1. Introduction
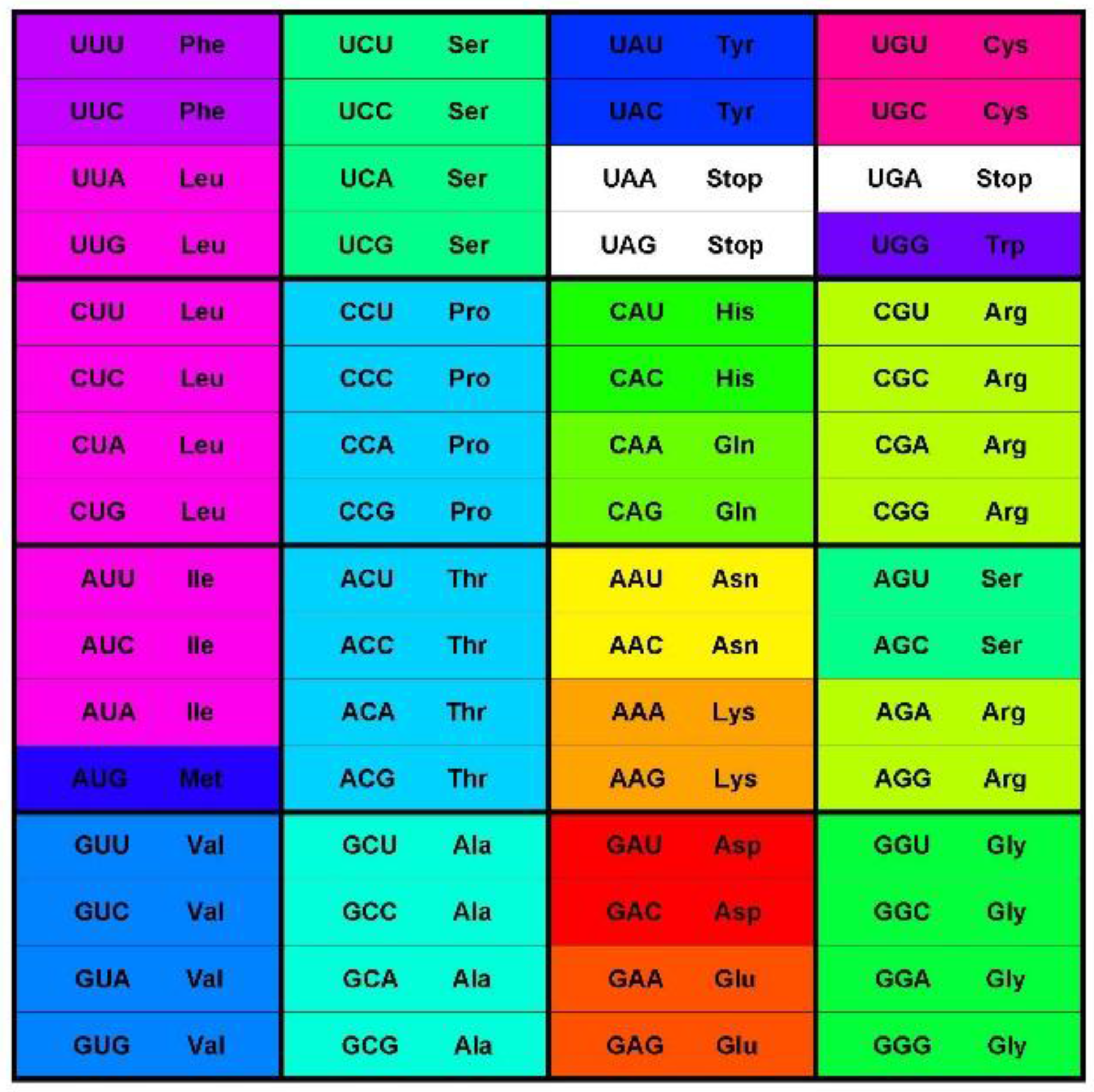
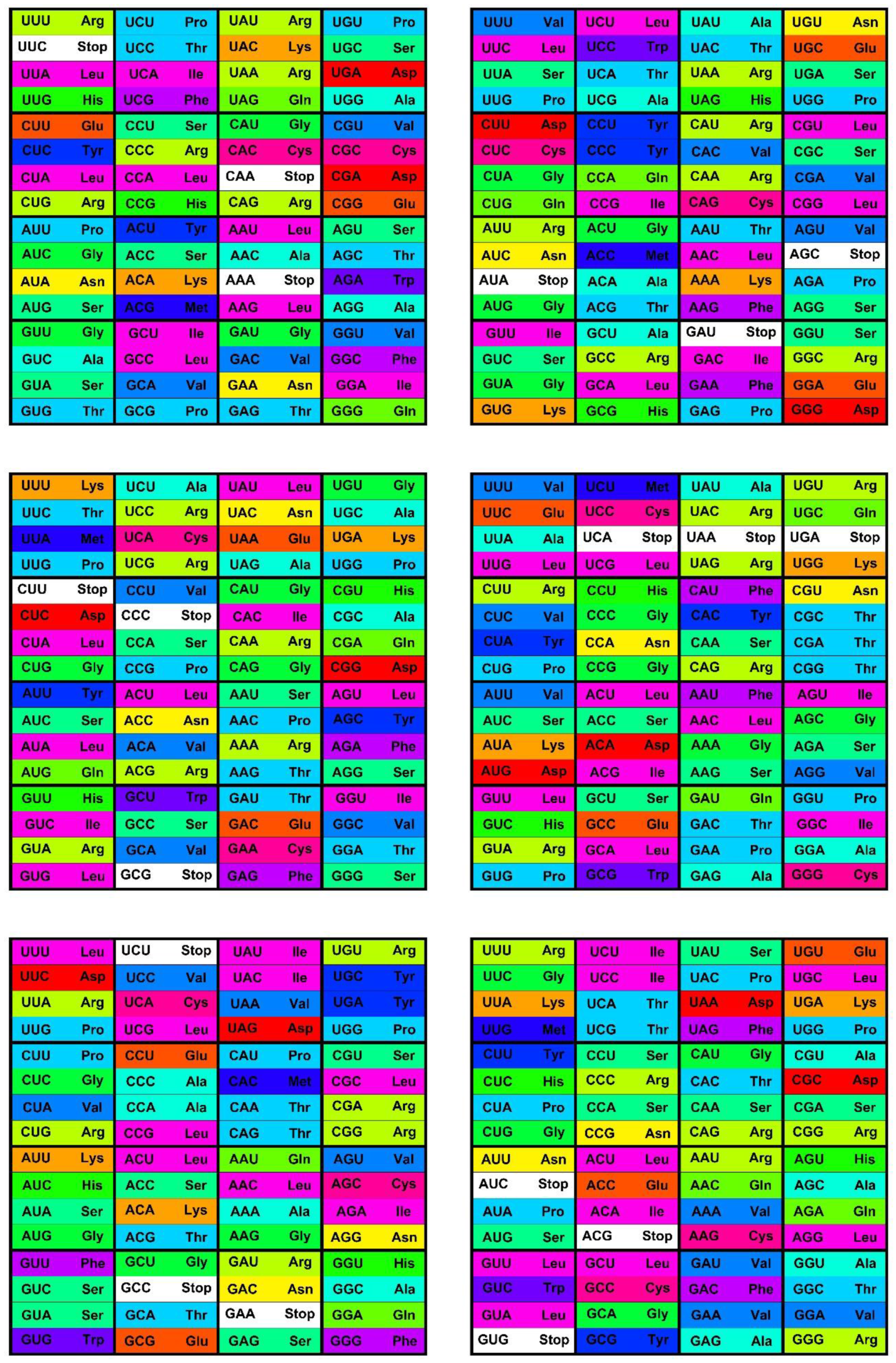
The canonical genetic code as summarized by the codon table (
Figure 1) consists of 64 codons or code words and each word encodes a single message—an amino acid or stop codon. The code is a mapping of the set of 64 codons onto 21 messages. All extant living organisms use this code, or minor variations thereof to synthesize the proteins encoded by their genomes [
1,
2]. This fact strongly argues in favor of the commonly held hypothesis that all known life evolved from a Last Universal Common Ancestor (LUCA), and that the code itself evolved in an RNA world inhabited by pre-LUCA organisms over 3.5 Billion years ago [
3,
4]. The code displays patterns of similarities [
5,
6]. Most messages are encoded by several synonymous codons (the degeneracy of the code), but the coloring of the codon table in
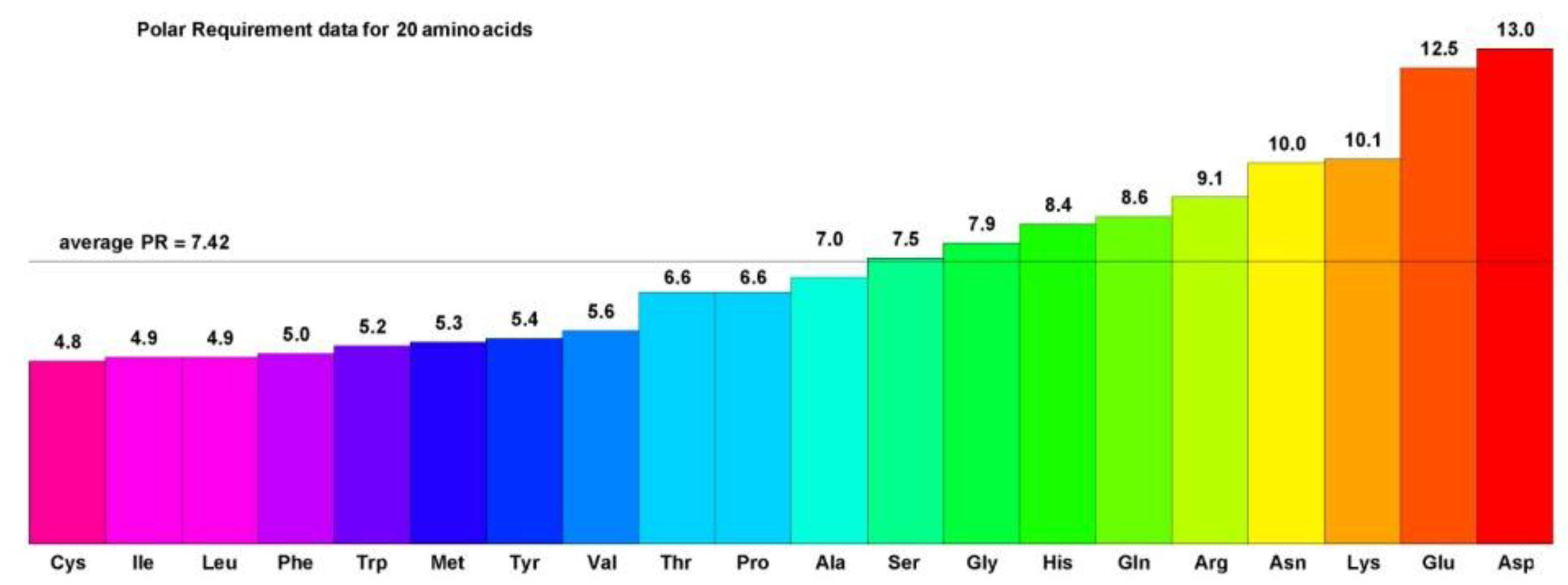
Figure 1 shows broader patterns of similarities as well. The color scheme shows the Polar Requirement of the amino acids (
Figure 2), a physicochemical characteristic frequently used for the analysis of the genetic code [
7]. In the codon table, amino acids with similar Polar Requirements tend to cluster together; that is, they tend to be encoded by similar codons. Simplified amino acid alphabets consist of various sets of similar amino acids, such as a size-2 alphabet composed of a set of hydrophobic and a set of hydrophilic amino acids, and as will be discussed in
Section 6, amino acids belonging to the same set are often grouped together in the codon table. Simplified amino acid alphabets based on physicochemical properties are not essentially different from those using protein sequence or structure information [
