Phosphorus Inactivation in Lake Sediments Using Calcite Materials and Controlled Resuspension—Mechanism and Efficiency

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental Design
2.2.1. Batch Experiments
2.2.2. Incubation Experiment
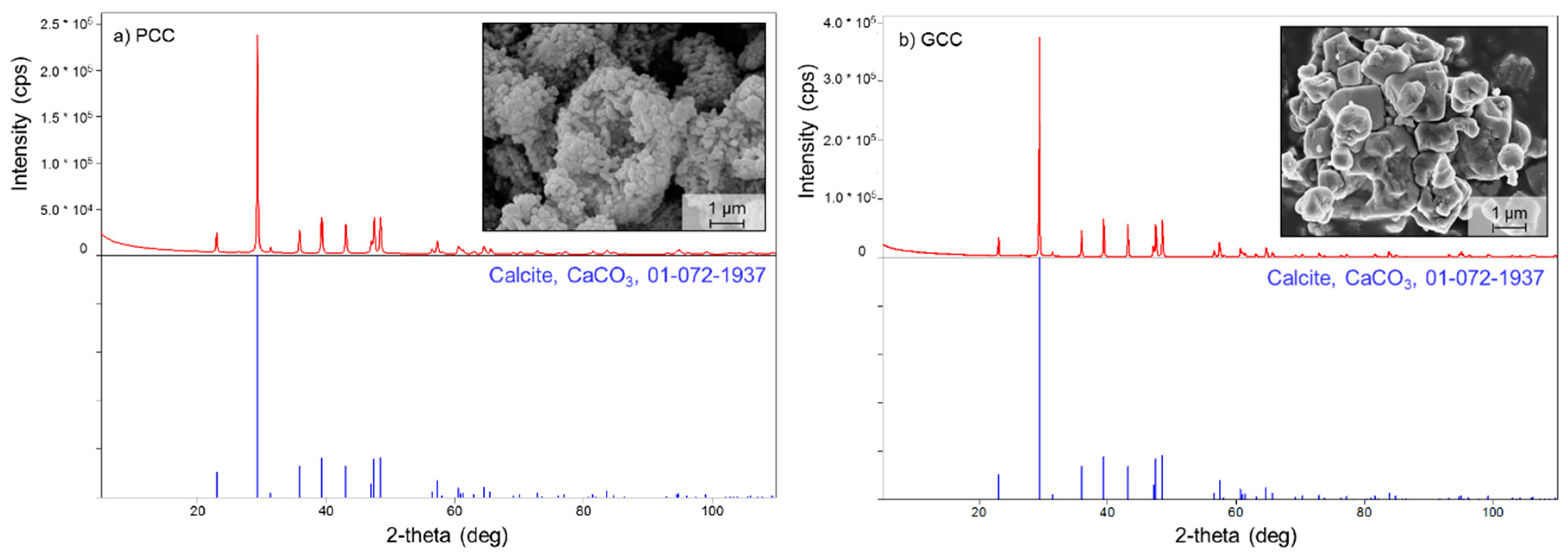
2.3. Characterization of Calcite Materials
2.4. Chemical Analysis
2.5. Data Analysis
3. Results
3.1. Batch Experiments
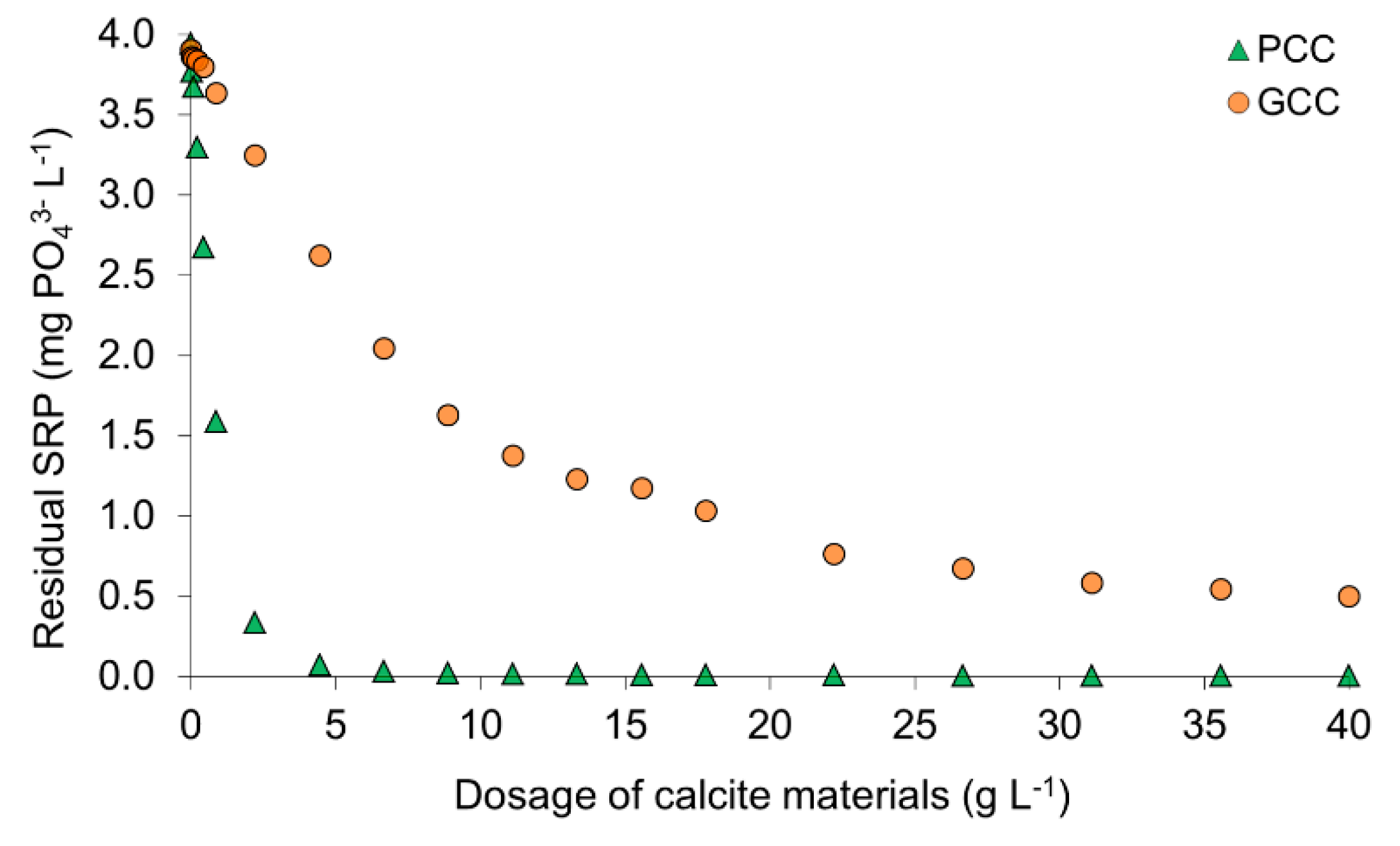
3.1.1. Sorption Capacity
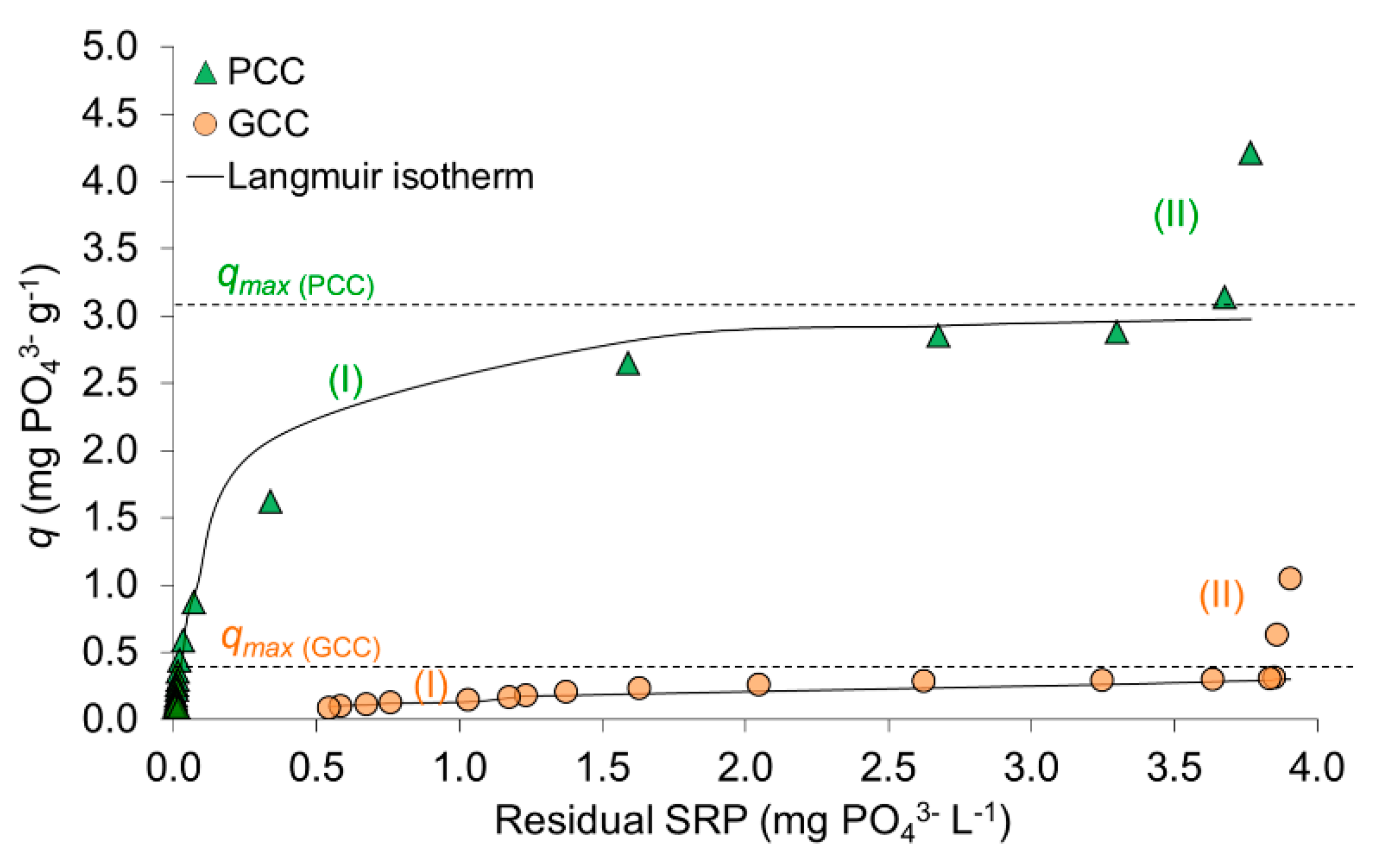
3.1.2. Sorption Isotherms
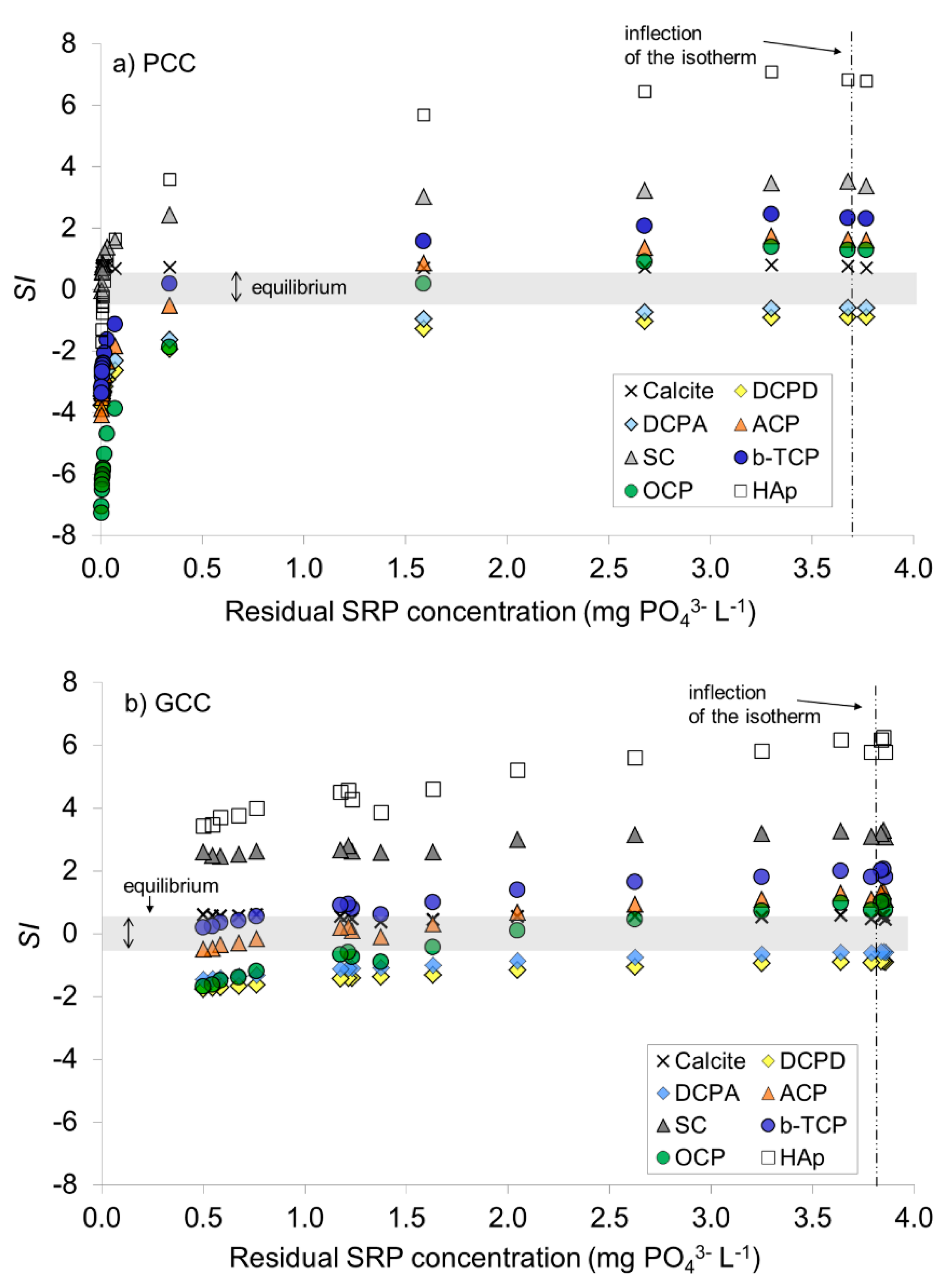
3.1.3. Saturation versus Ca-PO4 Compounds
3.2. Incubation Experiments
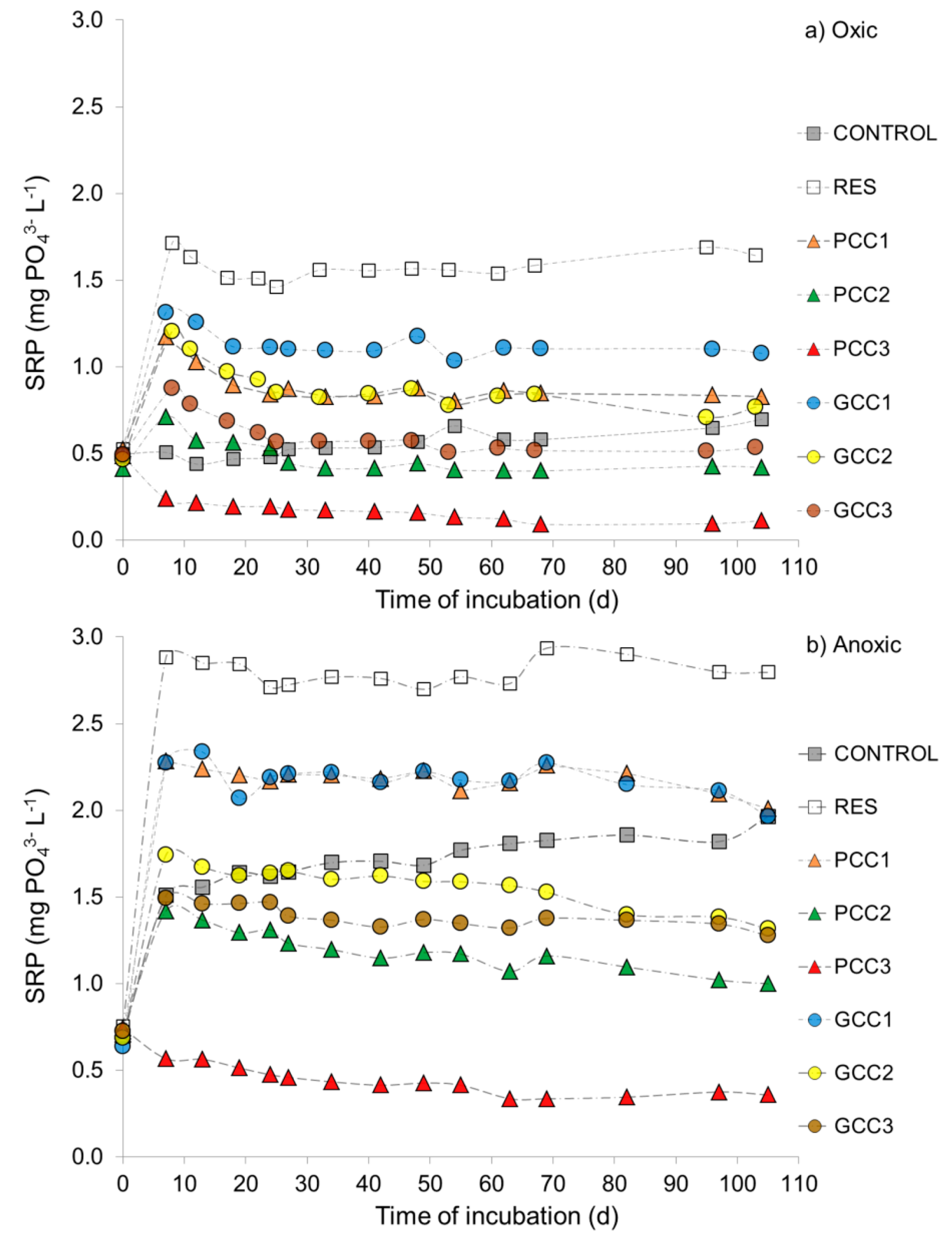
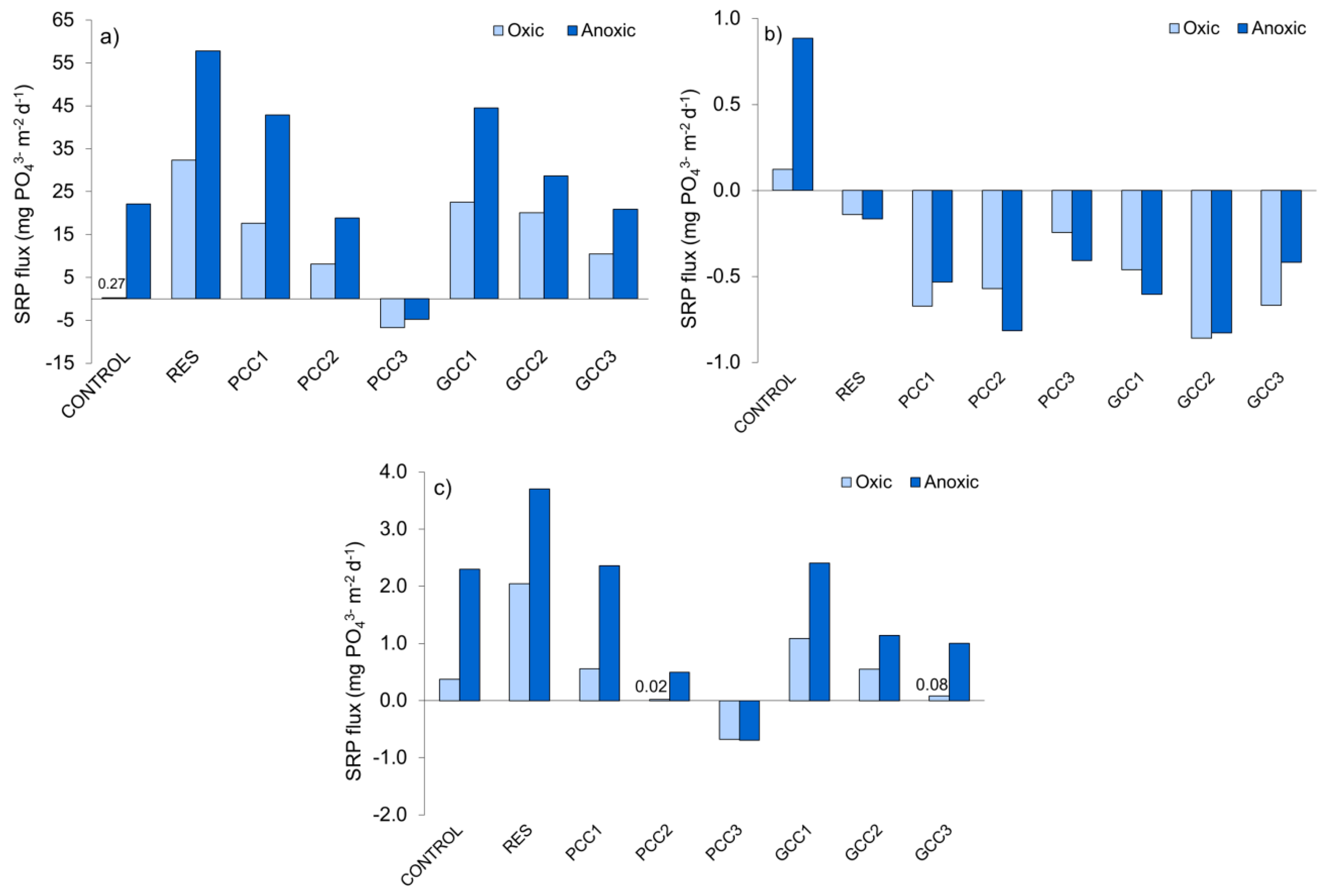
3.2.1. SRP in the Overlying Water and SRP Fluxes at the Sediment-Water Interface
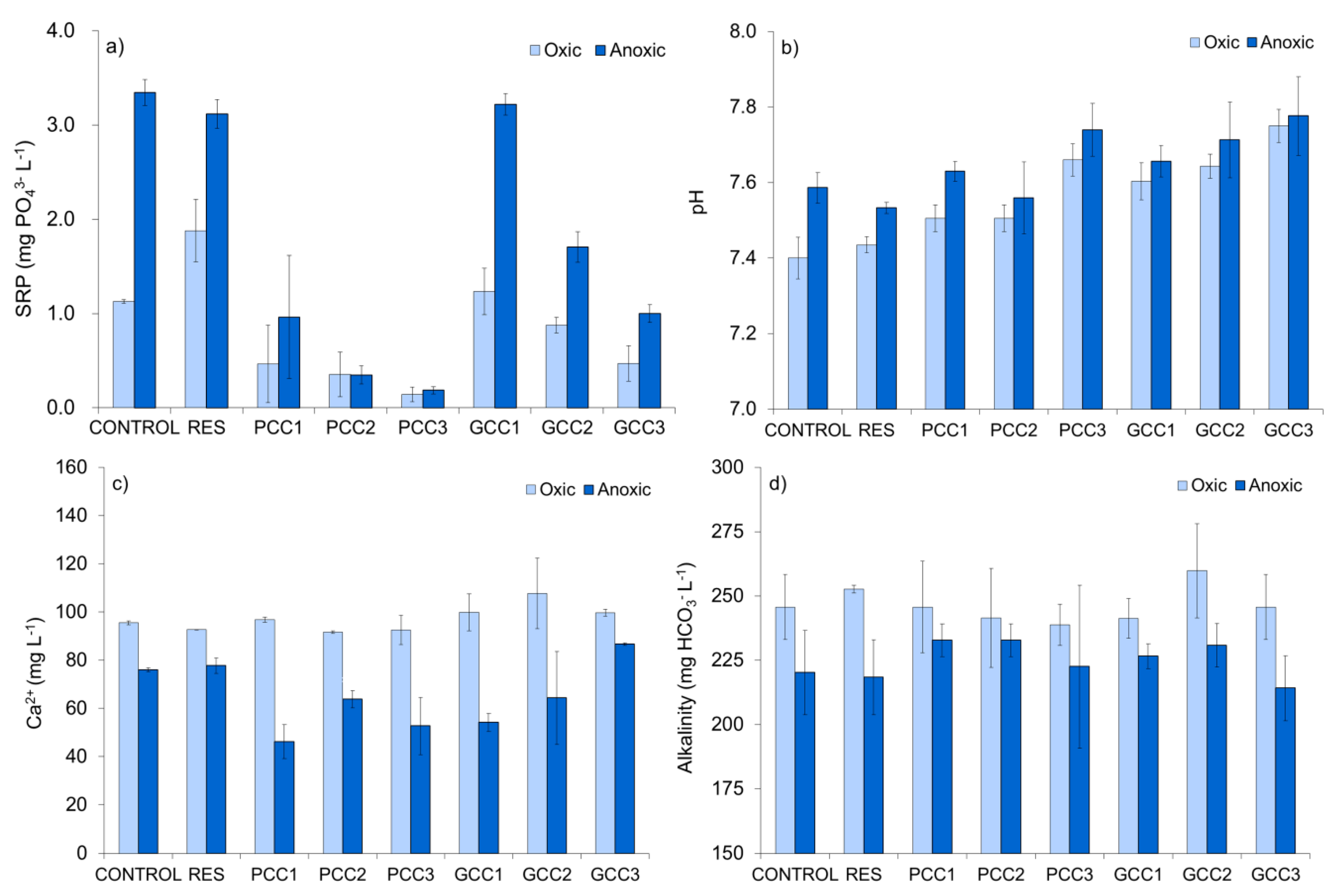
3.2.2. Physiochemical Conditions in the Interstitial Water
3.2.3. Saturation versus Calcite and Ca-PO4 Compounds
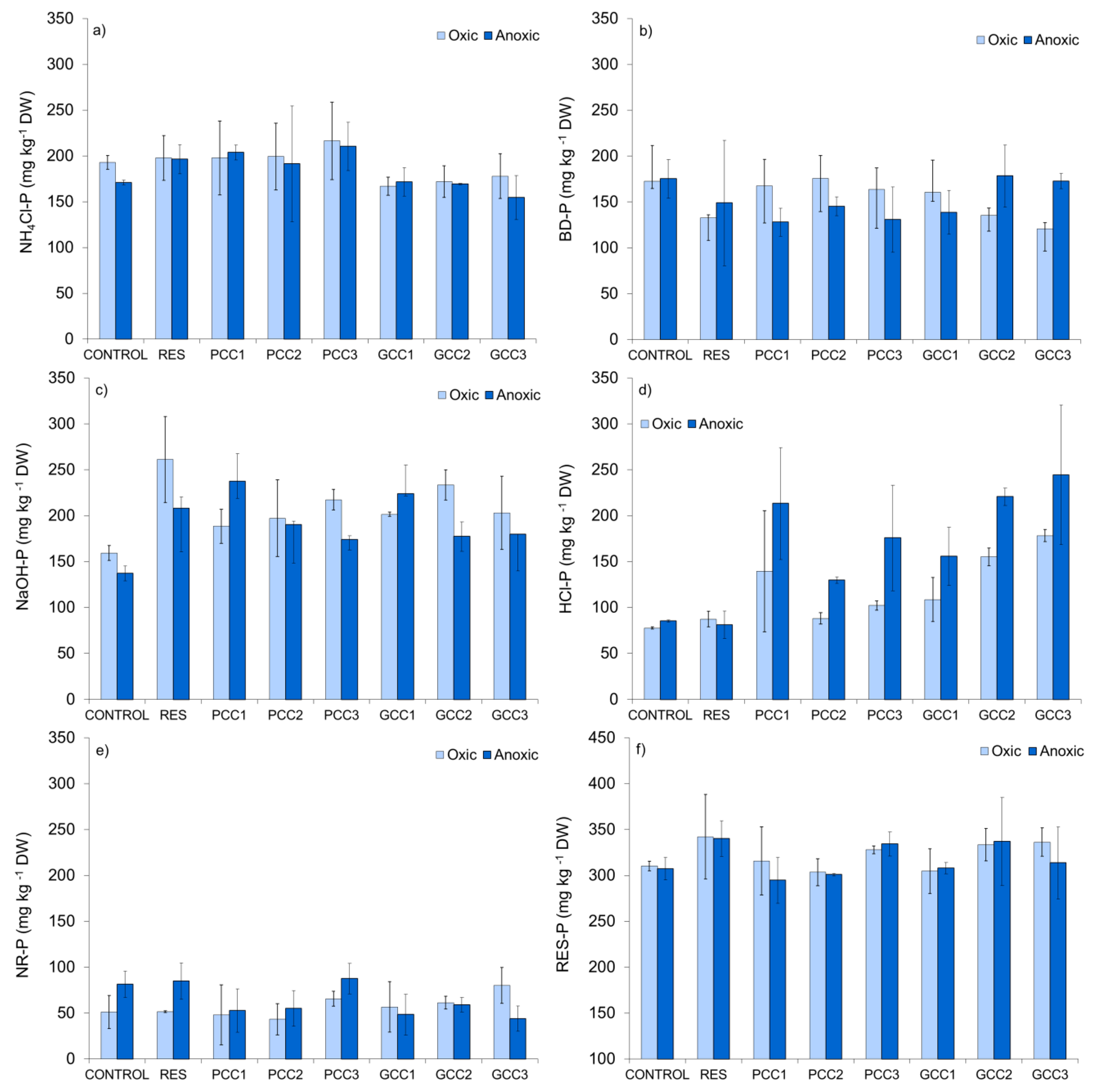
3.2.4. Phosphorus Fractions in Sediment
4. Discussion
4.1. Sorption Efficiency of Calcites as Revealed by Batch Experiments
4.2. SRP Removal Mechanism as Revealed by Sorption Isotherms
4.3. Mechanism of P Inactivation in Sediments
4.4. Efficiency and Stability of P Inactivation in Sediments
4.5. Possibilities of Method Implementation in Practice
5. Conclusions
Supplementary Materials
Author Contributions
Funding

Acknowledgments
Conflicts of Interest
References
- Cooke, G.D.; Welch, E.B.; Peterson, S.; Nichols, S.A. Restoration and Management of Lakes and Reservoirs, 3rd ed.; Taylor & Francis Group, LCC CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Cooke, G.D.; Welch, E.B.; Martin, A.B.; Fulmer, D.G.; Hyde, J.B.; Schrieve, G.D. Effectiveness of Al, Ca, and Fe salts for control of internal phosphorus loading in shallow and deep lakes. Hydrobiologia 1993, 253, 323–335. [Google Scholar] [CrossRef]
