4.1. Computational Analysis
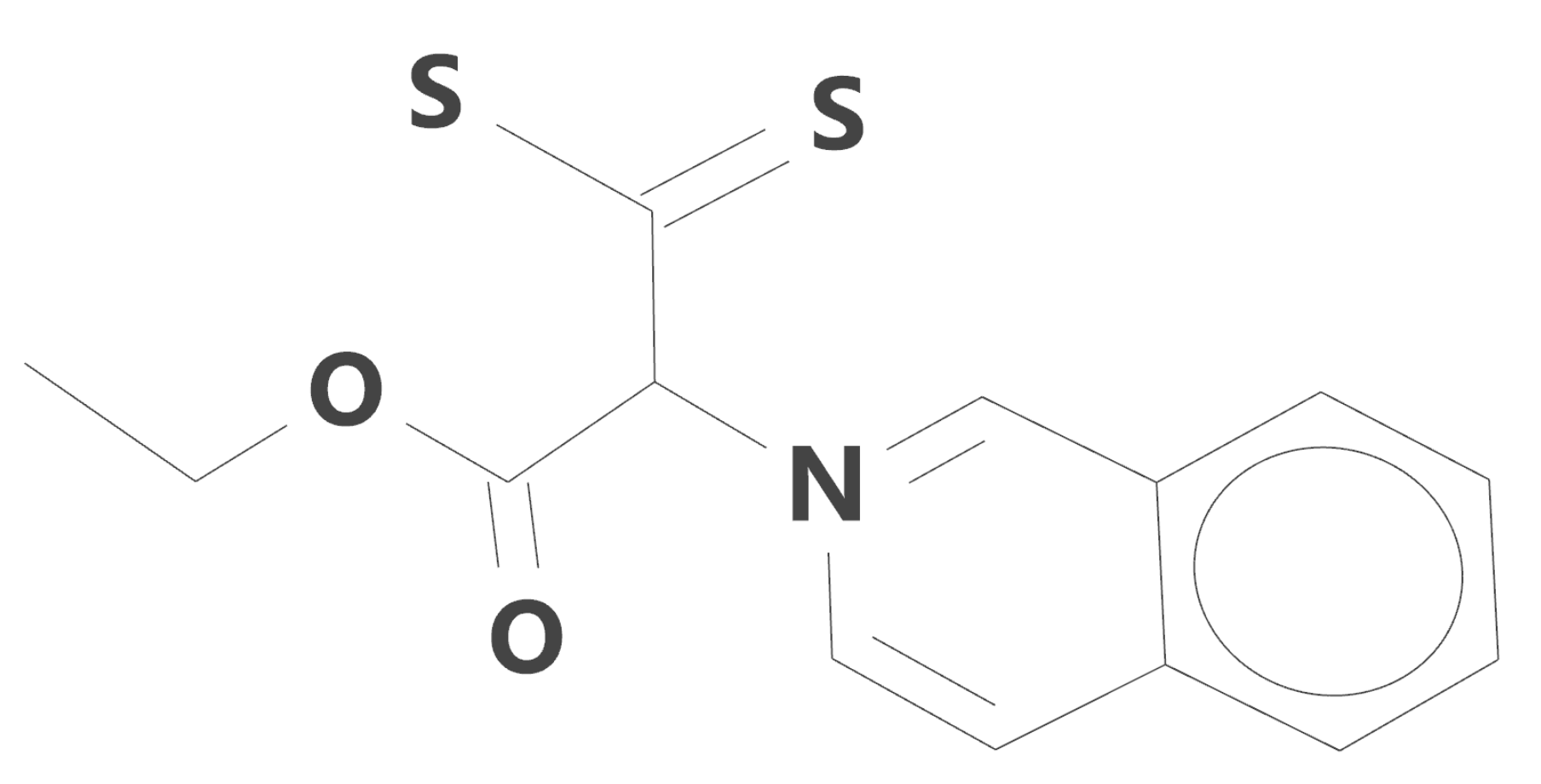
The optimized structure of the iQTCY molecule, obtained by Spartan’14 software [
33] with Density Functional EDF2 model [
34] and the basis set 6-31G*, is shown in
Figure 2.
The ylid bond N
+–C
− belongs to the plane of the iso-quinolinium heterocycle and the molecular dipole moment makes an angle of about 40° with the ylid bond. The iQTCY molecule possesses an asymmetric carbanion and the vector electric dipole moment is not parallel to the ylid bond as in the case of pyridinium dicarbethoxy methylid (PCCM) with carbanion symmetrically disubstituted, as previously studied [
36]. In the case of PCCM, the symmetric carbanion determines the symbiosis effect responsible for the small value of its molecular dipole moment. The symmetric distribution of the electronic charge on the carbanion of the PCCM molecule determines the parallelism between the ylid bond and electric dipole moment.
