Mineral-Inspired Materials: Synthetic Phosphate Analogues for Battery Applications



Abstract
:
1. Introduction
2. Background Information
3. Phosphate Minerals in Pegmatites
4. Mineral Prototypes for Battery Design
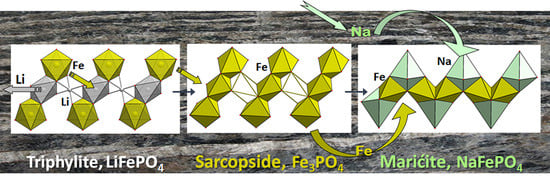
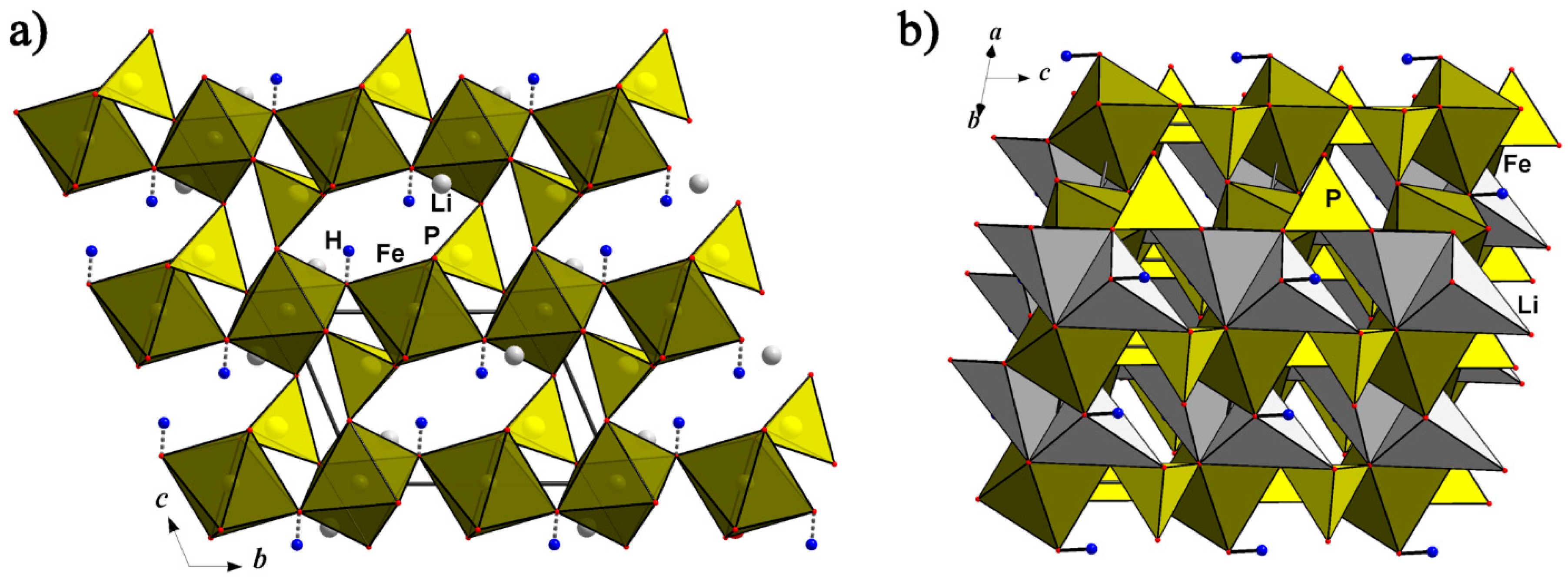
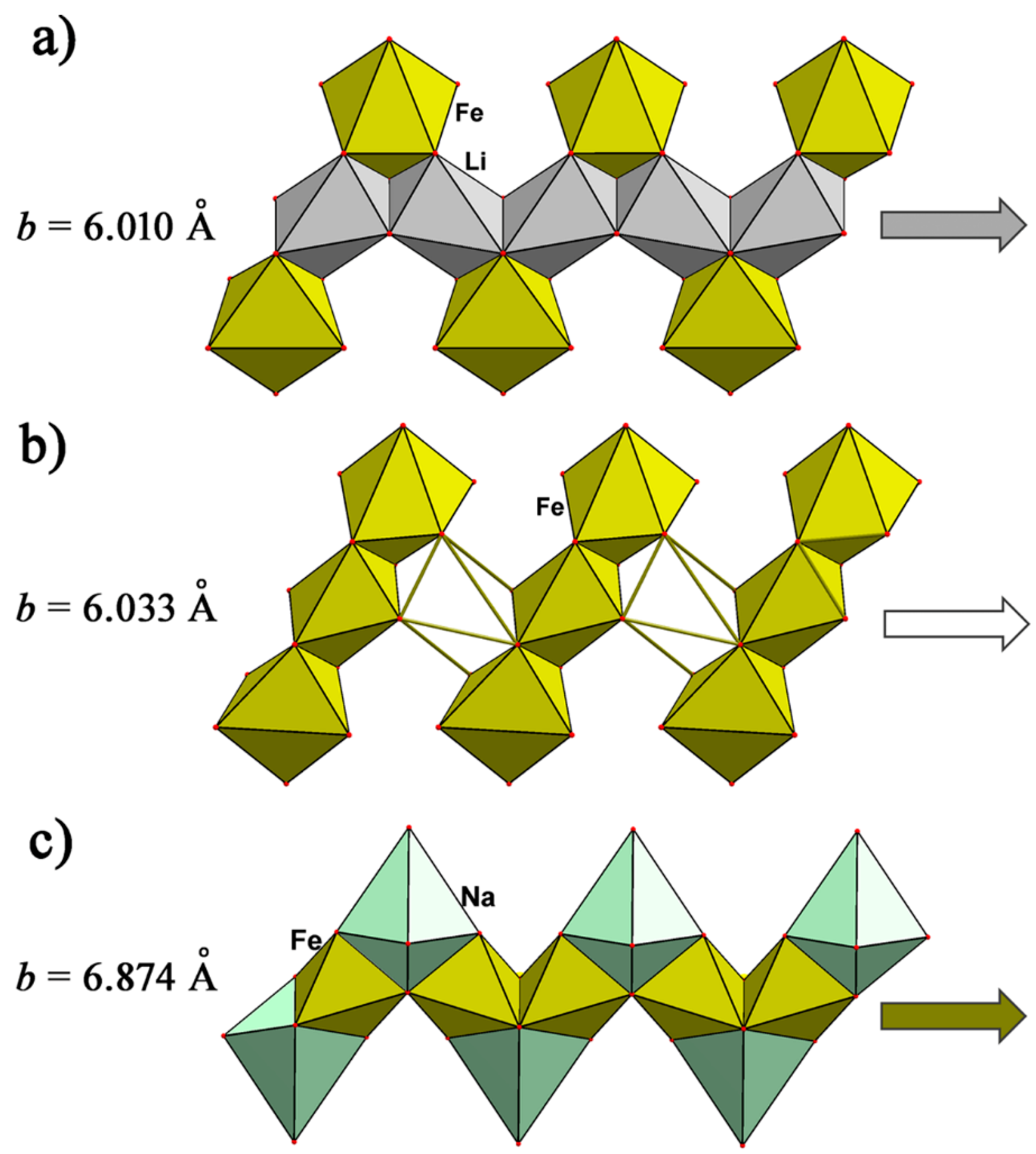
4.1. Phosphates of the Triphylite