérat, M.; Reshak, A.H.; Bouhafs, B.; Abid, H.; Driz, M. Structural, electronic and optical properties of fluorite-type compounds. Eur. Phys. J. B 2005, 47, 63–70. [Google Scholar] [CrossRef]
- Ivanovskikh, K.; Pustovarov, V.; Shulgin, B. Time-resolved luminescent VUV-spectroscopy of pure and doped by rare earth ions crystals of strontium fluoride. Nucl. Instruments Methods Phys. Res. Sect. A Accel. Spectrometers, Detect. Assoc. Equip. 2005, 543, 229–233. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.R.; Sawala, N.S.; Nagpure, P.A.; Barde, W.S.; Omanwar, S. The Highly Efficient Inorganic SrF2:Gd3+,Eu3+ Phosphor for Mercury Free Fluorescence Lamps. Adv. Mater. Res. 2022, 1171, 17–24. [Google Scholar] [CrossRef]
- Chaney, R.C. Self-consistent energy band structure of magnesium fluoride using the LCAO method. J. Phys. C Solid State Phys. 1980, 13, 5691–5699. [Google Scholar] [CrossRef]
- Vassilyeva, A.; Eglitis, R.; Kotomin, E.; Dauletbekova, A. Ab initio calculations of the atomic and electronic structure of MgF2 (011) and (111) surfaces. Open Phys. 2011, 9, 515–518. [Google Scholar] [CrossRef]
- Yi, Z.; Jia, R. Quasiparticle band structures and optical properties of magnesium fluoride. J. Phys. Condens. Matter 2012, 24, 085602(5pp). [Google Scholar] [CrossRef] [PubMed]
- Cappellini, G.; Bosin, A.; Serra, G.; Furthmüller, J.; Bechstedt, F.; Botti, S. Electronic and Optical Properties of Small Metal Fluoride Clusters. ACS Omega 2020, 5, 13268–13277. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.B.; Hargittai, M. Unusual Dimer Structures of the Heavier Alkaline Earth Dihalides: A Density Functional Study. J. Phys. Chem. A 2000, 104, 1950–1958. [Google Scholar] [CrossRef]
- Koput, J.; Roszczak, A. CaF2 As a Quasilinear Molecule: The Vibrational-Rotational Energy Levels Predicted by Ab Initio Quantum Chemistry Approach. J. Phys. Chem. A 2004, 108, 9267–9273. [Google Scholar] [CrossRef]
- Pandey, R.K.; Waters, K.; Nigam, S.; He, H.; Pingale, S.S.; Pandey, A.C.; Pandey, R. A theoretical study of structural and electronic properties of alkaline-earth fluoride clusters. Comput. Theor. Chem. 2014, 1043, 24–30. [Google Scholar] [CrossRef]
- Calder, V.; Mann, D.E.; Seshadri, K.S.; Allavena, M.; White, D. Geometry and Vibrational Spectra of Alkaline-Earth Dihalides. II. CaF2, SrF2, and BaF2. J. Chem. Phys. 1969, 51, 2093–2099. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Vinet, P.; Rose, J.H.; Ferrante, J.; Smith, J.R. Universal features of the equation of state of solids. J. Phys. Condens. Matter 1989, 1, 1941–1963. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 136406–1–136406–4. [Google Scholar] [CrossRef] [Green Version]
- Aroyo, M.I. (Ed.) Teaching Edition of International Tables for Crystallography: Crystallographic Symmetry; IUCr/Wilet: Chester, UK, 2021; ISBN 978-0-470-97422-3. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Armiento, R.; Mattsson, A.E. Functional designed to include surface effects in self-consistent density functional theory. Phys. Rev. B 2005, 72, 085108–1–085108–5. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5048–5079. [Google Scholar] [CrossRef] [Green Version]
- Haines, J.; Léger, J.M.; Gorelli, F.; Klug, D.D.; Tse, J.S.; Li, Z.Q. X-ray diffraction and theoretical studies of the high-pressure structures and phase transitions in magnesium fluoride. Phys. Rev. B 2001, 64, 134110–1–134110–10. [Google Scholar] [CrossRef]