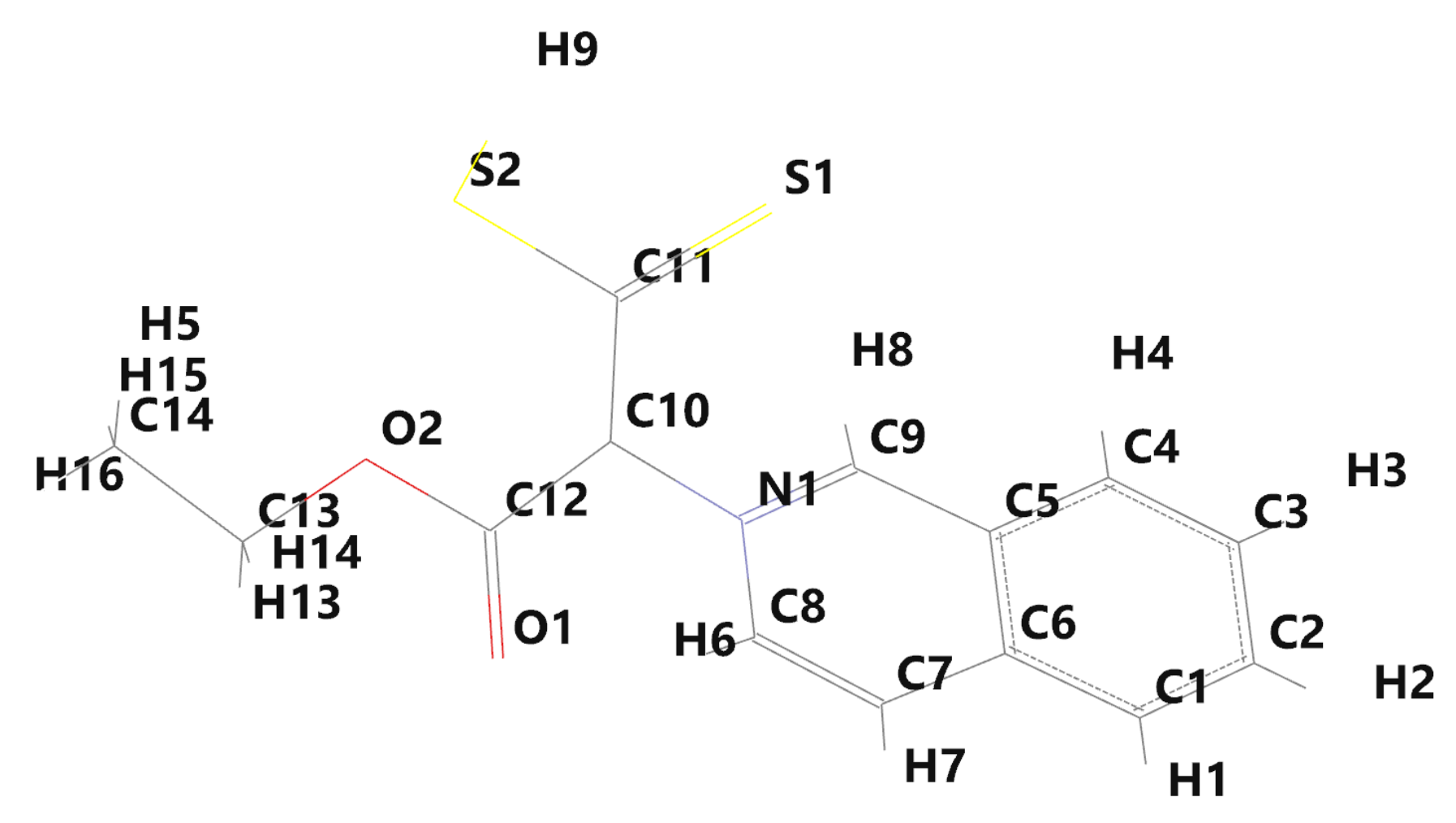
The labels of the molecular atoms are given in
Figure 3, while the electronic charges near the molecular atoms [
14], computed by Spartan’14, are given in
Table 1. A negative value of electronic charge indicates the excess of electrons near an atom, while the positive one shows a deficiency of electrons [
39,
40].
If one compares the electronic charges of the ylid iQTCY’ bond from
Table 1 with those computed for the symmetrically substituted methylid PCCM [
36], one can say that a high charge separation is characteristic for the last molecule in which the carbanion hybridization is
sp2 and its symmetry can be attributed to the C
2v point group of symmetry. In the iQTCY molecule, the carbanion hybridization is between
sp2 and
sp3, determining a 3D spatial distribution of the molecular atoms.
The separated charges on the ylid bond, C
−–N
+, give the zwitterionic character of the studied compound. The high dipole moment in the ground electronic state of iQTCY makes possible orientation–induction interactions between the ylid molecule and the solvent molecules. At the same time, the studied ylid has a basic character, being able to receive protons from the solvent molecules in specific interactions of the hydrogen bond type (its HBA count is equal to 4, while 1 is its HBD count in
Table 2).
The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) maps of iQTCY are illustrated in
Figure 4, together with the electronic levels participating in the spectral transition.
From
Figure 4, it can be seen that in the light absorption process, the valence electron cloud shifts from the ylid carbanion toward the heterocycle. Through the intramolecular charge transfer (ICT), the ylid heterocycle is enriched in electrons and the electric dipole moment of the molecule decreases.
Some energetic and electro-optic parameters of iQTCY in the isolated state, water, and ethanol, computed by Spartan’14, are listed in
Table 2.
Table 2 shows the results of the good stability of the iQTCY molecule (its energy is high in modulus). The molecule’s polar surface area (PSA) was small enough, showing the ability of the molecule to penetrate both the cell membrane and the blood–brain barrier [
41]. The molecule’s ovality (defined as the ratio between the surface of the molecule and the surface of a sphere with the same volume) was 1.44. This value demonstrates that the theory of the solvents’ influence on the wavenumbers in the maxima of the electronic spectra (developed in the hypothesis of spherical molecules [
18]) can be applied to the solutions of this molecule. The hydrogen bond acceptor (HBA) count was 4, showing the ability of the ylid to accept protons in the hydrogen bonds in four places, while its HBD was 1, characterizing the ylid’s low donating ability for protons. The ground state dipole moment of the isolated (in vacuum) iQTCY molecule had a considerable value (7.89 D), while for PCCM, the dipole moment was 2.87 D. In hydroxy solvents (water and ethanol), the computed ground state dipole moment increased compared with the isolated state of iQTCY, as shown in
Table 2. A considerable increase in the ground state dipole moment was established in [
36] for PCCM when this molecule was passed from the isolated state in hydroxy solutions achieved in water and in ethanol.