Li(Fe, Mn)PO4–Lithiophilite Li(Mn, Fe)PO4 Series and Their Derivatives
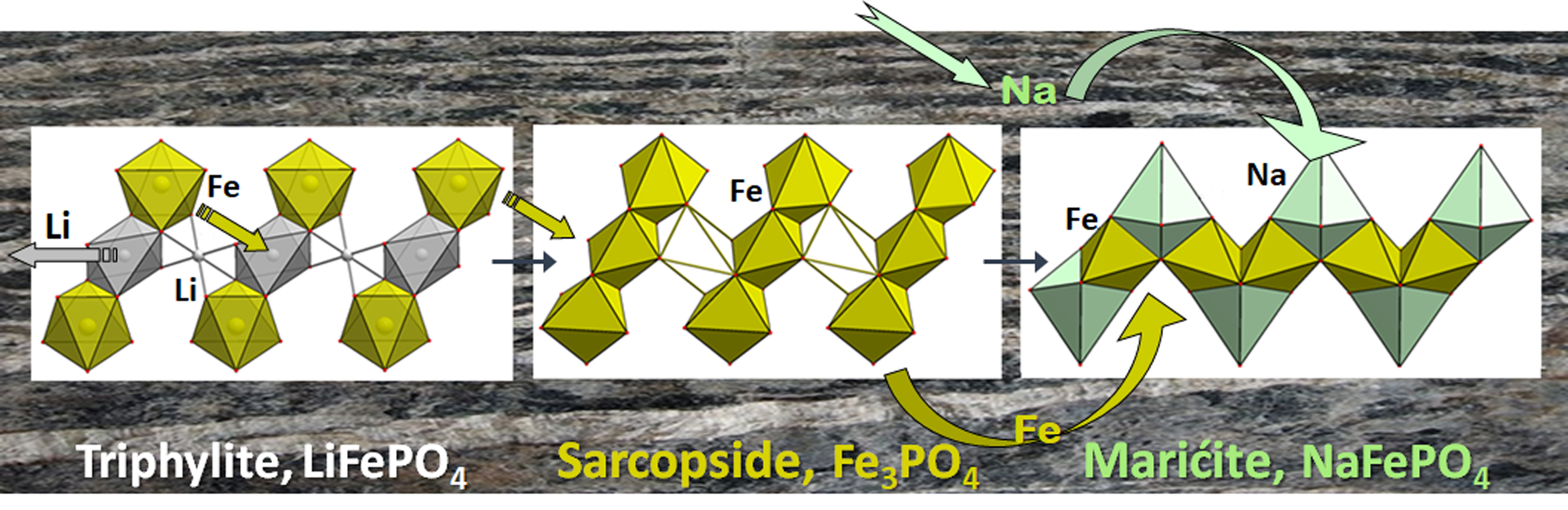
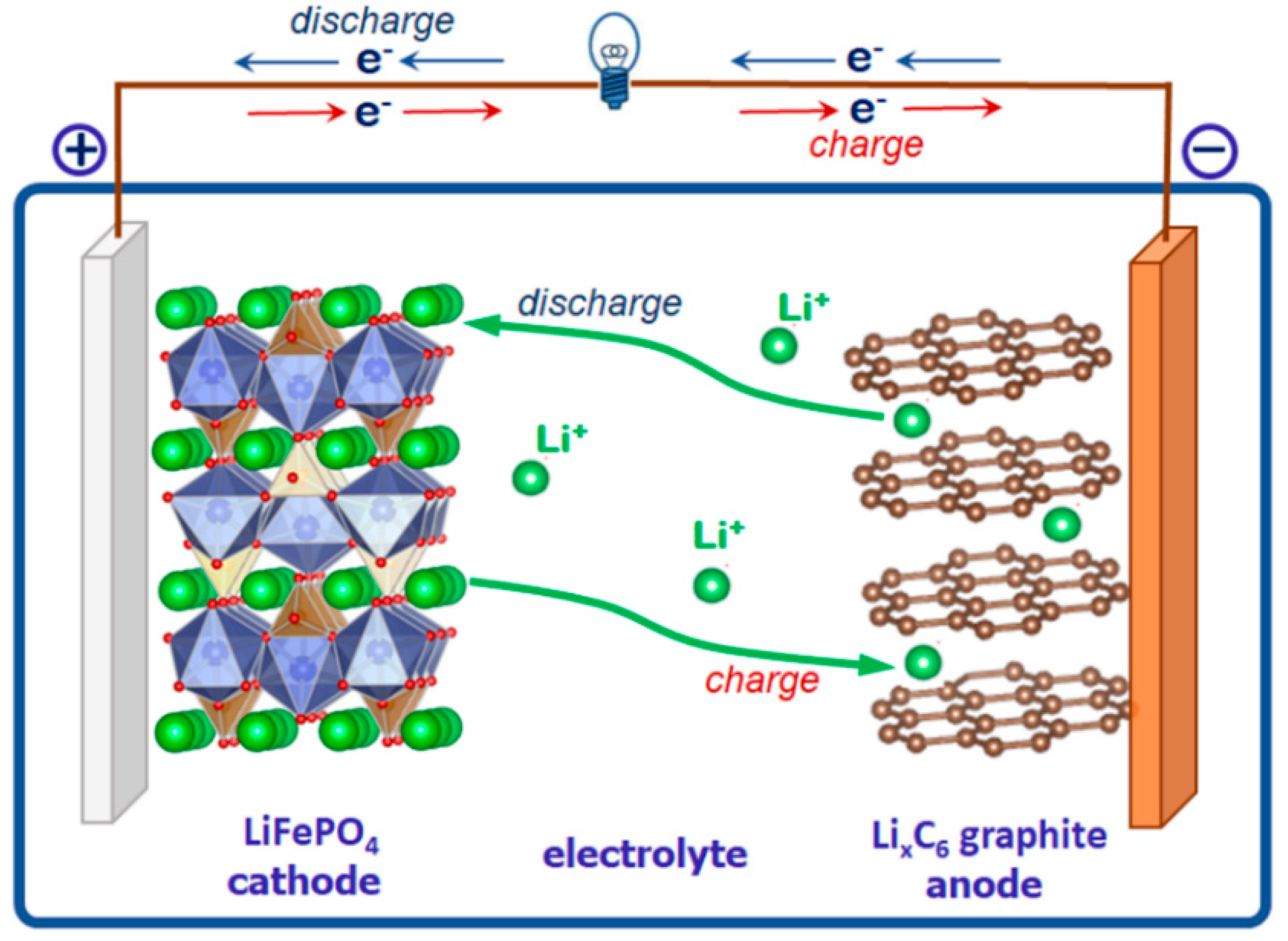
4.1.1. The Triphylite Li (Fe, Mn) PO4–Lithiophilite Li (Mn, Fe) PO4 Series: Occurrence, Crystal Chemistry, and Electrochemical Behavior
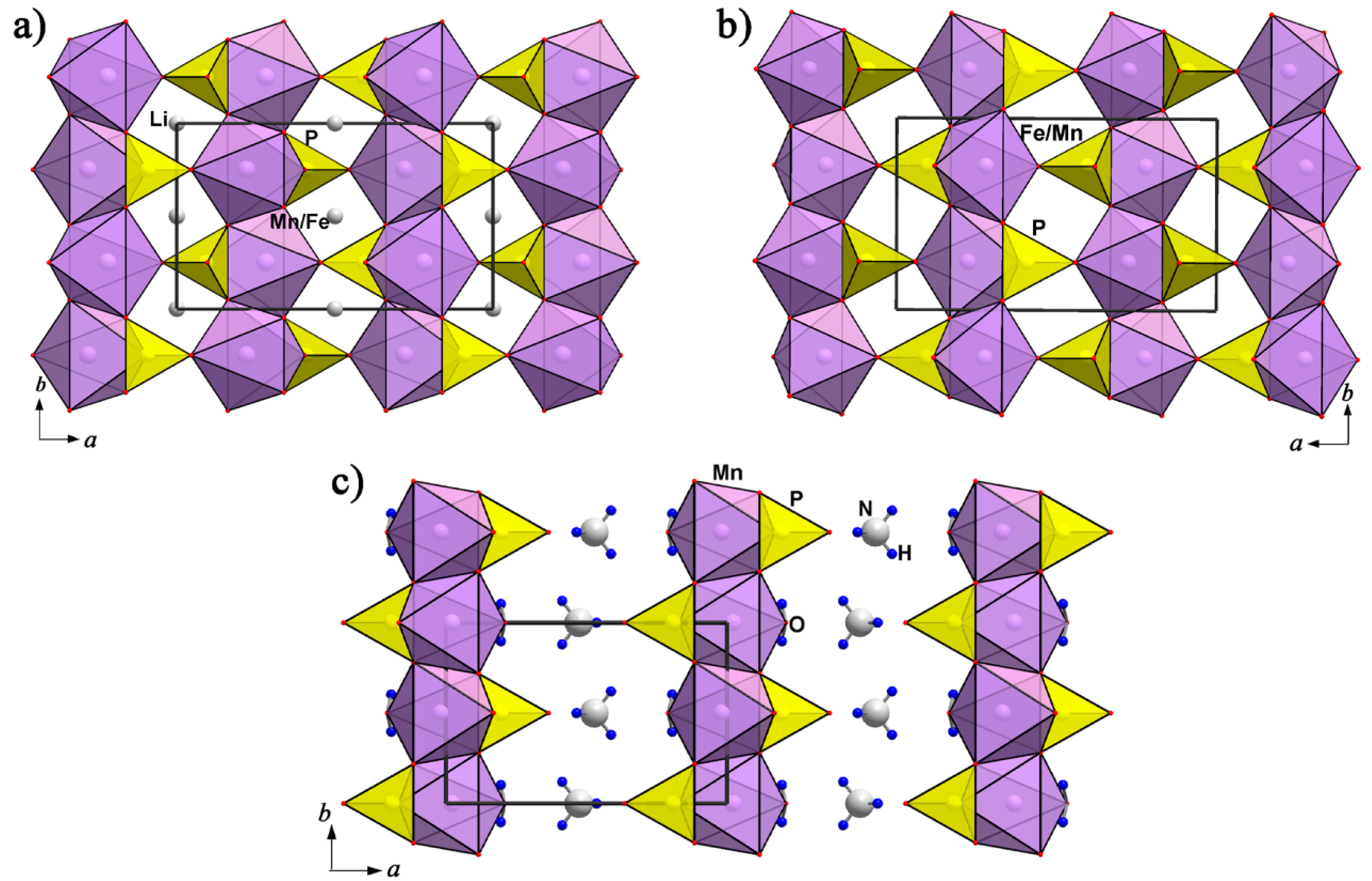
4.1.2. Lithiophilite—Purpurite—Niahite: Comparative Crystal Chemistry and Structure Transformation