- Subhadra, K.; Hussain, K.A.; Hussain, W.; Sirdeshmukh, D.B. Thermal expansion of strontium fluoride. J. Mater. Sci. Lett. 1985, 4, 777–778. [Google Scholar] [CrossRef]
- Gerlich, D. Elastic Constants of Strontium Fluoride between 4.2 and 300 °K. Phys. Rev. 1964, 136, A1366–A1368. [Google Scholar] [CrossRef]
- Hedin, L. New Method for Calculating the One-Particle Green’s Function with Application to the Electron-Gas Problem. Phys. Rev. 1965, 139, A796–A823. [Google Scholar] [CrossRef]
- Godby, R.W.; Schlüter, M.; Sham, L.J. Self-energy operators and exchange-correlation potentials in semiconductors. Phys. Rev. B 1988, 37, 10159–10175. [Google Scholar] [CrossRef]
- Bechstedt, F.; Sole, R.D.; Cappellini, G.; Reining, L. An efficient method for calculating quasiparticle energies in semiconductors. Solid State Commun. 1992, 84, 765–770. [Google Scholar] [CrossRef]
- Onida, G.; Reining, L.; Rubio, A. Electronic excitations: Density-functional versus many-body Green’s-function approaches. Rev. Mod. Phys. 2002, 74, 601–659. [Google Scholar] [CrossRef] [Green Version]
- Sangalli, D.; Ferretti, A.; Miranda, H.; Attaccalite, C.; Marri, I.; Cannuccia, E.; Melo, P.; Marsili, M.; Paleari, F.; Marrazzo, A.; et al. Many-body perturbation theory calculations using the yambo code. J. Phys. Condens. Matter 2019, 31, 325902. [Google Scholar] [CrossRef]
- Seidl, A.; Görling, A.; Vogl, P.; Majewski, J.A.; Levy, M. Generalized Kohn-Sham schemes and the band-gap problem. Phys. Rev. B 1996, 53, 3764–3774. [Google Scholar] [CrossRef]
- Bechstedt, F.; Fuchs, F.; Kresse, G. Ab-initio theory of semiconductor band structures: New developments and progress. Phys. Status Solidi (b) 2009, 246, 1877–1892. [Google Scholar] [CrossRef]
- Borlido, P.; Aull, T.; Huran, A.W.; Tran, F.; Marques, M.A.L.; Botti, S. Large-Scale Benchmark of Exchange–Correlation Functionals for the Determination of Electronic Band Gaps of Solids. J. Chem. Theory Comput. 2019, 15, 5069–5079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef] [Green Version]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106–1–224106–5. [Google Scholar] [CrossRef] [PubMed]
- Stankovski, M.; Antonius, G.; Waroquiers, D.; Miglio, A.; Dixit, H.; Sankaran, K.; Giantomassi, M.; Gonze, X.; Côté, M.; Rignanese, G.M. G0W0 band gap of ZnO: Effects of plasmon-pole models. Phys. Rev. B 2011, 84, 241201–1–241201–5. [Google Scholar] [CrossRef]
- Cappellini, G.; Sole, R.D.; Reining, L.; Bechstedt, F. Model dielectric function for semiconductors. Phys. Rev. B 1993, 47, 9892–9895. [Google Scholar] [CrossRef]
- Schmidt, W.G.; Glutsch, S.; Hahn, P.H.; Bechstedt, F. Efficient (N2) method to solve the Bethe-Salpeter equation. Phys. Rev. B 2003, 67, 085307–1–085307–7. [Google Scholar] [CrossRef]
- Caruso, F.; Rinke, P.; Ren, X.; Scheffler, M.; Rubio, A. Unified description of ground and excited states of finite systems: The self-consistentGWapproach. Phys. Rev. B 2012, 86, 081102–1–081102–5. [Google Scholar] [CrossRef] [Green Version]
- Vassilyeva, A.; Eglitis, R.; Kotomin, E.; Dauletbekova, A. Ab initio calculations of MgF2 (001) and (011) surface structure. Phys. B Cond. Matt. 2010, 405, 2125–2127. [Google Scholar] [CrossRef]
- Jia, R.; Shi, H.; Borstel, G. Ab initio calculations for SrF2 with F- and M-centers. Comp. Mat. Sci. 2008, 43, 980–988. [Google Scholar] [CrossRef]
- Yue, L.; Jia, R.; Shi, H.; He, X.; Eglitis, R.I. First-Principles Calculations for the H Center in SrF2 Crystals. J. Phys. Chem. A 2010, 114, 8444–8449. [Google Scholar] [CrossRef]