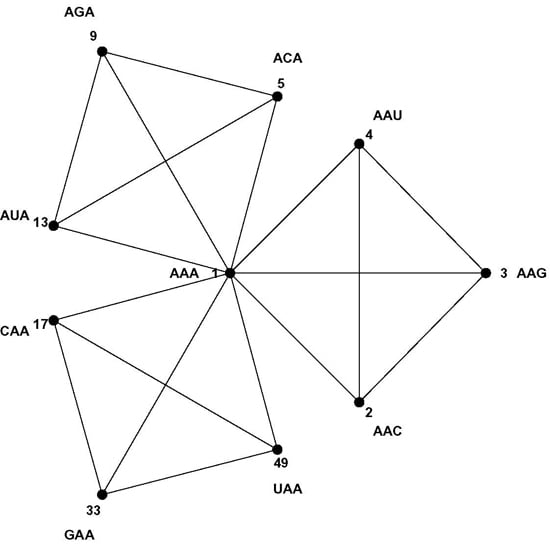
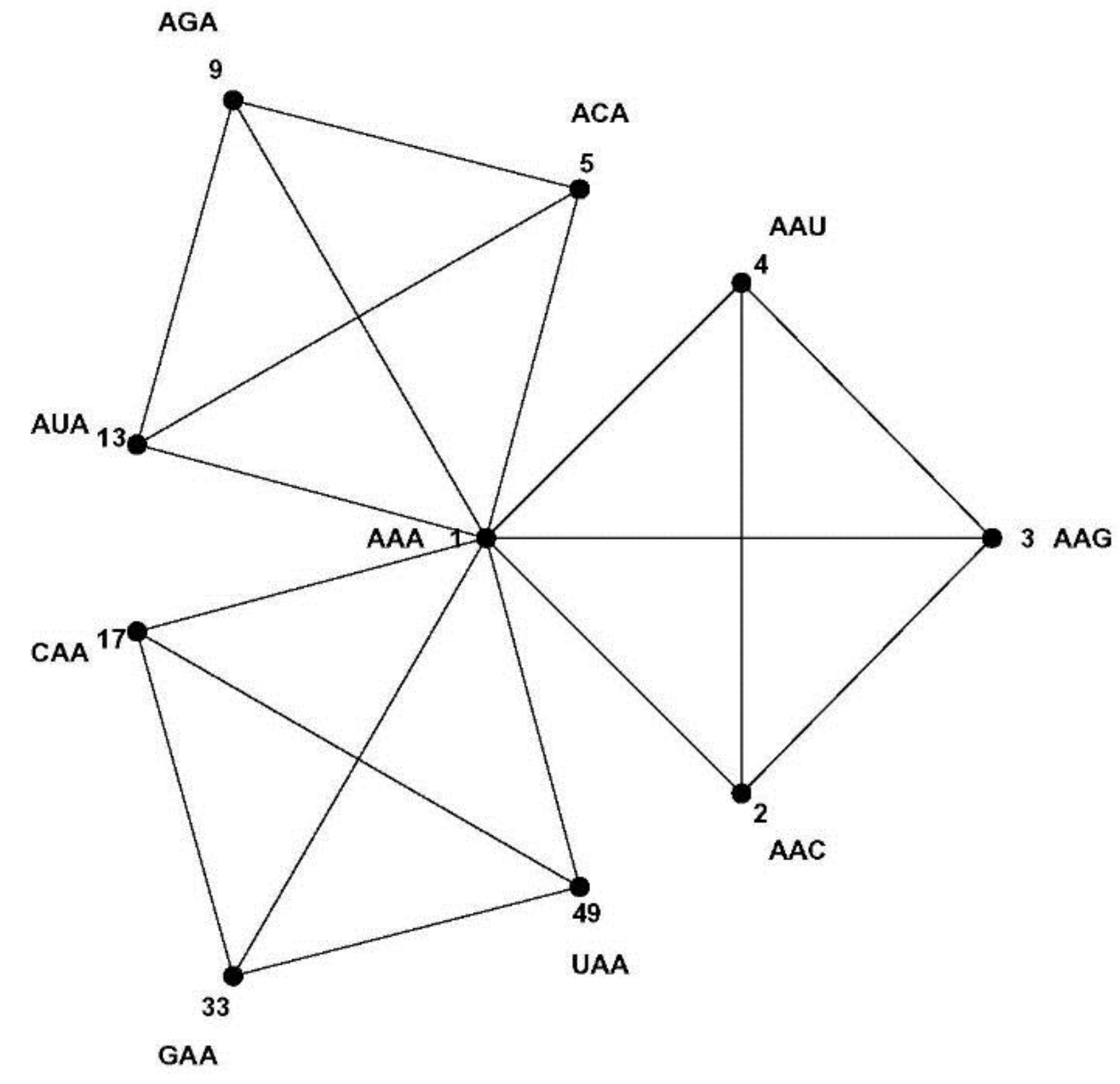
8]. Codons are three letter words made up from a four letter alphabet {A, C, G, U}, and codons encoding the same message most often differ in the third codon position only, while codons encoding similar amino acids usually differ only in one or two positions. The Hamming metric of mathematical coding theory measures these differences between code words: words differing at one, two or three positions are at Hamming 1-, 2-, or 3-distance, respectively [
9]. The codon set combined with the Hamming metric makes a
normed metric space of 64 points with well-defined distances between them. For example, each codon has nine nearest neighbor codons at 1-distance—nine codons differing at only one position, the most similar codons in the codon set by the Hamming metric; such as the nine nearest neighbors of codon AAA: AAC, AAG, AAU, ACA, AGA, AUA, CAA, GAA and UAA. The similarity patterns of the code result from the mapping of the codon space onto the message space and their mathematical analysis is the subject of this paper. As a first step we develop a geometric model of the code that faithfully maps Hamming distances onto Euclidian distances—the CodonPolytope (
Section 4). In this model the codons are represented by 64 points in Euclidian 9-space, the vertices of a 9-dimensional geometric object (containing lower dimensional objects such as cubes and tetrahedrons). While the codon set with Hamming metric possesses permutation symmetries that preserve this metric, the polytope displays Euclidian symmetries and space coordinates that greatly facilitate the (computational) analysis of the similarity patterns of the code (
Section 5,
Section 6 and
Section 7).