4.2. Spectral Analysis
The solvent parameters and the wavenumber in the maximum of the visible electronic absorption band of iQTCY are given in
Table 3. The electronic absorption band of iQTCY was recorded and they are in good accordance with those from [
14], where the solvent parameters can be found.
The spectral data from [
14] were verified for all solvents given in
Table 3 and used to compute in percent the contribution of each type of interaction taken into consideration in Equation (7) for the binary diluted solutions of iQTCY.
The solvent influence on the visible electronic absorption band of iQTCY can be described by a linear multi-parametric relation of the type (1) [
35,
36,
37,
38]. The statistical analysis of the multi-parametric Equation (1) with the data obtained in the quantum chemical computation (
Table 2) and solvatochromic data (from
Table 3) gives Equation (7).
The value of the multilinear correlation coefficient was R = 0.9195.
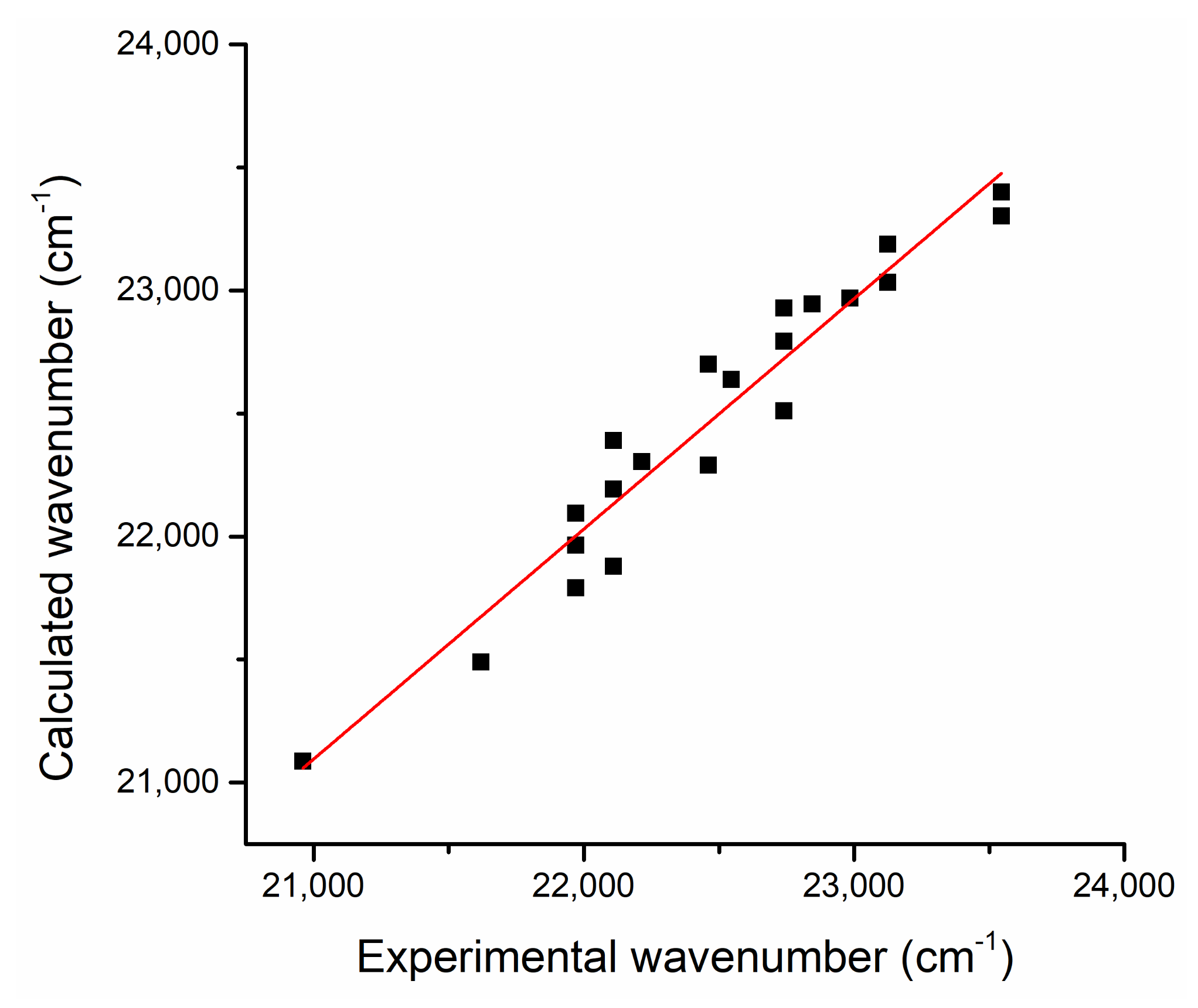
Figure 5 shows the calculated (by Equation (7)) versus experimental wavenumbers corresponding to the maximum of the electronic absorption band (
Table 3). A good correlation could be observed, the slope of the linear fit being 0.94, while the linear correlation coefficient had the value
R = 0.97.
The values of the regression coefficients
C1−
C4 from Equation (7) were used to compute the contributions of each type of intermolecular interaction to the total spectral shift of the visible electronic absorption band of iQTCY, given as a percent in
Table 3. It can be observed that the orientation–induction interactions were predominant, with a contribution of more than 50% for each of the solvents. The dispersion interactions were important for nonpolar solvents (e.g., 43.70% for benzene), while the specific interactions had a share of 20–25% for alcohols (HBD solvents). The specific interactions with the HBA solvents had the smallest contribution to the spectral shift, as shown in
Table 3, in accordance with the small value of the HBD count of iQTCY (see
Table 2).
The data related to the statistical cell model of ternary solutions iQTCY + water (1) + ethanol (2) and iQTCY + water (1) + methanol (2) are given in
Table 4 and
Table 5.