- Gibbs, M.M.; Hickey, C.W. Flocculants and Sediment Capping for Phosphorus Management. In Lake Restoration Handbook, A New Zealand Perspective, 1st ed.; Hamilton, D., Collier, K., Quinn, K., Howard-Williams, C., Eds.; Springer International Publishing AG: Cham, Switzerland, 2018. [Google Scholar] [CrossRef]
- Dunalska, J.A.; Wiśniewski, G. Can we stop the degradation of lakes? Innovative approaches in lake restoration. Ecol. Eng. 2016, 95, 714–722. [Google Scholar] [CrossRef]
- Huser, B.J.; Egemose, S.; Harper, H.; Hupfer, M.; Jensen, H.; Pilgrim, K.M.; Reitzel, K.; Rydin, E.; Futter, M. Longevity and effectiveness of aluminum addition to reduce sediment phosphorus release and restore lake water quality. Water Res. 2016, 97, 122–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parszuto, K.; Łopata, M.; Grochowska, J.; Tandyrak, R.; Augustyniak, R. Support of the Self-purification Processes in Lakes Restored in Poland. In Polish River Basins and Lakes—Part II, the Handbook of Environmental Chemistry 87, 1st ed.; Korzeniewska, E., Harnisz, M., Eds.; Springer Nature Switzerland AG: Cham, Switzerland, 2020. [Google Scholar] [CrossRef]
- Lewandowski, J.; Schauser, I.; Hupfer, M. Long term effects of phosphorus precipitations with alum in hypereutrophic Lake Susser See (Germany). Water Res. 2003, 37, 3194–3204. [Google Scholar] [CrossRef]
- Bakker, E.; Van Donk, E.; Immers, A.K. Lake restoration by in-lake iron addition: A synopsis of iron impact on aquatic organisms and shallow lake ecosystems. Aquat. Ecol. 2016, 50, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Augustyniak, R.; Grochowska, J.; Łopata, M.; Parszuto, K.; Tandyrak, R.; Tunowski, J. Sorption Properties of the Bottom Sediment of a Lake Restored by Phosphorus Inactivation Method 15 Years after the Termination of Lake Restoration Procedures. Water 2019, 11, 175. [Google Scholar] [CrossRef] [Green Version]