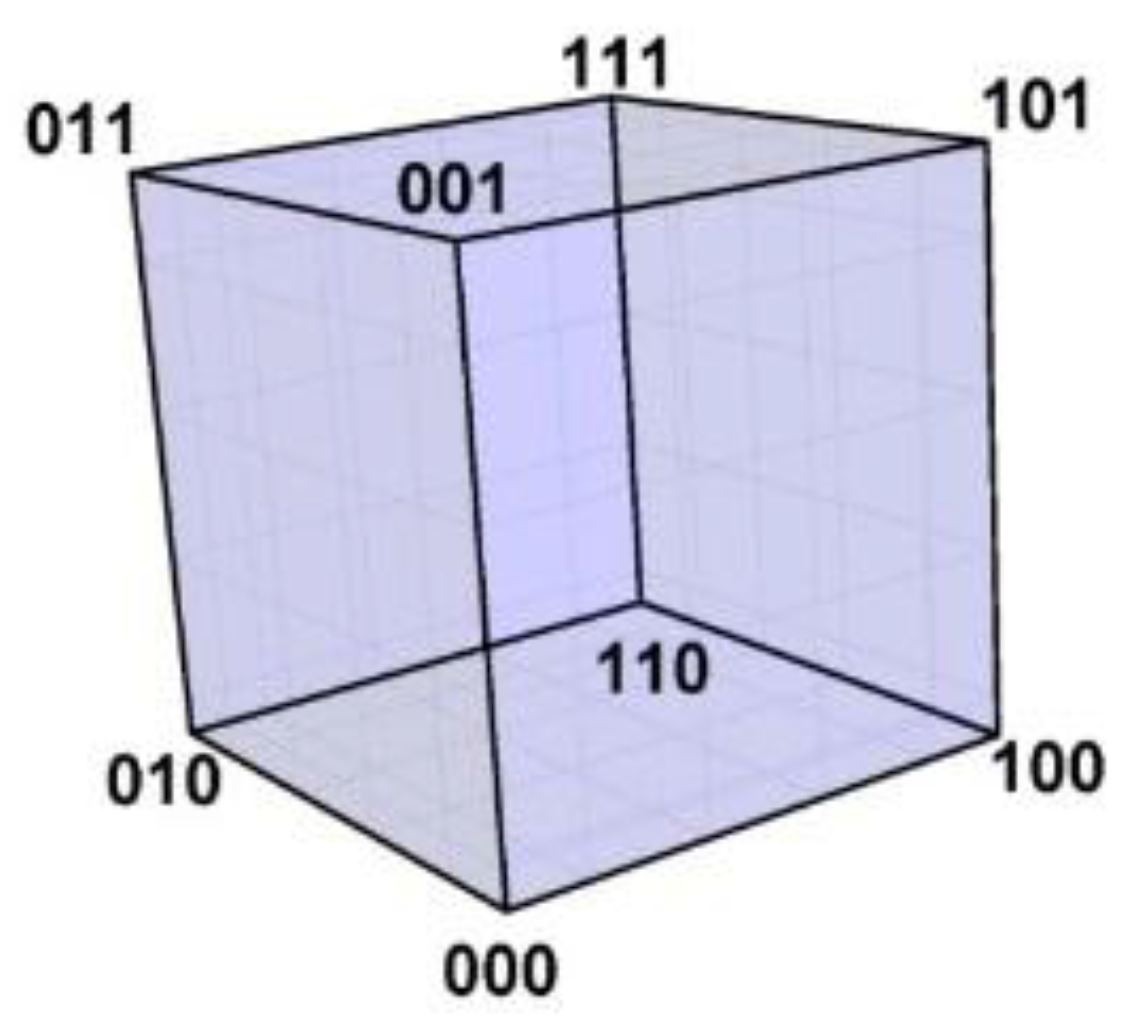
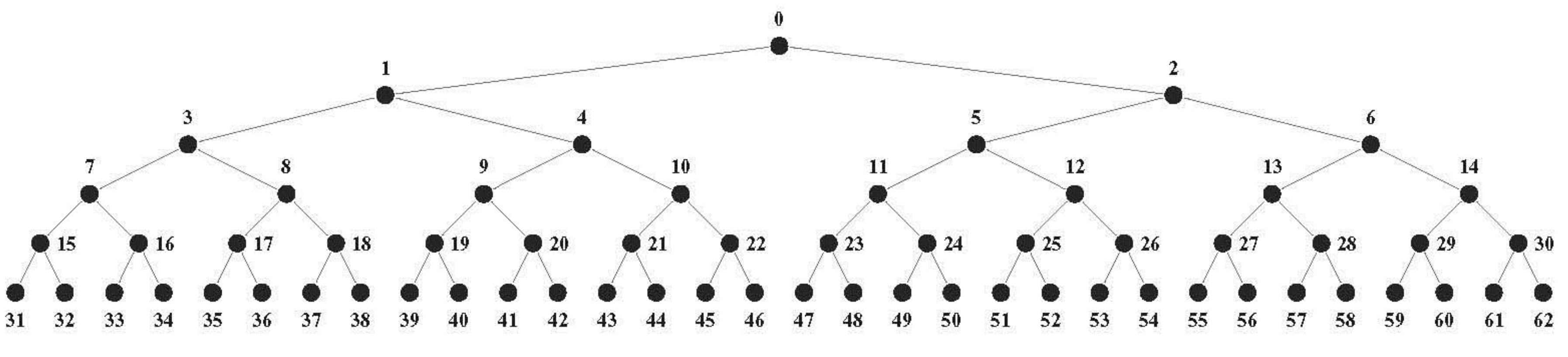
In 1950 Hamming published his now famous geometric cube model for binary codes: the code words are represented by vertices, and vertices representing code words differing at only one position are connected by edges [
10,
11]. The 3-bit binary code neatly illustrates this idea: the code word 000 is represented by a vertex with space coordinates (0,0,0) at the origin of a 3-dimensional Euclidian orthogonal space, 001 by a vertex with space coordinates (0,0,1), and so on for the remaining code words 010, 100, 011, 110, 101, 111; the eight vertices span a 3-cube with edges between vertices differing in just one coordinate, such as between (0,0,0) and (0,0,1). This binary Hamming cube is shown in
Figure 3. Vertices representing code words at Hamming 1-distance (1-HD), such as 000 and 001, are incident on the same edge (
at 1-edge distance) and at Euclidian 1-distance (1-ED), while vertices representing words at Hamming 2- and 3-distance are respectively at 2- and 3-
edge distances and at
Euclidian √2- and √3-distance.
Hamming distances are preserved in the cube model: they are mapped one-to-one onto Euclidian distances: 1-HD → 1-ED, 2-HD → √2-ED, and 3-HD → √3-ED. All
n-bit
binary codes can be mapped to
n-cubes this way; e.g., the 64 (=2
6) 6-bit binary words to a 6-cube with 192 (=64 × 6/2) edges connecting vertices representing words at 1-Hamming distance (each vertex is incident on six edges; each code word is at 1-HD of six other words.) Hamming distances and their geometric representation are basics tools of mathematical code analysis, with, among others, relevance to a code’s error detection and correction capacities [
9]. Hamming’s cube model has inspired similar models for the genetic code, but importantly the genetic code is
quaternary—it uses four symbols, {A, C, G, U}, and
not binary—{0,1}. This has implications for the geometric model of the code that hitherto have not been recognized to the best knowledge of this author.
Figure 1.
The standard codon table. The table orders the 64 codons into 16 blocks of four codons varying at the third position only—the family boxes. The nucleotides are ordered as in (U, C, A, G). The rows are in this order by the first, the columns by the second, and the blocks by third codon position. The 64 slots each contain a codon and the message encoded by this codon, an amino acid or stop signal. The stop codon slots are white; the amino acid slots are colored with the color code for the Polar Requirement of the amino acid shown in
Figure 2.
Figure 1.