Since the intermolecular interaction energies are proportional to
R−6 [
42,
43,
44], where
R is the distance between the molecules, in solvatochromism, in the first approximation, only the interactions between the spectrally active molecule and the solvent’s molecules from its first solvation shell are considered. In the case of the ternary solutions, the interaction strengths between the spectrally active molecule and the molecules of the two solvents are different, so the composition of the first solvation shell is different from that of the whole solution. Consequently, to study the shift of the electronic absorption spectral band in such solutions, it is very important to know the composition of the first solvation shell. There are several models [
30,
31,
45,
46,
47,
48] that can be used to estimate this composition.
The statistical cell model of ternary solutions [
30,
31] consider the system formed by the spectrally active molecule and the solvents’ molecules from its first solvation shell as a thermodynamic macro canonical ensemble, for which the reservoir is the rest of the solution. In such a frame, the model estimates the average statistical weights,
p1 and
p2, of the two solvents’ molecules in the first solvation shell function of the molecular fractions of the two solvents in the whole solution,
x1 and
x2, temperature
T, and the interaction energies,
w1 and
w2, in the pairs of molecules solute–solvent (1) and solute–solvent (2), respectively.
The Suppan model of preferential solvation through dielectric enrichment [
46] establishes a relation between the ratio of the mole fractions of the two solvents in solution
x2/
x1 and the ratio of the mole fractions of the two solvents in the first solvation shell
p2/
p1. This relation allows for the calculation of the mole fraction
p1 and
p2 by first determining the index of preferential solvation