- Van Hullebusch, E.; Deluchat, V.; Chazal, P.M.; Baudu, M. Environmental impact of two successive chemical treatments in a small shallow eutrophied lake: Part I. Case of aluminium sulphate. Environ. Pollut. 2002, 120, 617–626. [Google Scholar] [CrossRef]
- Egemose, S.; Wauer, G.; Kleeberg, A. Resuspension behaviour of aluminium treated lake sediments: Effects of ageing and pH. Hydrobiologia 2009, 636, 203–217. [Google Scholar] [CrossRef]
- Wiśniewski, R.; Ślusarczyk, J.; Kaliszewski, T.; Szulczewski, A.; Nowacki, P. “Proteus”, a new device for application of coagulants directly to sediment during its controlled resuspension. Verh. Int. Ver. Limnol. 2010, 30, 1421–1424. [Google Scholar] [CrossRef]
- Wagner, K.; Meringolo, D.; Mitchell, D.; Moran, E.; Smith, S. Aluminium treatments to control internal phosphorus loading in lakes on Cape Cod, Massachusetts. Lake Reserv. Manag. 2017, 33, 171–186. [Google Scholar] [CrossRef]
- Welch, E.B.; Gibbons, H.L.; Brattebo, S.K.; Corson-Rikert, H.A. Distribution of aluminium and phosphorus fractions following alum treatments in a large shallow lake. Lake Reserv. Manag. 2017, 33, 198–204. [Google Scholar] [CrossRef]
- Schumaker, R.J.; Funk, W.H.; Moore, B.C. Zooplankton responses to aluminium sulphate treatment of Newman Lake, Washington. J. Freshw. Ecol. 1993, 8, 375–387. [Google Scholar] [CrossRef]
- Randall, S.; Harper, D.; Brierley, B. Ecological and ecophysiological impacts of ferric dosing in reservoirs. Hydrobiologia 1999, 395–396, 355–364. [Google Scholar] [CrossRef]
- Immers, A.K.; Van der Sande, M.T.; Van der Zande, R.M.; Geurts, J.J.M.; Van Donk, E.; Bakker, E.S. Iron addition as a shallow lake restoration measure: Impacts on charophyte growth. Hydrobiologia 2013, 710, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Stabel, H.-H. Calcite precipitation in Lake Constance: chemical equilibrium, sedimentation, and nucleation by algae. Limnol. Oceanogr. 1986, 31, 1081–1093. [Google Scholar] [CrossRef] [Green Version]