The standard codon table. The table orders the 64 codons into 16 blocks of four codons varying at the third position only—the family boxes. The nucleotides are ordered as in (U, C, A, G). The rows are in this order by the first, the columns by the second, and the blocks by third codon position. The 64 slots each contain a codon and the message encoded by this codon, an amino acid or stop signal. The stop codon slots are white; the amino acid slots are colored with the color code for the Polar Requirement of the amino acid shown in
Figure 2.
Figure 2.
The Polar Requirements of 20 amino acids. Polar Requirement (PR) values for the 20 amino acids encoded by the canonical genetic code are listed by increasing value and color coded by a gradation of rainbow colors. Hydrophobic amino acids have PRs less than the PR of Ser and are colored with the purple to blue values, while hydrophilic amino acids have PRs greater than the PR of Ser and are colored with green to yellow and orange values.
Figure 2.
The Polar Requirements of 20 amino acids. Polar Requirement (PR) values for the 20 amino acids encoded by the canonical genetic code are listed by increasing value and color coded by a gradation of rainbow colors. Hydrophobic amino acids have PRs less than the PR of Ser and are colored with the purple to blue values, while hydrophilic amino acids have PRs greater than the PR of Ser and are colored with green to yellow and orange values.
Figure 3.
The Hamming 3-cube. The Hamming 3-cube is the geometric model for the 8-word, 3-length binary code, the Euclidian vertex coordinates correspond with the code words as indicated in the figure.
Figure 3.
The Hamming 3-cube. The Hamming 3-cube is the geometric model for the 8-word, 3-length binary code, the Euclidian vertex coordinates correspond with the code words as indicated in the figure.
Various mathematical models of the genetic code have been published. Many models ([
12,
13,
14,
15,
16] and references herein) use one of the 24 mappings of the four common nucleotides to 2-bit binary codes, such as (U, A, G, C) → (00, 10, 01, 11), so that codons are represented by 6-bit binary words. Importantly, these mappings do
not preserve the genetic code’s Hamming distances: 1-distances due to different nucleotides at the same codon position become either 1- or 2-distances in binary. To witness: the U-A, U-G, A-C and G-C nucleotide-to-nucleotide 1-distances map to binary 1-distances between, respectively, 00–10, 00–01, 10–11 and 01–11 (only one binary bit differs), while the U-C and A-G nucleotide 1-distances map to binary 2-distances between, respectively, 00–11 and 10–01 (both bits differ). The geometric model of this binary code is the Hamming 6-cube spanned by 64 vertices corresponding with the 64, 6-bit binary words representing the codons [
17,
18,
19]. In the 6-cube every 6-bit word has six nearest neighbors—not nine as in the genetic code, and the cube’s 192 length-1 edges correspond with the 192 binary Hamming 1-distances between the 64 6-bit words, but there are actually 288 Hamming 1-distances between the 64 codons, see
Section 2.3. Other mathematical models ignore the Hamming metric, but assume that the 64 codons form a mathematical group and analyze the code based on a quantum crystal basis [
20], or based on 64 dimensional irreducible representations of Lie groups [
21,
22] or finite groups [
23]. These group theoretical approaches are motivated by their successes in, among others, quantum and particle physics and by the thesis that breaking the code-group into smaller subgroups models the code’s evolution and explains its degeneracy patterns.
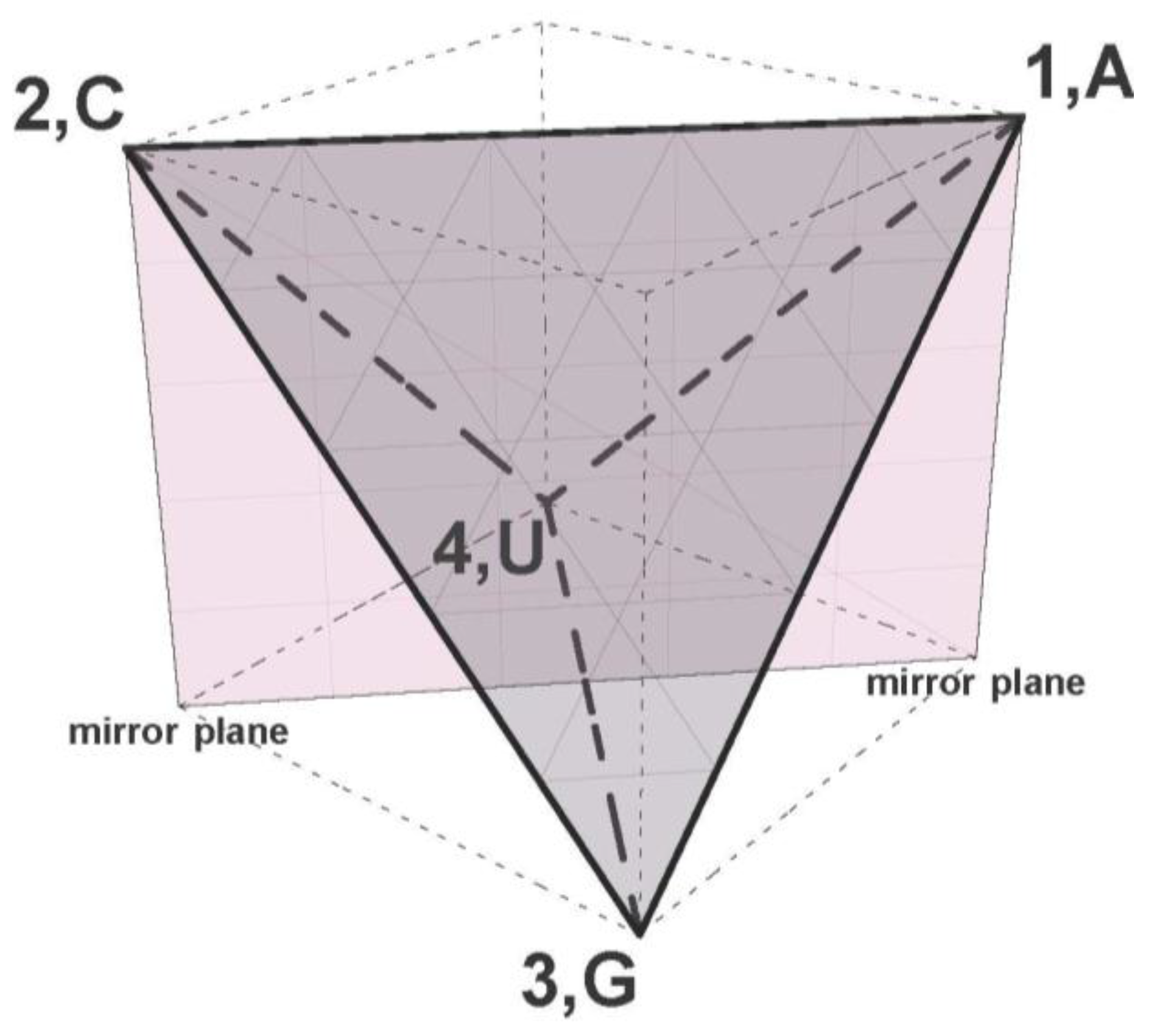
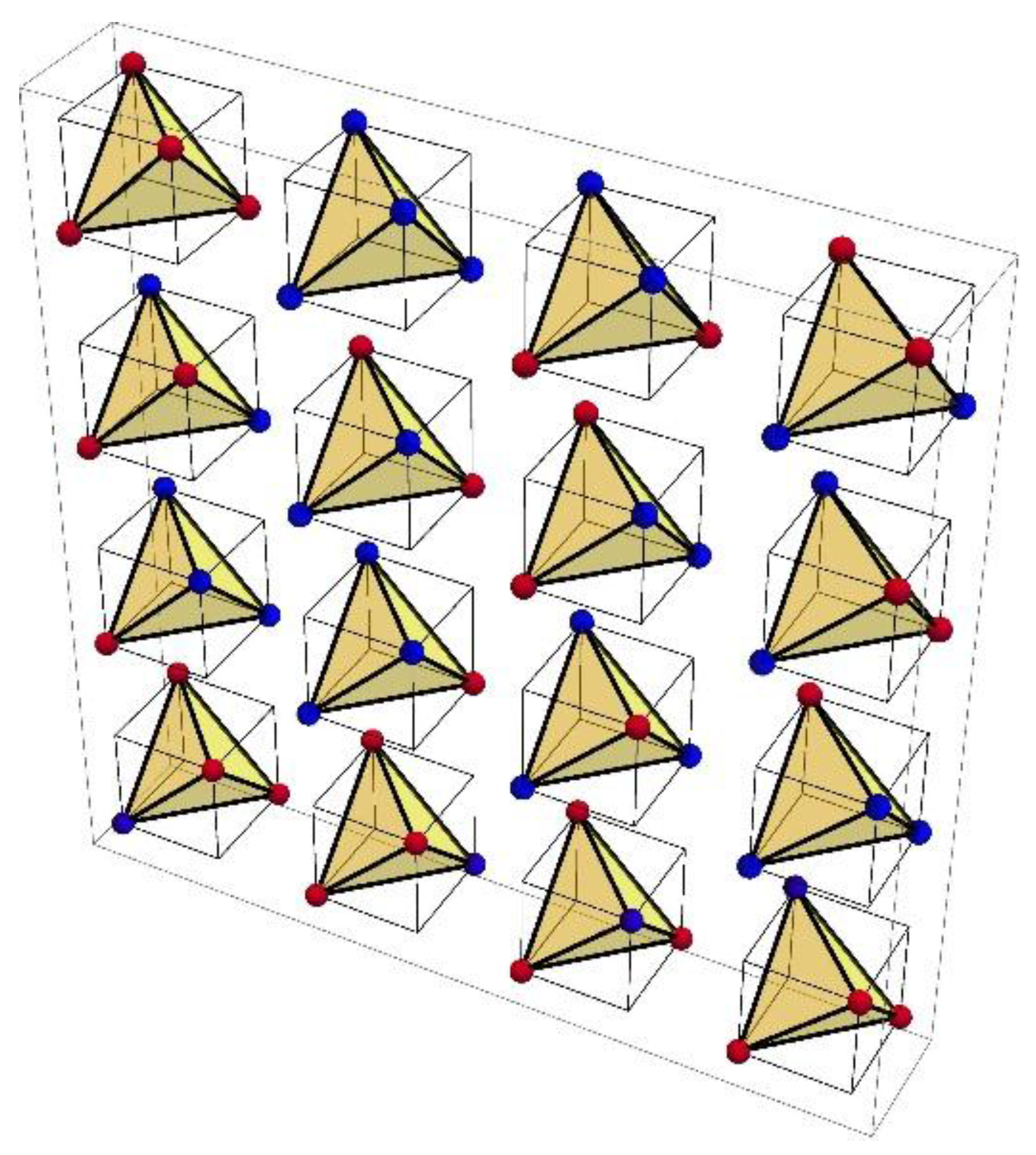
Our geometric model, a 9-polytope differs significantly from the 6-cube and the other mathematical models for the genetic code referenced above, but the polytope does uniquely correspond with the graph representation of the codon set with Hamming metric, the CodonGraph (
Section 2.3, [
24]). The symmetry group of the CodonPolytope is isomorphic to a product of small permutation groups acting on the codon set and to the symmetry group of the CodonGraph (
Section 4 and
Section 5). However the polytope symmetry group is very different from the 6-cube group and other mathematical groups mentioned above. Many
Euclidian symmetries of the polytope correspond with the similarity and degeneracy patterns of the genetic code, in other words,
these polytope symmetries identify code symmetries (