Z [
49].
Unlike the statistical cell model and Suppan model, the Bosch–Rosés model takes into consideration the formation of the 1:1 complex between the molecules of the two solvents. Based on the two-step solvent exchange model proposed by Skwierczynski and Connors [
50], the Bosch–Rosés model proposes a relation between the charge transfer energy at the solvation
ECT and the mole fraction of the second solvent
x2 in the whole solution. The nonlinear regression of this equation makes the determination of the mole fractions
p1,
p2, and
p12 of the two solvents and the complex, respectively, possible in the first solvation shell of the solute molecule.
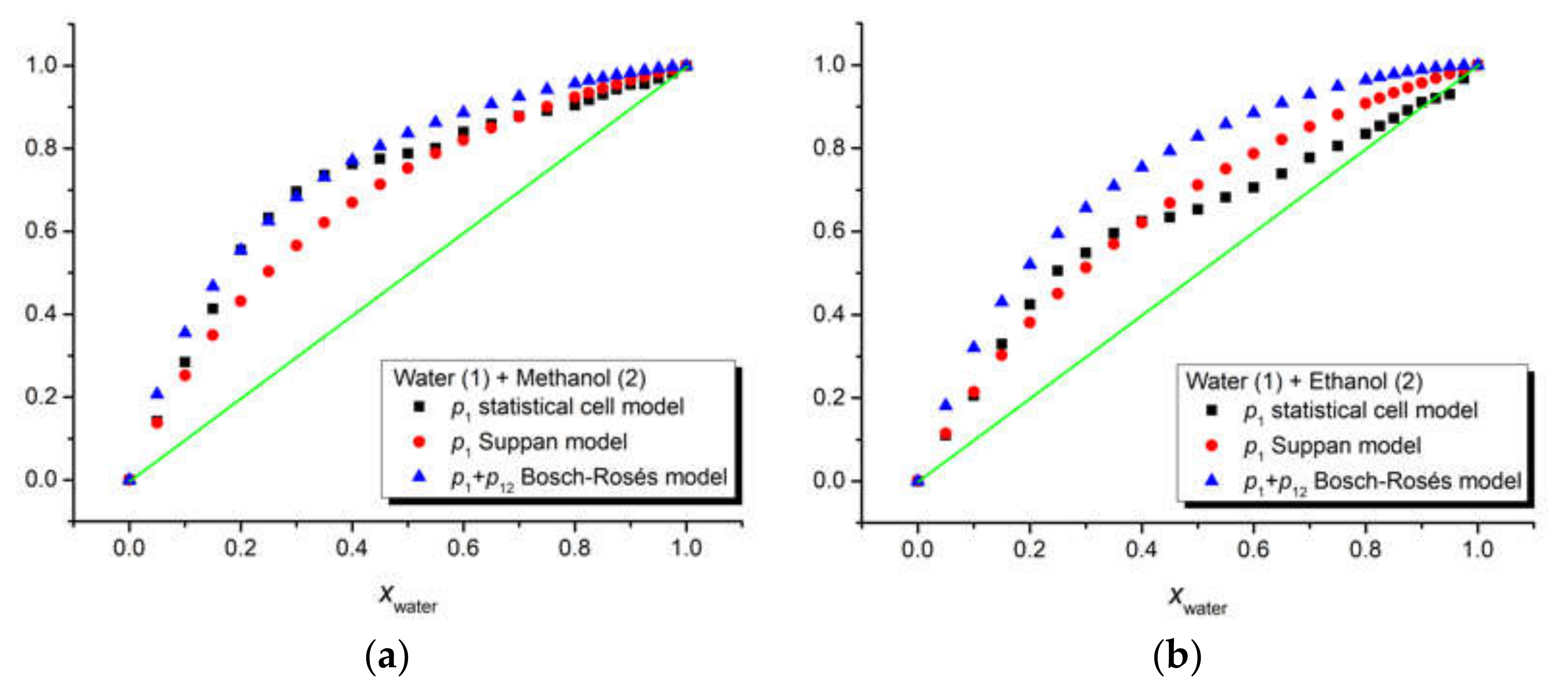
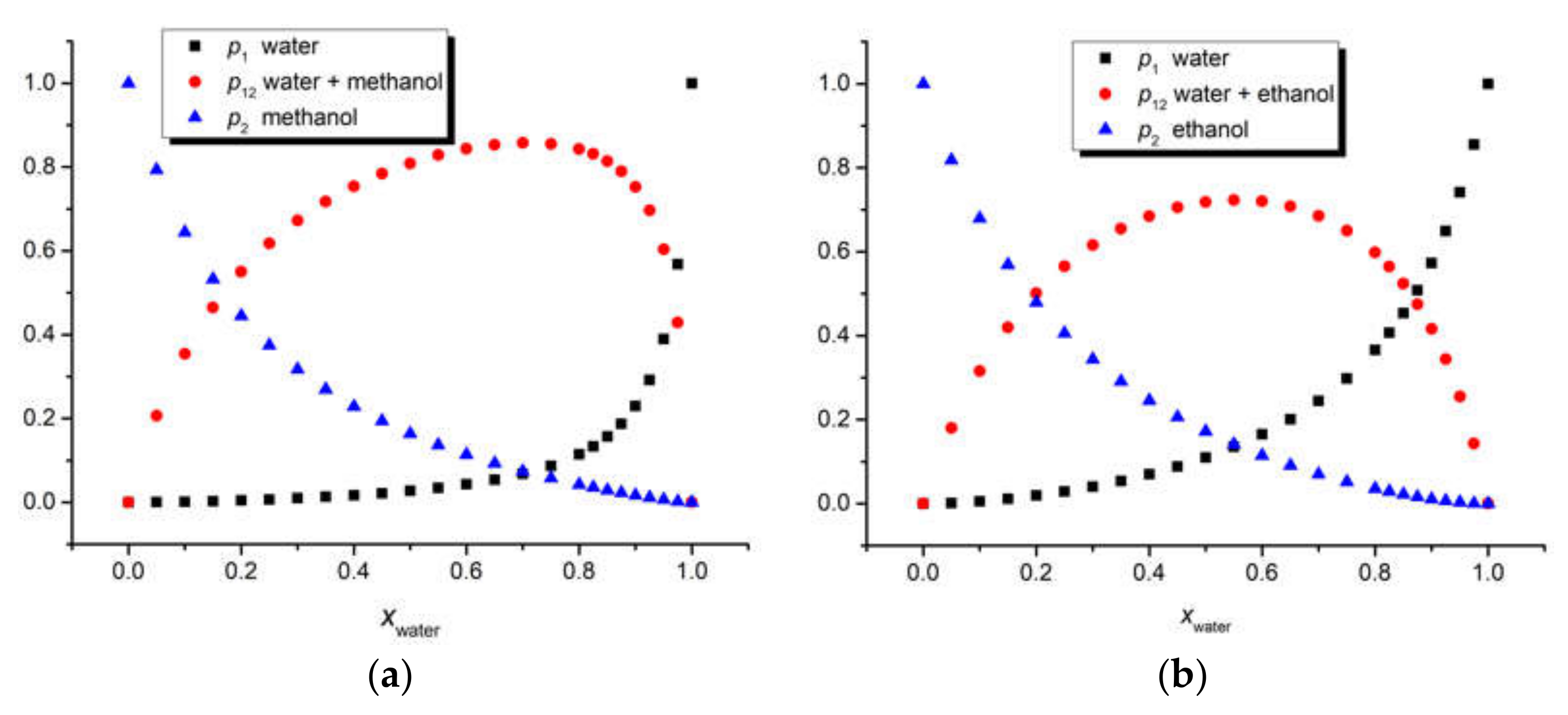
The three above-mentioned models were applied to our ternary solutions iQTCY + water (1) + methanol (2) and iQTCY + water (1) + ethanol (2). The comparative results of the estimation of the first solvation shell’s composition are shown in
Figure 6. It can be observed that water was the active solvent (i.e., the water molecules have stronger interactions with the ylid molecules compared with the alcohol molecules). It should be mentioned that, according to the literature [
51], in this comparison, the sum
p1 +
p12 is considered for the Bosch–Rosés model.
Figure 7 shows the dependence of the mole fractions of the two solvents and of their complex in the first solvation shell of the ylid molecule on the mole fraction of water in the whole solution, demonstrating that the mole fraction of the complex is not negligible at all, even being dominant for a large intermediate interval of the mole fractions of solvents in the whole solution.
The data related to the statistical cell model of ternary solutions iQTCY + water (1) + ethanol (2) and iQTCY + water (1) + methanol (2) are presented in
Table 4 and
Table 5.