- Danen-Louwerse, H.J.; Lijklema, L.; Coenraats, M. Coprecipitation of phosphate with calcium carbonate in Lake Veluwe. Water Res. 1995, 29, 1781–1785. [Google Scholar] [CrossRef]
- Dittrich, M.; Koschel, R. Interactions between calcite precipitation (natural and artificial) and phosphorus cycle in the hardwater lake. Hydrobiologia 2002, 469, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Murphy, T.P.; Hall, K.J.; Yesaki, I. Coprecipitation of phosphate with calcite in a naturally eutrophic lake. Limnol. Oceanogr. 1983, 28, 58–69. [Google Scholar] [CrossRef]
- Koschel, R. Pelagic calcite precipitation and trophic state of hardwater lakes. Arch. Hydrobiol. Beih. Ergebn. Limnol. 1990, 33, 713–722. [Google Scholar]
- De Vicente, I.; Cattaneo, K.; Cruz-Pizarro, L.; Brauer, A.; Guilizzoni, P. Sedimentary phosphate fractions related to calcite precipitation in an eutrophic hardwater lake (Lake Alserio, northern Italy). J. Paleolimnol. 2006, 35, 55–64. [Google Scholar] [CrossRef]
- Walsh, J.R.; Corman, J.R.; Munoz, S.E. Coupled long-term limnological data and sedimentary records reveal new control on water quality in a eutrophic lake. Limnol. Oceanogr. 2019, 64, S34–S48. [Google Scholar] [CrossRef] [Green Version]
- Klapper, H. Calcite covering of sediment as a possible way of curbing blue-green algae. In Eutrophication: Research and Application to Water Supply, 3rd ed.; Sutcliffe, D.W., Jones, J.G., Eds.; Freshwater Biological Association: Ambleside, UK, 1992; pp. 107–111. Available online: https://core.ac.uk/download/pdf/11020726.pdf (accessed on 13 December 2019).