Section 6). The lower dimensional faces of the polytope display the strongest code symmetries and this hierarchy of face symmetries suggests that the early evolution of the code in pre-LUCA organisms can be modeled by splitting the polytope progressively into its lower dimensional faces. This model evolves the characteristic symmetry patterns of the code, patterns most unlikely generated by random processes (
Section 6). An accurate geometric model for the genetic code can form the basis for further mathematical analysis. To illustrate: we applied Polya’s colorings enumeration to count the number of code (=colorings) classes. The polytope symmetries partition the astronomically large number of all possible codes mapping 64 codons onto 21 messages into symmetry equivalence classes. This classification quantifies the uniqueness of the genetic code and its symmetries (
Section 7). These findings and applications of the CodonPolytope for the analysis of the genetic code are discussed (
Section 8).
8. Discussion
The codon table,
Figure 2, reveals that similar codons encode similar amino acids and most “obvious” similarities were noted early on [
5,
6]. The table was even called a
periodic table by Jungck [
37] because he found that the Polar Requirement of the amino acids correlates with the hydrophylicity of the 3’dimer of their anticodons: a “direct interaction” possibly explaining the pattern of hydrophobic amino acids on the left side of the table, mostly hydrophilic ones on the right side ([
37],
Figure 5). In
Section 6 we map these and other familiar patterns from the codon table onto the polytope, a 9-dimensional geometric object of which the 64 vertices represent the codons in Euclidian space (
Section 4.1). Relevant similarity patterns of the codon table map to distinctive faces of the polytope and the Euclidian symmetries of the polytope characterize these patterns as code symmetries (
Section 6). For example, the 16 family boxes of the table correspond with 16 tetrahedron faces so that for the eight boxes encoding one amino acid the 24 tetrahedron symmetries are exact code symmetries, while for the six boxes encoding two amino acids they are conservative code symmetries (
Section 6). Why go to the trouble of using a geometric object, a rather abstract mathematical model? Why not use the codon table to find and analyze similarity patterns? Indeed many investigators use various rearrangements of the table to find new code patterns that are not obvious from the original layout and then analyze these patterns using distances and symmetries of the table. This use of the codon table is problematic as distances and symmetries of tables are not defined mathematically. There is no “table distance” and codons differing by one point mutation are found not only in the codon’s own family box, but in all four rows and columns of the table. And codons differing by two mutations are found in all other boxes outside the family box. Ignoring the variation at the third base, as is often done, there are 576 equally valid ways to display the layout of the codon table by permuting the order of the bases at the first and second position (576 = 4! × 4!). Certain particular layouts are said to exhibit symmetries, such as a “degenerate mirror symmetry” [
38] to name just one example, but tables are not rectangles on the Euclidian plane although our “intuitive eye” sees them as such. (This single “degenerate mirror symmetry” is generated in the polytope by two orthogonal Euclidian mirrors, G ↔ C and A ↔ U, and corresponds to a Euclidian rotation.) The “rectangle” view of the table is compatible with a continuous variation of parameters (such as Polar Requirement) “over” the table that can be displayed as a “continuous smooth surface” in 3-space “with chemical property as altitude” [
39]; however neither the table, nor the finite set of parameter values are continuous or smooth so this analysis is problematic, but nicely illustrates the issue at hand.
The Hamming metric of mathematical coding theory is a natural metric for the codon space as one Hamming distance corresponds with one point mutation, the commonly used
unit distance between nucleotide sequences. Other metrics define different spaces; for example, Dragovich and Dragovich [
40] use a p-adic, ultrametric norm; the four nucleotides are at one 5-adic and four codons differing only in the 3rd position are at 1/25 5-adic distance from each other. shCherbak [
41] assigns codons integer nucleon numbers of their encoded amino acids and develops a digital arithmetic code. Jungck [
42] reviews many of the “genetic code as mathematical code” models; the discussion below focuses on those most related to our approach.