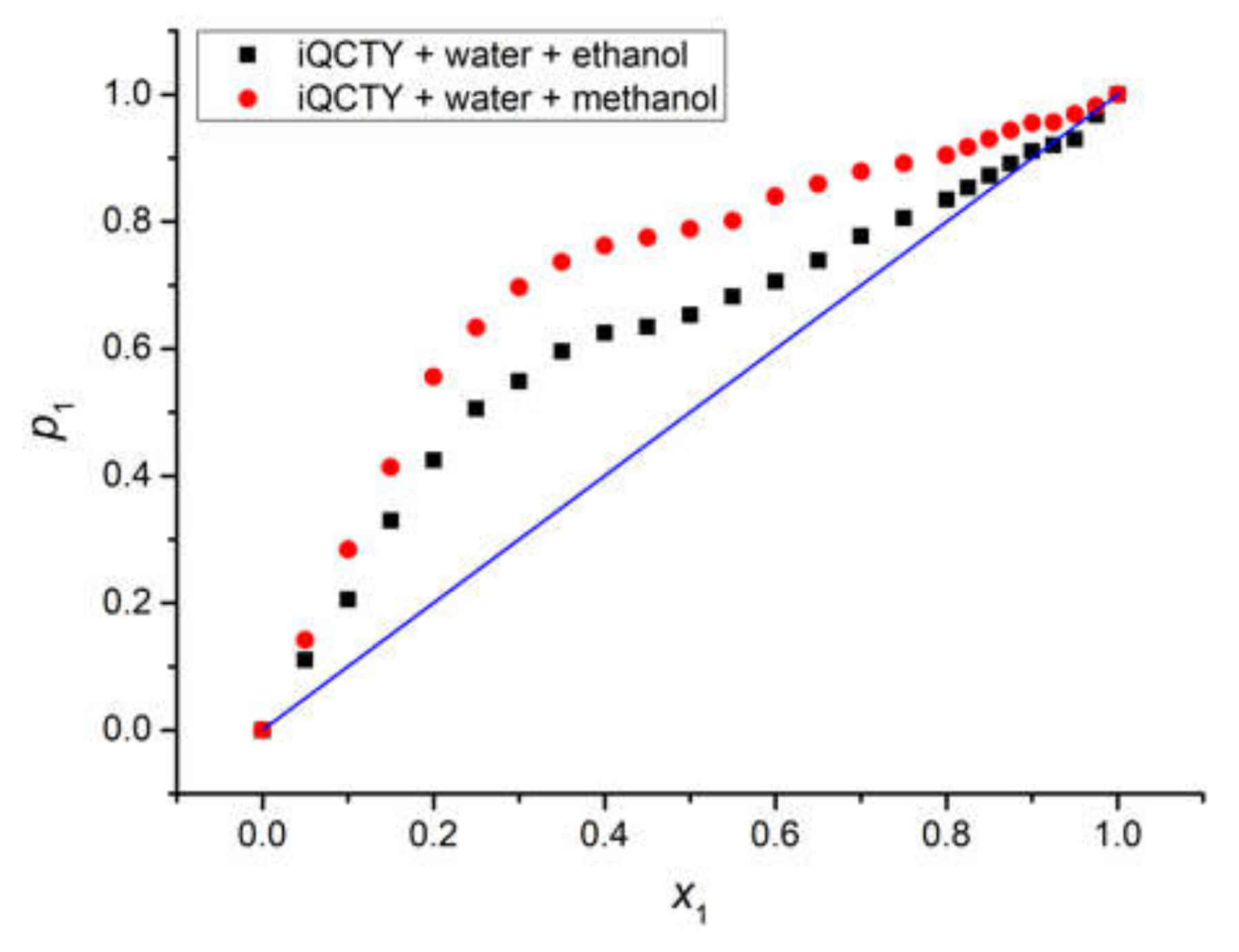
The graphical representation of
p1 versus
x1 for the water in the two binary solvents, shown in
Figure 8, clearly emphasizes that water was the active solvent, while the alcohols were less active solvents, since
p1 was higher than
x1. The average statistic weight of methanol,
p2, in the first solvation shell of the iQTCY molecule was smaller than that of ethanol, showing that ethanol was more active than methanol from the intermolecular interaction point of view.
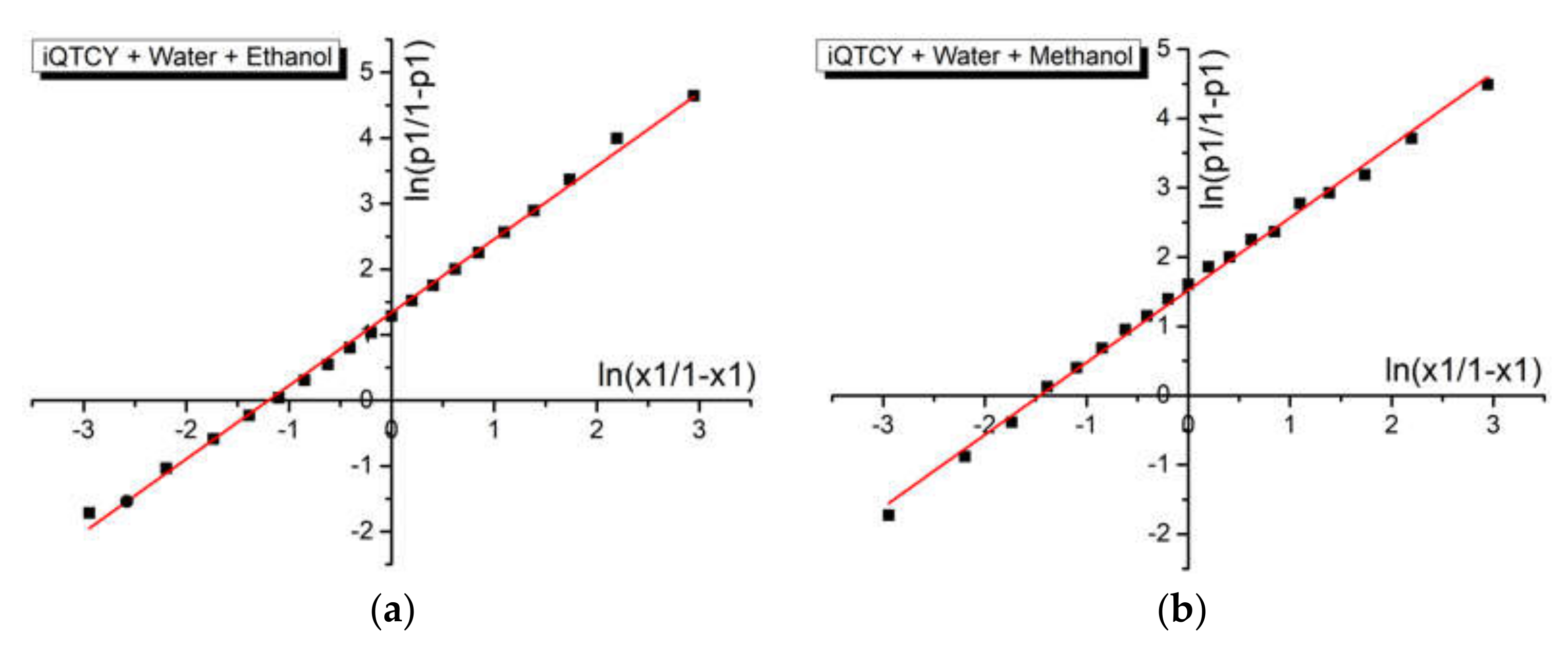
Figure 9 shows the graphical representation of data described by Equation (4) for the binary solvent mixtures water + ethanol and water + methanol, respectively. The linear fitting of these graphs allows for the estimation of the difference between the interaction’s energies in molecular pairs of the types of ylid–water and ylid–alcohol. The results are shown in
Table 6. There was good agreement between these results and those previously obtained for protic binary solvents [
52,
53] and similar zwitterionic spectrally active molecules.