- Stüben, D.; Walpersdorf, E.; Voss, K.; Rönicke, H.; Schimmele, M.; Baborowski, M.; Luther, G.; Elsner, W. Application of lake marl at Lake Arendsee, NE Germany: First results of a geochemical monitoring during the restoration experiment. Sci. Total Environ. 1998, 218, 33–44. [Google Scholar] [CrossRef]
- Walpersdorf, E.; Neumann, T.; Stüben, D. Efficiency of natural calcite precipitation compared to lake marl application used for water quality improvement in an eutrophic lake. Appl. Geochem. 2004, 19, 1687–1698. [Google Scholar] [CrossRef]
- Griffin, R.A.; Jurinak, J.J. The interaction of phosphate with calcite. Soil Sci. Soc. Am. J. 1973, 37, 847–850. [Google Scholar] [CrossRef]
- Holford, I.C.R.; Mattingly, G.E.G. Phosphate sorption by jurassic oolitic limestones. Geoderma 1975, 13, 257–264. [Google Scholar] [CrossRef]
- Freeman, J.S.; Rowell, D.L. The adsorption and precipitation of phosphate onto calcite. J. Soil Sci. 1981, 32, 75–84. [Google Scholar] [CrossRef]
- Hinedi, Z.R.; Goldberg, S.; Chang, A.C.; Yesinowski, J.P. A 31P and 1H MAS NMR study of phosphate sorption onto calcium carbonate. J. Colloid Interface Sci. 1992, 152, 141–160. [Google Scholar] [CrossRef]
- Berg, U. Die Kalzitapplikation als interne Restaurierungsmaβnahme für eutrophierte Seen—Ihre Optimierung und Bewertung. Karlsruher Geochem. H. 2001, 20. [Google Scholar]
- Brix, H.; Arias, C.A.; del Bubba, M. Media selection for sustainable phosphorus removal in subsurface flow constructed wetlands. Water Sci. Technol. 2001, 44, 47–54. [Google Scholar] [CrossRef]
- Donnert, D.; Berg, U.; Weidler, P.G.; Nüesch, R.; Song, Y.; Salecker, M.; Kusche, I.; Bumiller, W.; Friedrich, F. Phosphorus removal and recovery from waste water by crystallisation. Wasser Geotechnol. 2002, 3, 115–132. Available online: http://www.academia.edu/22247677/Phosphorus_removal_and_recovery_from_waste_water_by_crystallisation (accessed on 13 December 2019).