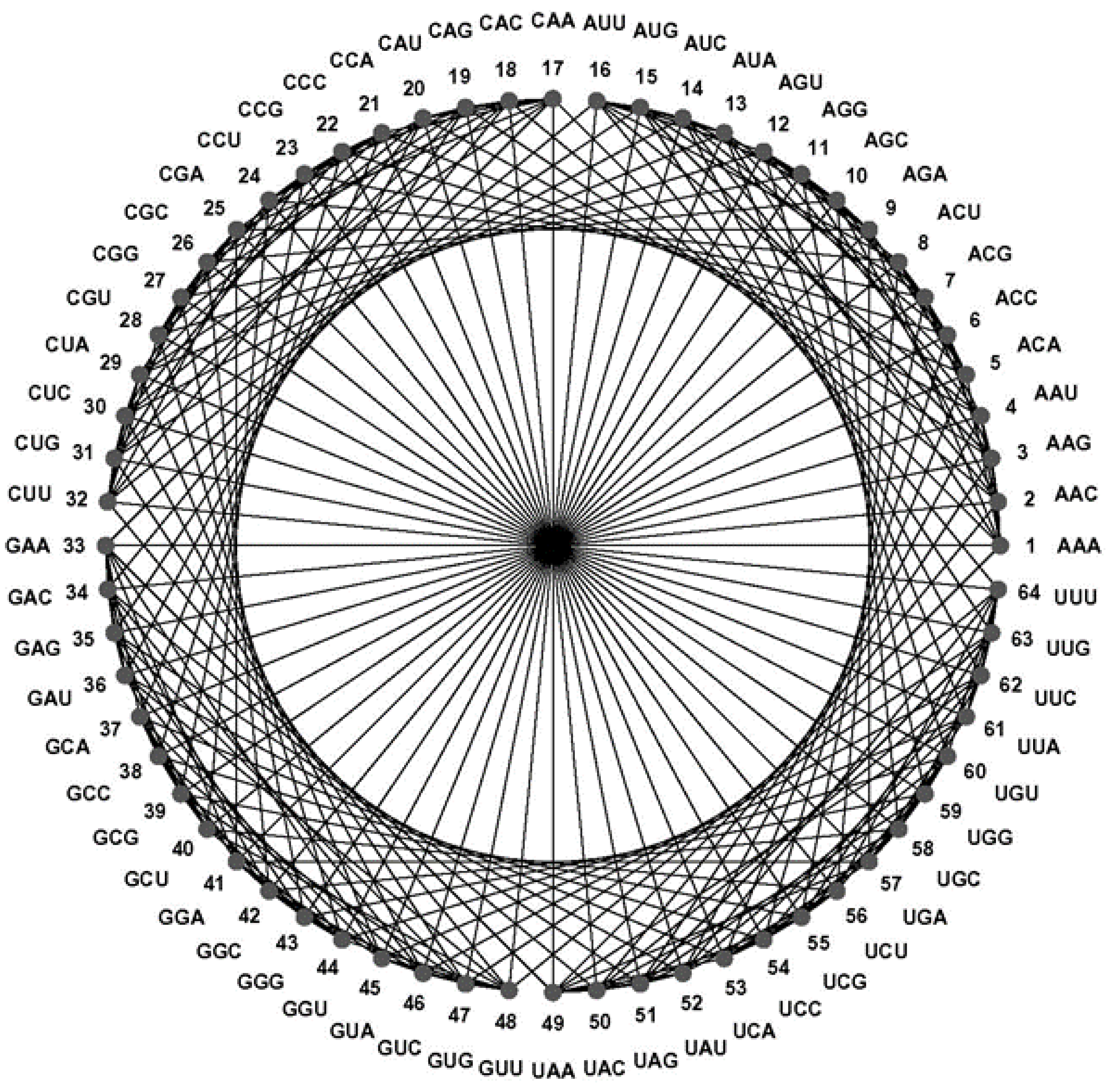
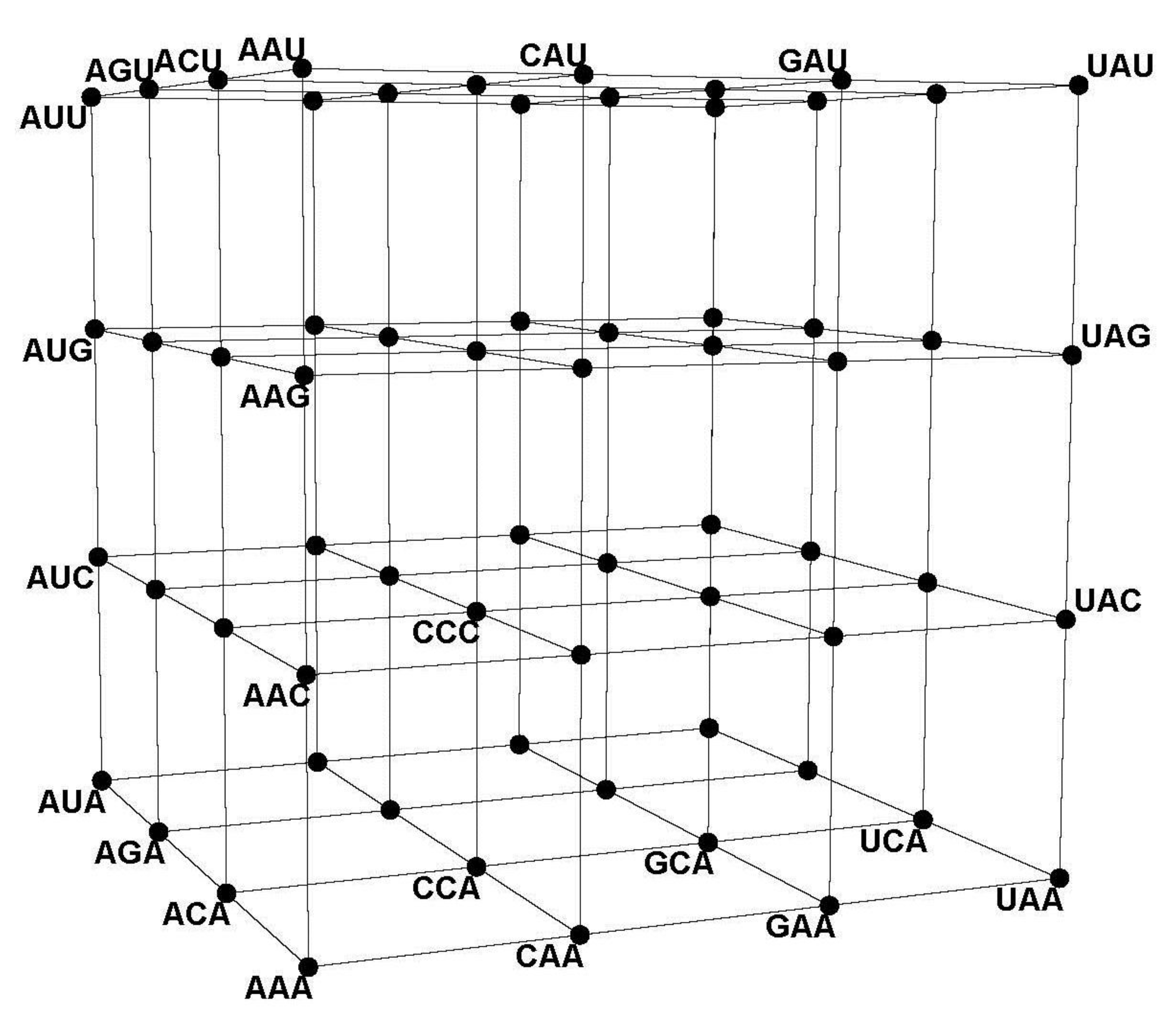
The CodonDistanceMatrix (
Figure 4) and the CodonGraph (
Figure 5 and
Figure 10) display the intercodon Hamming distances of the (
normed,
Hamming metric) codon space, and their symmetries are described by permutation groups (
Section 5), but they lack the mirror reflection and axial rotation symmetries of Euclidian space. (In Euclidian space an inner product defines the required angles and distances for these symmetries). In a non-Euclidian, graph topological approach, Tlusty [
43] embeds a codon graph with 48 vertices (synonymous codons ending in C or U are represented by the same vertex) and 192 edges on a 2-dimensional surface of genus 25 (25 holes are needed to avoid edges from crossing) and chromatic number 20 (20 colors are required to color all neighboring quadrilaterals differently); this coloring number (20) is the upper limit for encoded amino acids by this configuration and equals the number of amino acids of the genetic code. A similar embedding of a 64 vertex, 288 edges graph, defined like the CodonGraph, has genus 41 and chromatic number 25, a topology encoding up to 25 amino acids. (The CodonGraph, shown in circular and array embedding in
Figure 5 and
Figure 10, can encode up to 64 messages). Chechetkin [
44] uses a De Bruijn graph with 16 vertices representing the 16, 2-mers of the four nucleotides, and 64 directed edges representing the codons overlapping with both vertices (vertex AA is joined by edge AAC to vertex AC) to study the translational stability of the code. Tlusty [
43] noticed that the 64 vertex, 288 edge CodonGraph is “natural” to be used for this purpose as is discussed further below.
The CodonPolytope (
Section 4), a 9-dimensional geometric object in Euclidian space preserves the Hamming metric of the codon space: the intercodon Hamming 1-, 2- and 3-distances correspond one-to-one to the three particular Euclidian distances between the 64 vertices of the polytope representing the codons; in one realization of the polytope these distances are 2√2, 4, and √6, respectively (
Section 4.1 and
Section 4.2). The Euclidian reflection symmetry group of the polytope, the permutation symmetry group of the graph, and the largest permutation group that preserves the Hamming metric of the codon space, are all three isomorphic to S
3 x
wreath (S
4)
1 × (S
4)
2 × (S
4)
3 (Notation: the index-
i for
i = 1, 2, 3, in (S
4)
i corresponds with the codon position acted on by the S
4 group); S
4 permutes {A,C,GU}, S
3 the three codon positions (
Section 4.3 and
Section 5).
This group contains all 82,944 symmetries of the 64 codon set that preserve the intercodon Hamming distances (the distance between any two codons before and after the permutation is the same). The group induces permutations of the codon set and rearrangements of the codon table; for example, the two S
4 acting at the first two codon positions induce all 576 codon table layouts mentioned above.