Good values for the linear correlation coefficient (R = 0.997 for water + ethanol and R = 0.996 for water + methanol, see
Table 6) were obtained for both sets of ternary solutions.
As Equation (4) establishes, the intercept approximates the difference w2 − w1 between the interaction energies in molecular pairs of the types: iQTCY–water (1) and iQTCY–alcohol (2): where it was higher in the binary solvent water–methanol than in water–ethanol.
4.3. Excited State Dipole Moment of 1-Dithiocarboxy-2-ethoxy-1-(isoquinolin-2-yl)-2-oxoethan-1-ylid Determined by Variational Method
The values of the parameters C1 and C2 from Equations (1)–(3) are given in Equation (7). The ionization potential of cyclohexane was used in the computations due to its very small contribution to the orientation–induction interactions.
By using the sum
C1 +
C2 and the values of parameters
C1 and
C2 from Equation (7), and the molecular parameters of iQTCY from
Table 2, Equation (8) can be obtained:
In Equation (2), the parameters are introduced by their values; using Equation (8), one obtains:
The denominator of Equation (9) is:
Equation (9) has real solutions for the positive values of Δ and for the values of the angle φ smaller than 80.32°. The solutions of Equation (9) depend on the value of cos
φ:
To obtain information about the excited state dipole moment in the superior electronic state of the ICT transition responsible for the visible absorption band appearance, the variational method was applied. The angle
φ was varied in the range [0°–80.89°]. The dipole moment and the polarizability of the excited state of the ICT transition were computed based on Equations (11) and (8). The results of the computations are listed in
Table 7.
For the angle
φ equal to 20.3°, the electric polarizability in the excited state of iQTCY equalizes the ground state polarizability. In accordance with the McRae supposition, for this value of
φ, the visible photon absorption is realized. This fact can be demonstrated if one separates the contributions based only the dipole moment variation and those due to the electric polarization variation.
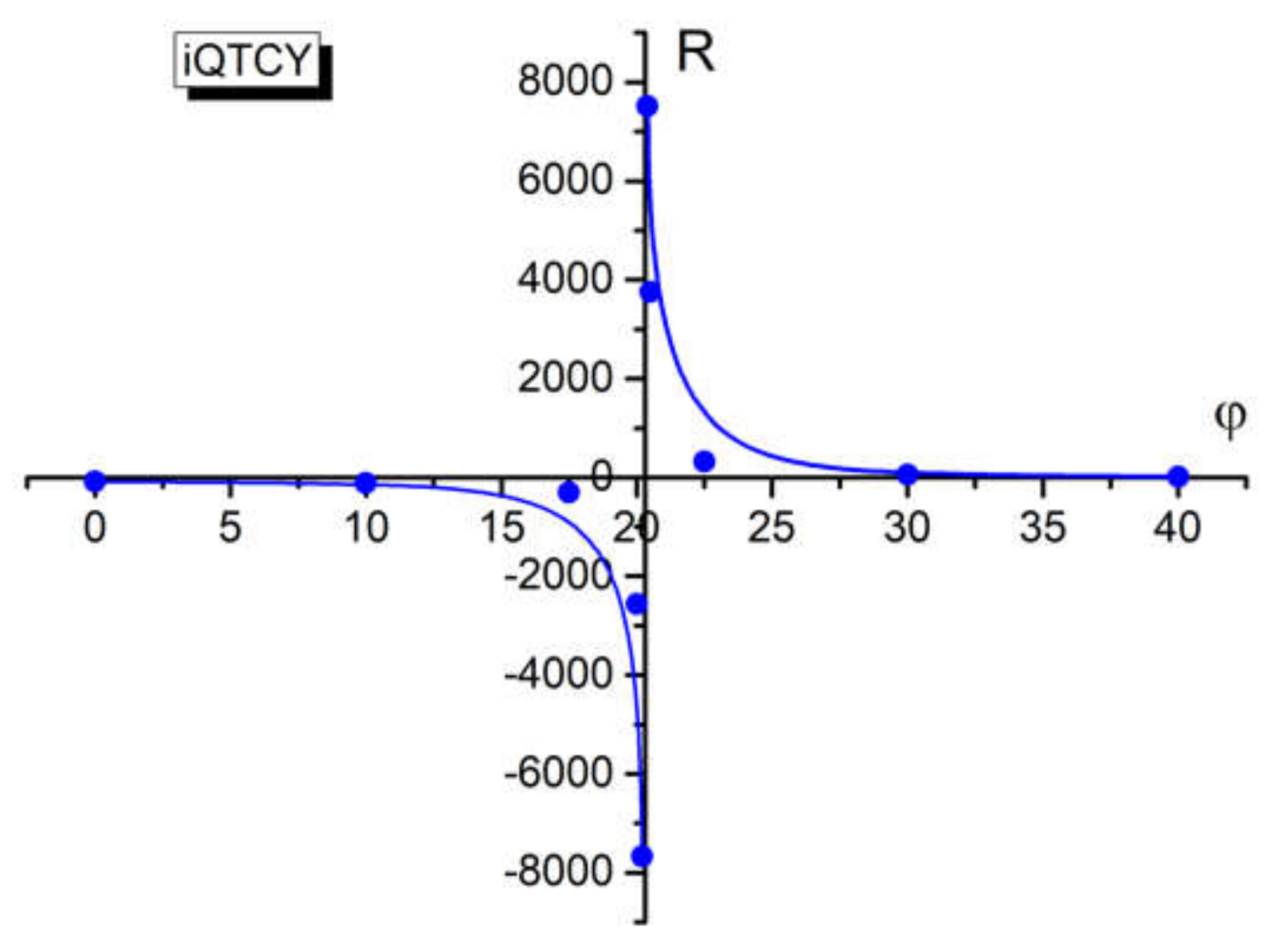
The ratio R between these terms of
C1, defined by Equation (14), supports a discontinuity near the angle 20.3° (angle for which the excited state polarizability equalizes the ground state polarizability of the studied molecule and the difference
αg −
αe changes its sign), as can be seen from
Table 7 and
Figure 10. The discontinuity in the graph
R vs.