- Jibran, M.; Murtaza, G.; Khan, M.; Khenata, R.; Muhmmad, S.; Ali, R. First principle study of MF2 ( M=Mg, Ca, Sr, Ba, Ra) compounds. Comp. Mater. Sci. 2014, 81, 575–581. [Google Scholar] [CrossRef]
- Kolobanov, V.N.; Mikhailin, V.V.; Chernov, S.P.; Spassky, D.A.; Makhov, V.N.; Kirm, M.; Feldbach, E.; Vielhauer, S. Luminescence of singlet self-trapped excitons in MgF2. J. Phys. Condens. Matter 2009, 21, 375501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisitsyn, V.; Lisitsyna, L.; Popov, A.; Kotomin, E.; Abuova, F.; Akilbekov, A.; Maier, J. Stabilization of primary mobile radiation defects in MgF2 crystals. Nucl. Instruments Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2016, 374, 24–28. [Google Scholar] [CrossRef]
- Shishkin, M.; Kresse, G. Self-consistent GW calculations for semiconductors and insulators. Phys. Rev. B 2007, 75. [Google Scholar] [CrossRef]
- Golze, D.; Keller, L.; Rinke, P. Accurate Absolute and Relative Core-Level Binding Energies from GW. J. Phys. Chem. Lett. 2020, 11, 1840–1847. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Jin, Y.; Rinke, P.; Yang, W.; Golze, D. Benchmark of GW Methods for Core-Level Binding Energies. J. Chem. Theory Comput. 2022, 18, 7570–7585. [Google Scholar] [CrossRef]
- Li, J.; Golze, D.; Yang, W. Combining Renormalized Singles GW Methods with the Bethe–Salpeter Equation for Accurate Neutral Excitation Energies. J. Chem. Theory Comput. 2022, 18, 6637–6645. [Google Scholar] [CrossRef]
- Eglitis, R.I.; Purans, J.; Jia, R. Comparative Hybrid Hartree-Fock-DFT Calculations of WO2-Terminated Cubic WO3 as Well as SrTiO3, BaTiO3, PbTiO3 and CaTiO3 (001) Surfaces. Crystals 2021, 11, 455. [Google Scholar] [CrossRef]
- Botti, S.; Marques, M.A.L. Strong Renormalization of the Electronic Band Gap due to Lattice Polarization in the GW Formalism. Phys. Rev. Lett. 2013, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambrecht, W.R.L.; Bhandari, C.; van Schilfgaarde, M. Lattice polarization effects on the screened Coulomb interaction W of the GW approximation. Phys. Rev. Mater. 2017, 1. [Google Scholar] [CrossRef]
- Cao, H.; Yu, Z.; Lu, P.; Wang, L.W. Fully converged plane-wave-based self-consistent GW calculations of periodic solids. Phys. Rev. B 2017, 95. [Google Scholar] [CrossRef] [Green Version]
- Albert, J.P.; Jouanin, C.; Gout, C. Electronic energy bands in the fluorite structure: CaF2 and CdF2. Phys. Rev. B 1977, 16, 4619–4629. [Google Scholar] [CrossRef]
- Fox, M. Optical Properties of Solids; Oxford University Press: Oxford, UK, 2001; Volume 384, p. 416. [Google Scholar]
- Wannier, G.H. The Structure of Electronic Excitation Levels in Insulating Crystals. Phys. Rev. 1937, 52, 191–197. [Google Scholar] [CrossRef]
- Satta, G.; Cappellini, G.; Olevano, V.; Reining, L. Many-body effects in the electronic spectra of cubic boron nitride. Phys. Rev. B 2004, 70, 195212–1–195212–13. [Google Scholar] [CrossRef] [Green Version]
- Kammerlander, D.; Botti, S.; Marques, M.A.L.; Marini, A.; Attaccalite, C. Speeding up the solution of the Bethe-Salpeter equation by a double-grid method and Wannier Wannier interpolation. Phys. Rev. B 2012, 86, 125203–1–125203–5. [Google Scholar] [CrossRef] [Green Version]
- Mocci, P.; Malloci, G.; Bosin, A.; Cappellini, G. Time-Dependent Density Functional Theory Investigation on the Electronic and Optical Properties of Poly-C,Si,Ge-acenes. ACS Omega 2020, 5, 16654–16663. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PBEsol | r-MgF | c-SrF |