To our knowledge, apart from the CodonPolytope, no published geometric model of the code preserves the Hamming metric. The often used 6-cube, 6-bit codon model ([
12,
13,
14,
15,
16,
17] and many references herein) does not preserve intercodon Hamming distances as discussed in the introduction. Moreover the symmetry group of the 6-cube of order 46,080 is smaller than the polytope group, but is
not a subgroup of the polytope group and therefore contains 6-cube symmetries that do not preserve intercodon Hamming distances: only the 384 symmetries of the 6-cube subgroup S
3 x
wreath (S
2 × S
2)
1 × (S
2 × S
2)
2 × (S
2 × S
2)
3 preserve these distances (
Section 4.3); the (S
2 × S
2) groups are isomorphic to the Klein-4 group, see further below. In general hypercube models of nucleotide sequences such as those used by Eigen ([
45], pp. 354–387) do not preserve the intercodon Hamming distances (as they do not contain triangles or tetrahedrons,
Section 2.5). Nonetheless the 6-cubes are frequently used for analysis of code patterns: Jiménez-Montaño
et al. [
17] hold that the cube structure explains amino acid substitution patterns, and according to Karasev and Stefanov [
18] the 6-cube’s topology encodes a 4-amino acid-arc helical protein topology—the “topological nature of the code”. Several investigators [
12,
15,
17,
46] derive genetic Gray codes based on the cube’s Hamming distances. The 3D “Genetic Hotels” [
16,
19,
47,
48] are projections of the 6-cube onto a “3-cube” in R
3 space, a 3D-version of the codon table with {C,U,A,G} mapped to {0,1,2,3} and the 1st, 2nd and 3rd codon positions plotted on the
x,
y and
z axes respectively, so CCC corresponds with (0,0,0) and GGG with (3,3,3). The hotel “cube” resembles the CodonArray graph,
Figure 10, but the hotel has only three edges per row or column, while the graph has six edges per row and is not a geometric object. In the hotel, the intercodon Hamming 1-distances vary from one to two to three cube edges; the hotel thus distorts this metric even more than the 6-cube. Moreover, the hotel 3-cube is
not Euclidian, as the 0, 1, 2, and 3 coordinates are projections of the values 0, 1, α, and 1 + α of the Galois 4-Field: the distance between 1 and α equals 1 + α (
not 1 as the cube suggests), and between 1 and 1 + α actually equals α (
not 2). The hotel cube can be manipulated with Galois 4-Field algebra (four additions, three multiplications), but does not possess Euclidian symmetries. Different projections onto the hotel result in different 3D-code geometries; for example (C,U,A,G) produces hotel-cube-edges for amino acids encoded by just two codons [
16], but (G,U,A,C) does not [
48]. Other three-dimensional geometries such as a simple tetrahedral construct with 20 lattice points representing 64 codons [
49] also do not preserve the Hamming metric. In fact any geometry preserving this metric in Euclidian space has to be isomorphic to the CodonPolytope (by a mathematical theorem,
Section 4.1), a
simple 9-polytope, and therefore no such object could exist in a Euclidian space of fewer than nine dimensions. This polytope thus provides a unique geometry for the identification of code symmetries by well-defined Euclidian symmetry transformations such as mirror reflections and rotations. The polytope has 82,944 symmetries, so one is bound to find a few interesting non-obvious code patterns and intriguing symmetry subgroups, but their biological significance might not be obvious either.
Jestin and Soulé [
50] identify three “base substitution symmetries” of the code; in our vocabulary these symmetries correspond with mirror symmetries of the codon polytope. As reviewed in [
14] the “Rumer transformations”, first reported by Rumer in Russian 1968, have been extensively analyzed, but their biological significance has not been identified. Of the 16 family boxes, eight each encode a single amino acid (the M1 set of boxes), and the other eight (the M2 set) encode two or three messages. The single “degenerate mirror symmetry” of the UCGA × UCGA layout of the codon table mentioned above [
38] or two “base substitution symmetries” G ↔ C and A ↔ U at the first codon position [
51] map the two sets of boxes onto themselves. The “Rumer transformations” exchange A ↔ C and G ↔ U at all codon positions and thereby exchange the 8-sets, M1 ↔ M2, as reviewed in [
14,
50]. Danckwerts and Neubert [
52] define three operators α, β, γ acting on nucleotide characters; α: Purine ↔ Pyrimidine, β: Weak ↔ Strong, or 2 H-bonds ↔ 3 H-bonds, and γ: Amino ↔ Keto; or α: (A ↔ C, G ↔ U), β: (A ↔ U, G ↔ C) and γ: (A ↔ G, C ↔ U). These plus an identity operator make up an operator group isomorphic to the Klein Four group that permutes the four nucleotides. The Rumer transformation corresponds with α acting on all three codon positions [
52], or with γ acting on the first, and α on the second and third codon positions as was found later by Jestin and Soulé [
50]. Jiménez-Montaño [
14] showed that a CGUA × CGUA table displays a “yin yang” pattern for the two 8-box sets M1 and M2, various “quadrant” patterns for the two nucleotide characteristics (R/Y and S/W), and Gray codes based on the 2-bit nucleotide representations. In a group theoretic approach Findley
et al. [
53] identify the four nucleotides with the four group elements of the Klein-4 or the Z4 group (cyclic group of order 4) and generate the 64 codons as product group K × K × K = K (three elements of K multiplied produce an element of K). Thus each codon identifies with an element of K and thereby with one of the nucleotides: the codons are partitioned in four blocks of 16 codons. The Klein-4 group induces a unique codon partition with degeneracies equivalent to those of the genetic code; the Z4 group induces six different partitions with different degeneracy patterns.
Historically various geometric models were used to analyze these patterns and symmetries. Danckwerts and Neubert [
52] illustrate the Klein-4 group (=K) with a square having the four nucleotides as vertices and the group elements (α, β and γ) as edges and diagonals, while Jiménez-Montaño [
14] uses a rectangle in an identical way; Bertman and Jungck [
54] showed a tesseract or 4-cube representation of K × K having the 16 dinucleotides as vertices, and two group elements (α and β) as edges so that all eight vertices of the set M1 (dimers of 4× degenerate codons) lie in three connected planes (2D-square faces, in our parlance). The Klein-4 group is isomorphic to the bitwise addition group for {00, 01, 10, 11} [
14,
16]—the very definition of a Hamming square, as well as to the rectangle symmetry group, and therefore investigators often use the rectangle and square models simultaneously. Remarkably the isomorphism with the Euclidian rectangle mirror symmetry group (identity, two mirrors bisecting the opposite sides, and one 180 degree rotation) was never used explicitly. The Klein-4 group also is isomorphic to the rotation symmetry subgroup of order 4 (identity, 180 degree rotations on three orthogonal axes) of A3—the tetrahedron reflection symmetry group; A3 is isomorphic to S
4, and Klein-4 is isomorphic to S
2 × S
2, a small subgroup of S
4. These isomorphisms show that the symmetries and similarity patterns discussed above can all be expressed as Euclidian symmetries of the tetrahedron and CodonPolytope. The other geometries have drawbacks because the 24 ways to label the four vertices of a square or rectangle with {A,C,G,U} are
not equivalent. The non-equivalent ways of labeling correspond with
different neighborhoods for each label. Depending on the labeling of the vertices, different nucleotides are non-adjacent (diagonally opposite vertices are separated by
two edges,
not one), or if on a rectangle, adjacent via long or short edges. Therefore the edge or Euclidian distance between the vertices in these models
cannot coincide with the “one point mutation” distance between the four nucleotides, and this is also why n-cube models
cannot preserve the intercodon Hamming distances. In contrast, all ways of labeling the regular tetrahedron are equivalent: all four vertices are at 1-edge and at identical Euclidian distance in correspondence with the Hamming 1-distance between the nucleotides. Moreover the tetrahedron A3 reflection symmetry group contains subgroups isomorphic to the rectangle and square symmetry groups, and the polytope as product of three tetrahedrons contains all products of these symmetries, such as K × K × K and K × K mentioned above, in its direct product symmetry subgroup A3 × A3 × A3, isomorphic to (S
4)
1 × (S
4)
2 × (S
4)
3.