- Hart, B.; Roberts, S.; James, R.; Taylor, J.; Donnert, D.; Furrer, R. Use of active barriers to reduce eutrophication problems in urban lakes. Water Sci. Technol. 2003, 47, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Lyngsie, G.; Penn, C.J.; Hansen, H.C.B.; Borggaard, O.K. Phosphate sorption by three potential filter materials as assessed by isothermal titration calorimetry. J. Environ. Manag. 2014, 143, 26–33. [Google Scholar] [CrossRef]
- Wan, B.; Yan, Y.; Liu, F.; Tan, W.; Chen, X.; Feng, X. Surface adsorption and precipitation of inositol hexakisphosphate on calcite: A comparison with orthophosphate. Chem. Geol. 2016, 421. [Google Scholar] [CrossRef]
- Li, Z.; Sun, X.; Huang, L.; Liu, D.; Yu, L.; Wu, H.; Wei, D. Phosphate adsorption and precipitation on calcite under calco-carbonic equilibrium condition. Chemosphere 2017, 183, 419–428. [Google Scholar] [CrossRef]
- Berg, U.; Neumann, T.; Donnert, D.; Nüesch, R.; Stüben, D. Sediment capping in eutrophic lakes—Efficiency of undisturbed calcite barriers to immobilize phosphorus. Appl. Geochem. 2004, 19, 1759–1771. [Google Scholar] [CrossRef]
- Eiche, E.; Berg, U.; Song, Y.; Neumann, T. Fixation and phase transformation of phosphate at calcite surfaces—Implications for eutrophic lake restoration. In Proceedings of the Ninth International Congress for Applied Mineralogy (1–10), Brisbane, QLD, Australia, 8–10 September 2008. [Google Scholar]
- Sø, H.U.; Postma, D.; Jakobsen, R.; Larsen, F. Sorption of phosphate onto calcite: results from batch experiments and surface complexation modelling. Geochim. Cosmochim. Acta 2011, 75, 2911–2923. [Google Scholar] [CrossRef]
- Wang, L.; Ruiz-Agudo, E.; Putnis, C.V.; Menneken, M.; Putnis, A. Kinetics of calcium phosphate nucleation and growth on calcite: implications for predicting the fate of dissolved phosphate species in alkaline soils. Environ. Sci. Technol. 2012, 46, 834–842. [Google Scholar] [CrossRef]
- Klasa, J.; Ruiz-Agudo, E.; Wang, L.J.; Putnis, C.V.; Valsami-Jones, E.; Menneken, M.; Putnis, A. An atomic force microscopy study of the dissolution of calcite in the presence of phosphate ions. Geochim. Cosmochim. Acta 2013, 117, 115–128. [Google Scholar] [CrossRef]
- Suzuki, T.; Inomata, S.; Sawada, K. Adsorption of phosphate onto calcite. J. Chem. Soc. Faraday Trans. Phys. Chem. Condens. Phases 1986, 82, 1733–1743. [Google Scholar] [CrossRef]
- Plant, L.J.; House, W.A. Precipitation of calcite in the presence of inorganic phosphate. Colloids Surf. A Phys. Eng. Asp. 2002, 203, 143–153. [Google Scholar] [CrossRef]
- Eiche, E.; Berg, U.; Neumann, T.; Nüesch, R.; Stüben, D. Multilayer fixation of dissolved phosphate on natural calcites derived from sorption experiments. Geochim. Cosmochim. Acta 2007, 71, 251. Available online: https://www.goldschmidtabstracts.info/abstracts/abstractView?id=2007004069 (accessed on 13 December 2019).
- Sø, H.U. Adsorption of Arsenic and Phosphate Onto the Surface of Calcite as Revealed by Batch Experiments and Surface Complexation Modelling. Ph.D. Thesis, Technical University of Denmark, Lyngby, Denmark, 2011. Available online: http://orbit.dtu.dk/files/5511322/Helle%20Ugilt%20Soe%20PhD-thesis%20WWW-Version.pdf (accessed on 13 December 2019).
- Valsami-Jones, E. Mineralogical controls on phosphorus recovery from wastewaters. Miner. Mag. 2001, 65, 611–620. [Google Scholar] [CrossRef]
- Mekmene, O.; Quillard, S.; Rouillon, T.; Bouler, J.-M.; Piot, M.; Gaucheron, F. Effects of pH and Ca/P molar ratio on the quantity and crystalline structure of calcium phosphates obtained from aqueous solutions. Dairy Sci. Technol. 2009, 89, 301–316. [Google Scholar] [CrossRef] [Green Version]
- Naidu, S.; Scherer, G.W. Nucleation, Growth and Evolution of Calcium Phosphate Films on Calcite. J. Colloid Interface Sci. 2014, 435, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zhan, Y.; Zhu, Z. Evaluation of sediment capping with active barrier systems (ABS) using calcite/zeolite mixtures to simultaneously manage phosphorus and ammonium release. Sci. Total Environ. 2011, 409, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Qin, B.; Xu, H.; Dong, B.; Brookes, J.D. Comparison of efficacy of two P-inactivation agents on sediments from different regions of Lake Taihu: sediment core incubations. Fundam. Appl. Limnol. 2012, 181, 271–281. [Google Scholar] [CrossRef]
- Galvez-Cloutier, R.; Saminathan, S.K.M.; Boillot, C.; Triffaut-Bouchet, G.; Bourget, A.; Soumis-Dugas, G. An evaluation of several in-lake restoration techniques to improve the water quality problem (eutrophication) of Saint-Augustin Lake, Quebec, Canada. Environ. Manag. 2012, 49, 1037–1053. [Google Scholar] [CrossRef]
- Yu, X.; Grace, M.R.; Sun, G.; Zou, Y. Application of ferrihydrite and calcite as composite sediment capping materials in a eutrophic lake. J. Soils Sediments 2018, 18, 1185–1193. [Google Scholar] [CrossRef]
- Ripl, W. Biochemical oxidation of polluted lake sediment with nitrate—A new lake restoration method. Ambio 1976, 5, 132–135. Available online: www.jstor.org/stable/4312194 (accessed on 13 December 2019).
- Wiśniewski, R. Phosphate inactivation with iron chloride during sediment resuspension. Lakes Reserv. Res. Manag. 1999, 4, 65–73. [Google Scholar] [CrossRef]
- Schütz, J.; Rydin, E.; Huser, B.J. A newly developed injection method for aluminum treatment in eutrophic lakes: Effects on water quality and phosphorus binding efficiency. Lake Reserv. Manag. 2017, 33, 152–162. [Google Scholar] [CrossRef]
- Wei, S.-H.; Mahuli, S.K.; Agnihotri, R.; Fan, L.-S. High Surface Area Calcium Carbonate: Pore Structural Properties and Sulfation Characteristics. Ind. Eng. Chem. Res. 1997, 36, 2141–2148. [Google Scholar] [CrossRef]
- Xu, N.; Chen, M.; Zhou, K.; Wang, Y.; Yin, H.; Chen, Z. Retention of phosphorus on calcite and dolomite: Speciation and modeling. RSC Adv. 2014, 4, 3525–35214. [Google Scholar] [CrossRef]
- Chang, R.; Kim, S.; Lee, S.; Choi, S.; Kim, M.; Park, Y. Calcium Carbonate Precipitation for CO2 Storage and Utilization: A Review of the Carbonate Crystallization and Polymorphism. Front. Energy Res. 2017, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Bańkowska, A.; Sawa, K.; Popek, Z.; Wasilewicz, M. The analysis of the Zdworskie Lake water supply in aspect of the restoration programme (in Polish). Sci. Rev. Eng. Environ. Sci. 2011, 20, 84–96. Available online: http://iks_pn.sggw.pl/z52/art2.pdf (accessed on 13 December 2019).
- Popek, Z.; Wasilewicz, M.; Bańkowska-Sobczak, A. Qualitative and Quantitative Monitoring of the Zdworskie Lake Water Supply. Tech. Rep. (Polish); Warsaw University of Life Sciences: Warsaw, Poland, 2017. [Google Scholar]
- Wiśniewski, R. Properties of the Sediment and Water of the Zdworskie Lake. Tech. Rep. (Polish); PROTE; Technologies for the Environment: Poznań, Poland, 2006. [Google Scholar]
- Bus, A.; Karczmarczyk, A. Kinetic studies on removing phosphate from synthetic solution and river water by reactive material in a form of suspended reactive filters. Desalin. Water Treat. 2018, 136, 237–244. [Google Scholar] [CrossRef]
- PN-EN ISO 6878. Water Quality. Determination of Phosphorus. Ammonium Molybdate Spectrometric Method; PKN: Warsaw, Poland, 2004. [Google Scholar]
- Heiri, O.; Lotter, A.F.; Lemcke, G. Loss on ignition as a method for estimating organic and carbonate content in sediments: reproducibility and comparability of results. J. Paleolimnol. 2001, 25, 101–110. [Google Scholar] [CrossRef]
- PN-EN ISO 7980:2002. Water Quality—Determination of Calcium and Magnesium—Atomic Absorption Spectrometric Method; PKN: Warsaw, Poland, 2002. [Google Scholar]
- U.S. EPA. Method 3051A (SW-846): Microwave Assisted Acid Digestion of Sediments, Sludges, Soils, and Oils, Revision 1; U.S. EPA: Washington, DC, USA, 2007.