|---|---|---|
| a [Å] | 4.6313 | 5.7744 |
| c [Å] | 3.0558 | — |
| 0.6598 | — | |
| B [MPa] | 97.1 | 72.8 |
| dB/dp | 4.69 | 4.71 |
| PBEsol | r-MgF | c-SrF |
|---|---|---|
| E [eV] | 1020 | 640 |
| E [eV] | 1700 | 1640 |
| E [eV] | −30.1122 | −16.3389 |
| k-point set | 12 × 12 × 18 | 12 × 12 × 12 |
| a[Å] | 4.6313 | 4.6928 | 4.6649 | 4.5638 | 4.6249 |
| c[Å] | 3.0558 | 3.0875 | 3.0741 | 3.0194 | 3.0520 |
| 0.6598 | 0.6579 | 0.6590 | 0.6616 | 0.6599 | |
| x | 0.3033 | 0.3035 | 0.3037 | 0.3030 | 0.3027 |
| B[GPa] | 97.1 | 90.1 | 91.6 | 111.2 | 101 ± 3 |
| dB/dP | 4.69 | 4.74 | 4.73 | 4.64 | 4.2 ± 1.1 |
| E[eV] | −30.1122 | −28.7552 | −29.7466 | −33.0805 | |
| a[Å] | 5.7744 | 5.8712 | 5.8094 | 5.6813 | 5.7994 |
| B[GPa] | 72.8 | 64.5 | 67.5 | 84.9 | 67.1 − 74.6 |
| dB/dp | 4.71 | 4.73 | 4.74 | 4.61 | 4.2 ± 1.1 |
| E[eV] | −16.3389 | −15.6630 | −16.0187 | −17.8951 |
| Direct Gap | ||
|---|---|---|
| [eV] | [eV] | |
| PBEsol | 6.921 | −0.320 |
| HSE06 | 9.433 | −0.289 |
| GW | 12.800 | −0.291 |
| GW | 13.243 | −0.285 |
| scQP-GW | 13.945 | −0.277 |
| B3PW | 9.48 | - |
| Other | 12.17 | - |
| Exp. | 12.4 | −0.2 |
| Direct Gap | Indirect Gap | |
| [eV] | [eV] | |
| PBEsol | 6.932 | 6.827 |
| HSE06 | 9.172 | 9.072 |
| GW | 11.437 | 11.316 |
| GW | 11.820 | 11.700 |
| scQP-GW | 12.490 | 12.375 |
| B3PW | 11.306/10.96 | - |
| Other | 11.24 | 11.20(7.55) |
| Exp. | 11.25 | — |
| BSE (GW) | BSE (GW) | BSE (scQP-GW) | ||
|---|---|---|---|---|
| E[eV] | 11.37 | 11.81 | 12.23 | 11.6 |
| E[eV] | 11.76 | 12.21 | 12.62 | 12.1 |
| E[eV] | 1.14 | 1.14 | 1.43 | 0.8 |
| E[eV] | 1.04 | 1.03 | 1.32 | 0.5 |
| 1.88 | 1.85 | 1.84 | 1.67 | |
| 1.91 | 1.890 | 1.87 | 1.68 | |
| BSE (GW) | BSE (GW) | BSE (scQP-GW) | ||
| E[eV] | 10.01 | 10.40 | 10.83 | 10.6 |
| E[eV] | 1.43 | 1.42 | 1.66 | 0.65 |
| 2.18 | 2.15 | 2.13 | 2.08 |
| [eV] | [eV] | [eV] | |
|---|---|---|---|
| Clusters (MgF) | 11.45–12.49 | 6.56–6.78 | 4.49–5.71 |
| Solid r-MgF (Present) | 13.24 (12.4) | 11.8 (11.6) | 1.4 (0.8) |
| Solid r-MgF (Other) | 12.17 | 10.90 | 1.13 |
| Clusters (SrF) | 9.33–10.16 | 5.10–5.26 | 4.23–4.9 |
| Solid c-SrF (Present) | 11.82 (11.25) | 10.4 (10.6) | 1.4(0.65) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cappellini, G.; Furthmüller, J.; Bechstedt, F.; Botti, S. Electronic and Optical Properties of Alkaline Earth Metal Fluoride Crystals with the Inclusion of Many-Body Effects: A Comparative Study on Rutile MgF2 and Cubic SrF2. Symmetry 2023, 15, 539. https://doi.org/10.3390/sym15020539
Cappellini G, Furthmüller J, Bechstedt F, Botti S. Electronic and Optical Properties of Alkaline Earth Metal Fluoride Crystals with the Inclusion of Many-Body Effects: A Comparative Study on Rutile MgF2 and Cubic SrF2. Symmetry. 2023; 15(2):539. https://doi.org/10.3390/sym15020539
Chicago/Turabian StyleCappellini, Giancarlo, Jürgen Furthmüller, Friedhelm Bechstedt, and Silvana Botti. 2023. "Electronic and Optical Properties of Alkaline Earth Metal Fluoride Crystals with the Inclusion of Many-Body Effects: A Comparative Study on Rutile MgF2 and Cubic SrF2" Symmetry 15, no. 2: 539. https://doi.org/10.3390/sym15020539
APA StyleCappellini, G., Furthmüller, J., Bechstedt, F., & Botti, S. (2023). Electronic and Optical Properties of Alkaline Earth Metal Fluoride Crystals with the Inclusion of Many-Body Effects: A Comparative Study on Rutile MgF2 and Cubic SrF2. Symmetry, 15(2), 539. https://doi.org/10.3390/sym15020539