The CodonPolytope and its faces display many code symmetries (
Section 6): exact symmetries permuting synonymous codons, conservative symmetries permuting codons encoding similar amino acids, anti-symmetries that exchange codons encoding amino acids with opposite characteristics (such as hydrophobic and hydrophilic) and near (not perfect) symmetries of all three kinds. The larger polytope faces display weaker, less perfect code symmetries than the smaller faces, which display stronger, more perfect and exact symmetries. This hierarchy of faces and symmetries suggests that the stepwise splitting of the polytope into its smaller faces models an early evolution of the code that generates the similarity patterns of the extant codes (
Section 6.5). This model is fully compatible with a more abstract symmetry breaking model [
24]. Both models partition the 64-codon set along a binary tree in five steps to 32, 2-codon blocks; at each step the codon blocks correspond with the polytope faces generated by the splitting process. Increasing codon-anticodon base pairing stringencies from none to wobble to Watson-Crick, initially at the middle codon position, subsequently at the first, and lastly at the third position (but only to wobble at the third position) as in the sequential “2-1-3” model [
55], partitions the codons in exactly this manner. At each stage each codon block (polytope face) encodes a class of similar amino acids; the members of this class are not differentiated from each other by the cellular machinery so that the face symmetries correspond with exact code symmetries. Only during the final fifth step is the class size reduced to one specific amino acid for all codon blocks. This process generates the extant mitochondrial codes and nearly the canonical code. Early codes are ambiguous as each codon block encodes similar amino acids and these similarity patterns are (near) conservative symmetries in the extant codes. For example, almost all codons represented by a polytope 6A-face (corresponding with the left column of the codon table) encode aliphatic hydrophobic amino acids, a remnant of an early code that neither distinguishes between the 16 NUN codons of the left column, nor between these very similar amino acids. The polytope model makes no particular assumptions about the presence or absence of particular amino acids during its evolution, but most likely amino acid repertoires increased over time [
56]. If at the four 6A-faces stage only Val is present, and the other aliphatic amino acids, Ile and Leu have not yet been generated, then all codons of the 6A-face block encode Val and only at the next, eight 4A-face stage are the codons of one of the 4A-blocks reassigned to Leu, while the codons of the other block encode Val and Ile. Thus, the polytope model is compatible with Higgs’s “four column model” [
57] and Wong’s and Di Giulio’s coevolutionary theory [
58,
59] that assign amino acids sequentially to specific codon blocks. Such assignments also depend on the evolving specificities of primitive RNA-based aminoacyl-tRNA synthetases (aaRSs) for blocks of tRNAs and classes of amino acids [
60]. For example, the early aaRS for Val evolves to three aaRSs differentiating between the three aliphatic amino acids and the seven tRNAs complementary to different codon blocks of the extant code [
24].
Delarue [
61] found an “asymmetric pattern” in the AGCU × CUGA layout of the codon table that depends on whether the encoded amino acid is recognized by a Class I aaRS or a Class II aaRS. Rodin and Rodin [
62] rearrange the table differently with “complementary” codons “face to face” to show a “latent mirror symmetry,” while Jestin and Soulé [
50] describe this pattern by six “base substitution symmetries”. The pattern maps to distinct polytope faces and “recapitulates” the progression of splits of our evolution model: the two NUN and NCN 6A faces (table columns) are Class I and Class II respectively, in correspondence with a NUN ↔ NCN = Class I ↔ Class II anti-symmetry mirror in our nomenclature. The two NRN columns are split into upper and lower 4A faces, in correspondence with a NYN ↔ NRN = Class I ↔ Class II anti-symmetry mirror of the 6A faces,
etc., which leads to a binary partition of the codon set [
61] and decision tree [
14] much like our polytope splitting model (
Section 6.5).
The clustering of same and similar amino acids on the faces of the polytope (with vertices representing encoded amino acids) and the related exact and conservative code symmetries (
Section 6, and discussed above) confers on the code a degree of robustness to mutations and reading errors because single errors correspond with polytope symmetries (mirror reflections of the affected codon). As the genetic code displays many near-symmetries this robustness seems suboptimal, and indeed codes less prone to errors have been found, as reviewed by Santos and Monteaguado [
63]. The error-sensitivity of the code defined as the average variation in Polar Requirement of the amino acids encoded caused by all single point mutations affecting all codons [
63], can be computed as the average edge weight of the CodonPolytope/Graph with edge weights reflecting the difference in Polar Requirement between the neighboring amino acid vertices on polytope (or graph); Tlusty [
43] considered the graph a natural way to study the impact of mutations, and Buhrman
et al. [
64] explicitly constructed the graph for their computations. Polytope/graph faces with exact code symmetries contribute nil to the total edge weight; those with conservative symmetries less than those with near conservative or anti-symmetries. On the one hand symmetry breaking [
21,
22,
23,
24,
65], code trees [
14,
61] and polytope splitting generate symmetric codes with error robustness, but on the other hand selection for codes with low error loads during the pre-LUCA evolution of early codes, the error minimization hypothesis [
66], should generate codes with many symmetries.
Several group theoretical models [
21,
22,
23], reviewed in [
65], evolve the code from an initial group with a 64-dimensional irreducible representation (irrep) under which all 64 codons are equivalent, via a series of symmetry breaking steps, into subgroups with smaller dimensional irreps, corresponding exactly with the codon degeneracies (the block partition 1
22
93
24
56
3 of the codon set,
Section 7). Antoneli
et al. [
21] claim to have screened all algebraic models within this approach, whether based on Lie groups/Lie algebras, on Lie superalgebras or on finite groups ([
21] reviews all earlier work in this area). They identified several Symplectic groups, but foremost Sp(6) and its supersymmetric version as “unique solution”. The Sp(6) model evolves the canonical code from the initially non-coding 64 codons in at most four steps (
Figure 5 in [
21]). The first step generates six subgroups comprised of 20, 16, 10, 4 and two codons, encoding respectively Gly, Ala, Ser, Val, Asp, and His; in the second step the 20 Gly codons split into eight Gly, six Cys, and six Ile codons (and all other groups other than the two His codons split up as well); and in a third step the six Ile codons split in three Ile and three Stop codons, and most other codon blocks split as well. This splitting scheme generates the codon partition of the canonical code per force—some subgroups are artificially “frozen” (
partial symmetry breaking) as otherwise blocks of synonymous codons are split into smaller sets. The similarity patterns of the early codes do not seem to match those of the codon table at all (for example, the three Ile and three Stop codons do not form one block of six related codons), and the fixation of the two His codons in the earliest code seems particular. Only three finite Symplectic groups [
23] of orders 103,608; 2,903,040; and 4,245,696 evolve the code under partial symmetry breaking; these groups do not break in a predetermined order and, thus, generate a variety of early codes. Bashford
et al. [
22,
65] find that the Lie superalgebra A(5,0) ≈
sl(6.1) can evolve the
anticodon genetic code (anticodon as opposed to amino acid assignments to codon blocks) through partial symmetry breaking in five to six steps in three different ways, one of which resembles the binary tree models mentioned above ([
14,
24,
61] and the polytope model). In a different approach, Sciarino
et al. [
20,
67] assign the four nucleotides to the irreps of the quantum group
Uq(
su(2)⊕
su(2)) in the limit
q → 0 (the crystal basis), obtain the codons as tensor products of the nucleotides, and their amino acid assignments as eigenvalues of a codon reading operator; different genetic codes correspond with different operators. A more recent application of this Crystal Basis Model [
68] determines codon-anticodon pairing based on the “minimum principle” of the mitochondrial code: anticodon wobble base U is identified for family boxes of four synonymous codons, and G and U for boxes with two pairs of synonymous codons. This quantum group does not evolve codes, but computes similar wobble bases for early codes such as the “Archetypal” code (15 family boxes encoding a single amino acid, one box encoding Tyr and Stop, each by two codons) [
69], which closely resembles the 16 tetrahedron stage of the polytope splitting model. These group theoretical models are mathematically ambitious, but none evolves
ab inito with
complete symmetry breaking the genetic code from a primordial non-differentiated codon set (no such group was found). The riddle as to how the code itself evolved in pre-LUCA organisms is thus alive and well.
The canonical genetic code is unique, but how unique? Taking the 64 triplet codons as a given, 64! ≈1.3 × 10
89 different codes are possible; these codes assign up to 64 messages to the 64 codons. Assuming the 21 messages of the canonical code, the literature sometimes uses 21
64 ≈ 4.2 × 10
84 as “first approximation,” but there are 1.5 × 10
84 (≈36% of 21
64) codes that convey exactly 21 messages, or [64] → [21] surjections [
24]. If one further assumes the number of codons assigned to each message by the canonical code as fixed,
i.e., one codon to Met, six codons to Ser, etc, then the number of codes is given by the multinomial formula M(64, 1
22
93
24
56
3) ≈ 2.3 × 10
69 (
Appendix A. An often quoted formula from ([
70], p. 96) is incorrect.) None of these calculations takes the 82,944 symmetries of the polytope into account. Polya’s formula enumerates colorings classes of geometric objects—in each class all colorings are equivalent (essentially the same) under the symmetry group of the object [
36]. We adapted Polya’s enumeration to count only colorings using exactly 21 colors (and not fewer), the colorings then correspond with codes. When applied to the CodonPolytope with 20 amino acids and one stop signal as 21 colors the canonical code represents a class of 41,472 equivalent codes, and the mitochondrial code a class of 20,736 codes (
Section 7.2). The ≈1.5 × 10
84, [64] → [21] surjections are partitioned into ≈1.8 × 10
79 code classes with an average class size of ≈82,944 (within rounding), the maximum class size as virtually all codes, unlike the canonical and mitochondrial codes, lack exact symmetries. (
Section 7.3). The ≈2.3 × 10
69 codes that assign the same number of codons to same message as the canonical code are partitioned into ≈ 2.8 × 10
64 classes of size ≈82,944 (
Section 7.4); These spaces of code classes are too vast to find even a single representative of any of the classes of the few extant codes among billions of randomly generated codes; in other words, there is no chance of generating codes like the genetic code by chance.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