φ suggests an electronic discontinuity between the two electronic states participating in the absorption process.
By using the values of the dipole moment in the ground state, (7.89 D) and in the first excited state of ylid (7.78 D) and the angle φ determined in the McRae hypothesis (that the photon absorption takes place when the excited state polarizability equalizes the ground state polarizability of the ylid molecule), one can determine the dipole moment variation in the absorption process (2.76 D).
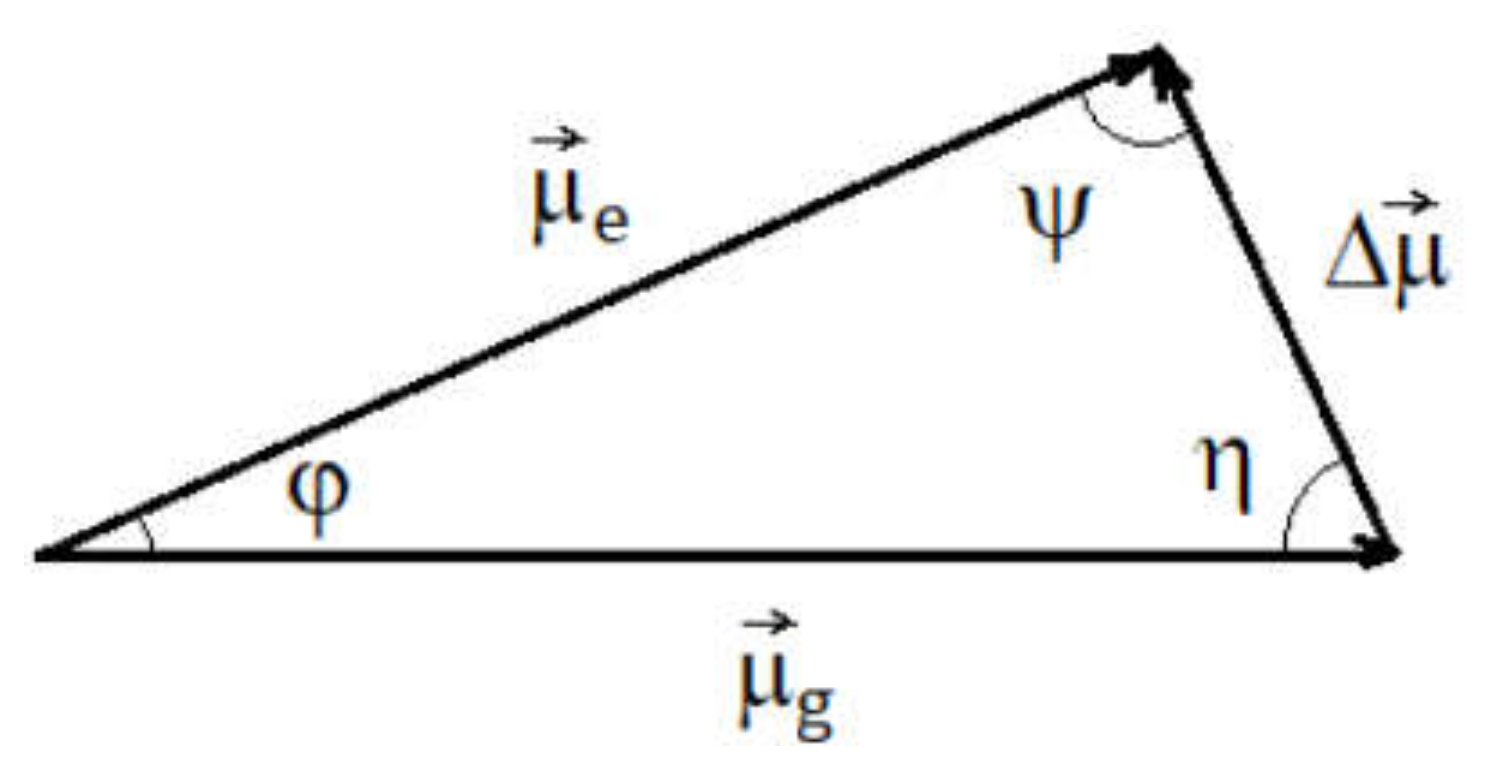
The value of the variation in the molecular dipole moment in the absorption process was obtained by solving the triangle of the electric dipole moments of iQTCY corresponding to the electronic states participating in the ICT (see
Figure 11).
One results the values ψ = 82.09 and η = 77.61 and Δμ = 2.76 D. The values obtained in this paragraph demonstrate the decrease in polarity of iQTCY by absorption in the visible range.















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}