- Hupfer, M.; Gächter, R.; Giovanoli, R. Transformation of phosphorus species in settling seston and during early sediment diagenesis. Aquat. Sci. 1995, 57, 305–324. [Google Scholar] [CrossRef]
- Lewandowski, J. Untersuchungen Zum Einfluss Seeinterner Verfahren auf Die Phosphor-Diagenese in Sedimenten. Ph.D. Thesis, HU Berlin, Institut für Biologie, Berlin, Germany, 2002. [Google Scholar] [CrossRef]
- Hupfer, M.; Zak, D.; Roßberg, R.; Herzog, C.; Pöthig, R. Evaluation of a well-established sequential phosphorus fractionation technique for use in calcite-rich lake sediments: identification and prevention of artifacts due to apatite formation. Limnol. Oceanogr. Methods 2009, 7, 399–410. [Google Scholar] [CrossRef]
- Limousin, G.; Gaudet, J.-P.; Charlet, L.; Szenknect, S.; Barthès, V.; Krimissa, M. Sorption isotherms: A review on physical bases, modeling and measurement. Appl. Geochem. 2007, 22, 249–275. [Google Scholar] [CrossRef]
- Deutsch, W.J.; Siegel, R. Groundwater Geochemistry: Fundamentals and Applications to Contamination, 1st ed.; CRC Press LCC: Boca Raton, FL, USA, 1997. [Google Scholar] [CrossRef]
- Macioszczyk, A.; Dobrzyński, D. Hydrogeochemia Strefy Aktywnej Wymiany Wód Podziemnych; Wydawnictwo PWN: Warszawa, Polska, 2002. (In Polish) [Google Scholar]
- Upchurch, S.; Scott, T.M.; Alfieri, M.; Fratesi, B.; Dobecki, T.L. The Karst Systems of Florida, Understanding Karst in a Geologically Young Terrain, 1st ed.; Springer International Publishing AG: Cham, Switzerland, 2019. [Google Scholar] [CrossRef]
- Song, Y.; Hahn, H.H.; Hoffmann, E. The effect of carbonate on the precipitation of calcium phosphate. Environ. Technol. 2002, 23, 207–215. [Google Scholar] [CrossRef]
- Recillas, S.; Rodríguez-Lugo, V.; Montero, M.L.; Viquez-Cano, S.; Hernandez, L.; Castano, V.M. Studies on the precipitation behaviour of calcium phosphate solutions. J. Ceram. Process. Res. 2012, 13, 5–10. Available online: http://jcpr.kbs-lab.co.kr/file/JCPR_vol.13_2012/JCPR13-1/5_10.pdf (accessed on 13 December 2019).
- Avnimelech, Y. Phosphorus and calcium carbonate solubilities in Lake Kinneret. Limnol. Oceanogr. 1983, 28, 640–645. [Google Scholar] [CrossRef]
- Sanciolo, P.; Zou, L.; Gray, S.; Leslie, G.; Stevens, D. Accelerated seeded precipitation pre-treatment of municipal wastewater to reduce scaling. Chemosphere 2008, 72, 243–249. [Google Scholar] [CrossRef] [Green Version]
- Stumm, W.; Morgan, J. Aquatic Chemistry: Chemical Equilibria and Rates. In Natural Waters, 3rd ed.; Wiley: New York, NY, USA, 1996. [Google Scholar]
- Yagi, S.; Fukushi, K. Removal of phosphate from solution by adsorption and precipitation of calcium phosphate onto monohydrocalcite. J. Colloid Interface Sci. 2012, 384, 128–136. [Google Scholar] [CrossRef]
- Zhou, M.; Li, Y. Phosphorus-sorption characteristics of calcareous soils and limestone from the Southern Everglades and adjacent farmlands. Soil Sci. Soc. Am. J. 2001, 65, 1404–1412. [Google Scholar] [CrossRef] [Green Version]
- House, W.A. The physico-chemical conditions for the precipitation of phosphate with calcium. Environ. Technol. 1999, 20, 727–733. [Google Scholar] [CrossRef]
- Reddy, K.R.; Fisher, M.M.; Ivanoff, D. Resuspension and diffusive flux of nitrogen and phosphorus in a hypereutrophic lake. J. Environ. Qual. 1996, 25, 363–371. [Google Scholar] [CrossRef]
- Rosell, E.A. Influence of Resuspension on Sediment-Water Solute Exchange and Particle Transport in Marine Environments. Ph.D. Thesis, University of Gothenburg, Gothenburg, Sweden, 2011. Available online: https://gupea.ub.gu.se/bitstream/2077/27061/1/gupea_2077_27061_1.pdf (accessed on 13 December 2019).
- Fan, C.; Zhang, L.; Qu, W.C. Lake sediment resuspension and caused phosphate release – A simulation study. J. Environ. Stud. (China) 2001, 13, 406–410. [Google Scholar]
- Kuo, S.; Lotse, E.G. Kinetics of phosphate adsorption by calcium carbonate and Ca-kaolinite. Soil Sci. Soc. Am. J. 1972, 36, 725–729. [Google Scholar] [CrossRef]
- Lin, Y.; Singer, P.C. Inhibition of calcite precipitation by orthophosphate: speciation and thermodynamic considerations. Geochim. Cosmochim. Acta 2006, 70, 2530–2539. [Google Scholar] [CrossRef]
- Koschel, R.; Casper, P.; Gonsiorczyk, T.; Rossberg, R.; Wauer, G. Hypolimnetic Al- and CaCO3-treatments and aeration for restoration of a stratified eutrophic hardwater lake (Lake Tiefwarensee, Mecklenburg-Vorpommern, Germany). Verh. Des Int. Ver. Limnol. 2006, 29, 2165–2171. [Google Scholar]
- Staudinger, B.; Peiffer, S.; Avnimelech, Y.; Berman, T. Phosphorus mobility in pore waters of sediments in Lake Kinneret, Israel. Hydrobiologia 1990, 207, 167–177. [Google Scholar] [CrossRef]
- Müller, B.; Wang, Y.; Wehrli, B. Cycling of calcite in hard-water lakes of different trophic states. Limnol. Oceanogr. 2006, 51, 1678–1688. [Google Scholar] [CrossRef]
- Golterman, H.L. The Chemistry of Phosphate nnd Nitrogen Compounds in Sediments, 1st ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2004. [Google Scholar]
- Søndergaard, M. Nutrient dynamics in lakes—With emphasis on phosphorus, sediment and lake restoration. Ph.D. Thesis, (DSc), National Environmental Research Institute of Denmark, University of Aarhus, Aarhus, Denmark, 2007. Available online: https://www2.dmu.dk/pub/dsc_ms_uk.pdf (accessed on 13 December 2019).
- Golterman, H.L. The calcium- and iron-bound phosphate phase diagram. Hydrobiologia 1988, 159, 149–151. [Google Scholar] [CrossRef]
- Gonsiorczyk, T.; Casper, P.; Koschel, R. Phosphorus-binding forms in the sediment of an oligotrophic and an eutrophic hardwater lake of the Baltic Lake District (Germany). Water Sci. Technol. 1998, 37, 51–58. [Google Scholar] [CrossRef]
- Ann, Y.; Reddy, K.R.; Delfino, J.J. Influence of redox potential on phosphorus solubility in chemically amended wetland organic soils. Ecol. Eng. 1999, 14, 169–180. [Google Scholar] [CrossRef]
- Giguet-Covex, C.; Poulenard, J.; Chalmin, E.; Arnaud, F.; Rivard, C.; Jenny, J.P.; Dorioz, J.-M. XANES spectroscopy as a tool to trace phosphorus transformation during soil genesis and mountain ecosystem development from lake sediments. Geochim. Cosmochim. Acta 2013, 118, 129–147. [Google Scholar] [CrossRef]
- Adam, N. A Wet–Chemical and Phosphorus K-edge X-ray Absorption Near Edge Structure Investigation of Phosphate Adsorption on Binary Mixtures of Ferrihydrite and Calcite: Implications for Phosphorus Bioavailability. Soil Sci. Soc. Am. J. 2017, 81, 1079–1087. [Google Scholar] [CrossRef]
- Gomez, E.; Durillon, C.; Rofes, G.; Picot, B. Phosphate adsorption and release from brackisch lagoons: pH, O2 and loading influence. Water Res. 1999, 33, 2437–2447. [Google Scholar] [CrossRef]
- Song, Y.; Weidler, P.G.; Berg, U.; Nüesch, R.; Donnert, D. Calcite-seeded crystallization of calcium phosphate for phosphorus recovery. Chemosphere 2006, 63, 236–243. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Calcite Materials | Physio-Chemical Parameters | ||||
|---|---|---|---|---|---|
| SRP (mg PO43− L−1) | Ca2+ (mg L−1) | Alkalinity (mg HCO3− L−1) | pH | SICC | |
| PCC | 4.10 | 176 | 160 | 7.9 | 0.8 |
| GCC | 4.10 | 181 | 165 | 7.7 | 0.6 |
| Solution Type | Physio-Chemical Parameters | |||||
|---|---|---|---|---|---|---|
| SRP (mg PO43− L−1) | Ca2+ (mg L−1) | Alkalinity (mg HCO3− L−1) | pH | SICC | ORP [mV] | |
| Pore water | 11.58 | 112 | 409 | 7.8 | 0.8 | −165 |
| Lake water | 0.02 | 68 | 107 | 8.3 | 0.5 | 386 |
| Treatment Type | Denotations for Treatments | |||||||
|---|---|---|---|---|---|---|---|---|
| CONTROL | RES | PCC1 | PCC2 | PCC3 | GCC1 | GCC2 | GCC3 | |
| Resuspension | − | + | + | + | + | + | + | + |
| Calcite materials dosage (kg m−2) | − | − | 0.25 (low) | 0.75 (medium) | 1.50 (high) | 1.50 (low) | 3.00 (medium) | 6.00 (high) |
| Solid Phases with Denotation (in Brackets) | Stoichiometry | Solubility Product |
|---|---|---|
| Dicalciumphosphate dihydrate (DCPD) | CaHPO4·2H2O | 10−6.59 [76] |
| Dicalcium phosphate anhydrous (DCPA) | CaHPO4 | 10−6.9 [77] |
| Calcite (CC) | CaCO3 | 10−8.48 [wateq4f.dat database] |
| Amorphous calcium phosphate (ACP) | Ca3(PO4)2 | 10−28.25 [38] |
| Surface complex (SC) | Ca3(HCO3)3PO4 | 10−28.5 [78] |
| ß-tricalcium phosphate (β-TCP) | Ca3(PO4)2 | 10−28.92 [76] |
| Octacalcium phosphate (OCP) | Ca4H(PO4)3 | 10−47.8 [76] |
| Hydroxyapatite (HAp) | Ca5OH(PO4)3 | 10−54.0 [wateq4f.dat database] |
| Treatment | SICC | SIDCPD | SIDCPA | SIACP | SISC | SIß– TCP | SIOCP | SIHAp |
|---|---|---|---|---|---|---|---|---|
| Oxic | ||||||||
| CONTROL | 0.1–0.5 | −1.3–−1.5 | −1.0–−1.1 | −1.1–−0.9 | 2.5–3.0 | 0.0–0.8 | −1.7–−1.0 | 2.0–3.4 |
| RES | 0.1–0.1 | −1.6 | −1.3 | −0.8–−0.3 | 2.4 | −0.4– −0.10 | −2.2–−2.0 | 1.4–1.8 |
| PCC1 | 0.2–0.3 | −2.3–−1.6 | −1.3–−2.0 | −0.8–−0.2 | 1.8–2.5 | −1.5–−0.1 | −4.1–−2.0 | −0.1–2.0 |
| PCC2 | 0.2–0.3 | −1.8–−2.6 | −1.5–−2.3 | −0.6–−0.5 | 1.5–2.4 | −2.1–−0.2 | −5.0–−2.4 | −1.2–1.8 |
| PCC3 | 0.3–0.4 | −2.3–−2.8 | −2.0–−2.5 | −1.3–−0.8 | 1.4–1.8 | −2.1–−1.2 | −5.2–−3.8 | −1.0–0.3 |
| GCC1 | 0.3–0.4 | −1.4–−1.6 | −1.1–−1.3 | −2.3–−0.9 | 2.4–2.8 | 0.0–0.6 | −1.9–−1.1 | 2.1–3.1 |
| GCC2 | 0.3–0.4 | −1.6 | −1.3 | −2.9–−2.0 | 2.6–2.8 | 0.2 | −1.7 | 2.4–2.5 |
| GCC3 | 0.4–0.5 | −1.8–−2.1 | −1.5–−1.8 | −5.3–−3.5 | 2.3–2.5 | −0.5–0.1 | −3.0–−2.0 | 1.5–2.3 |
| Anoxic | ||||||||
| CONTROL | 0.1–0.2 | −1.1 | −0.8 | −0.1–0.3 | 2.6–2.8 | 0.9–1.1 | −0.5–−0.4 | 3.4–3.7 |
| RES | 0.1–0.1 | −1.2–−1.1 | −0.9–−0.8 | 0.0–0.1 | 2.5–2.7 | 0.7–0.9 | −0.8–−0.6 | 3.1–3.3 |
| PCC1 | 0.0 – 0.1 | −2.3–−1.5 | −2.0–−1.2 | −0.2–−0.1 | 1.1–2.2 | −1.6–0.1 | −4.3–−1.7 | −0.4–2.2 |
| PCC2 | 0.0 – 0.2 | −2.6–−2.0 | −2.0–−1.7 | −0.6–−0.2 | 1.3–1.8 | −1.8–−0.7 | −4.4–−3.1 | −0.8–1.0 |
| PCC3 | 0.0 – 0.3 | −1.3–−2.4 | −2.3–−2.1 | −0.5–−0.2 | 0.9–1.5 | −1.9–−1.3 | −4.8–−4.0 | −0.8–0.2 |
| GCC1 | 0.0 – 0.2 | −1.5–−1.2 | −0.9 | −2.4–−0.7 | 2.3–2.6 | 0.6–0.9 | −1.0–−0.6 | 2.9–3.5 |
| GCC2 | 0.1 – 0.3 | −1.6–−1.3 | −1.2–−1.0 | −2.6–−1.5 | 2.1–2.7 | 0.2–0.6 | −1.7–−1.1 | 2.3–3.2 |
| GCC3 | 0.2 – 0.5 | −1.5 | −1.3–−1.2 | −2.8–−2.1 | 2.3–2.6 | 0.3–0.6 | −1.6–−1.3 | 2.6–3.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bańkowska-Sobczak, A.; Blazejczyk, A.; Eiche, E.; Fischer, U.; Popek, Z. Phosphorus Inactivation in Lake Sediments Using Calcite Materials and Controlled Resuspension—Mechanism and Efficiency. Minerals 2020, 10, 223. https://doi.org/10.3390/min10030223
Bańkowska-Sobczak A, Blazejczyk A, Eiche E, Fischer U, Popek Z. Phosphorus Inactivation in Lake Sediments Using Calcite Materials and Controlled Resuspension—Mechanism and Efficiency. Minerals. 2020; 10(3):223. https://doi.org/10.3390/min10030223
Chicago/Turabian StyleBańkowska-Sobczak, Agnieszka, Aurelia Blazejczyk, Elisabeth Eiche, Uwe Fischer, and Zbigniew Popek. 2020. "Phosphorus Inactivation in Lake Sediments Using Calcite Materials and Controlled Resuspension—Mechanism and Efficiency" Minerals 10, no. 3: 223. https://doi.org/10.3390/min10030223
APA StyleBańkowska-Sobczak, A., Blazejczyk, A., Eiche, E., Fischer, U., & Popek, Z. (2020). Phosphorus Inactivation in Lake Sediments Using Calcite Materials and Controlled Resuspension—Mechanism and Efficiency. Minerals, 10(3), 223. https://doi.org/10.3390/min10030223