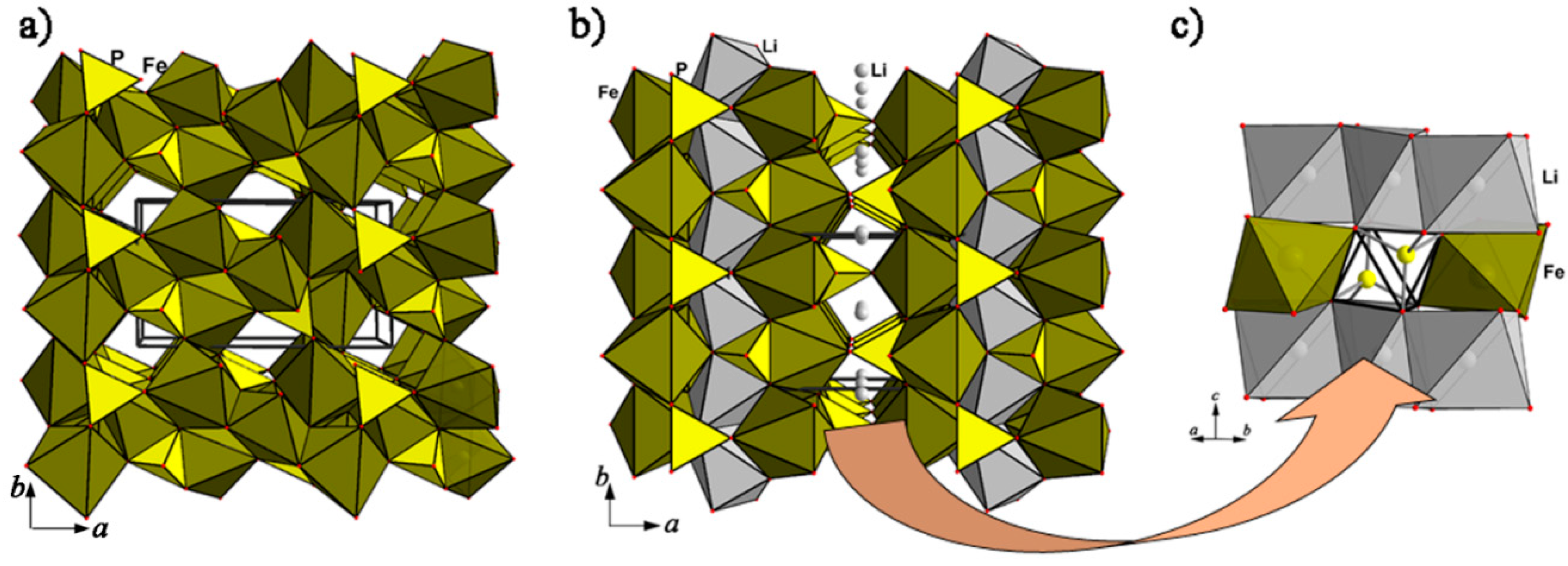
4.1.3. Simferite, Li(Mg,Fe3+,Mn3+)2(PO4)2 and Tavorite, LiFe(PO4)(OH,F) Based Materials and Their Electrochemical Potential
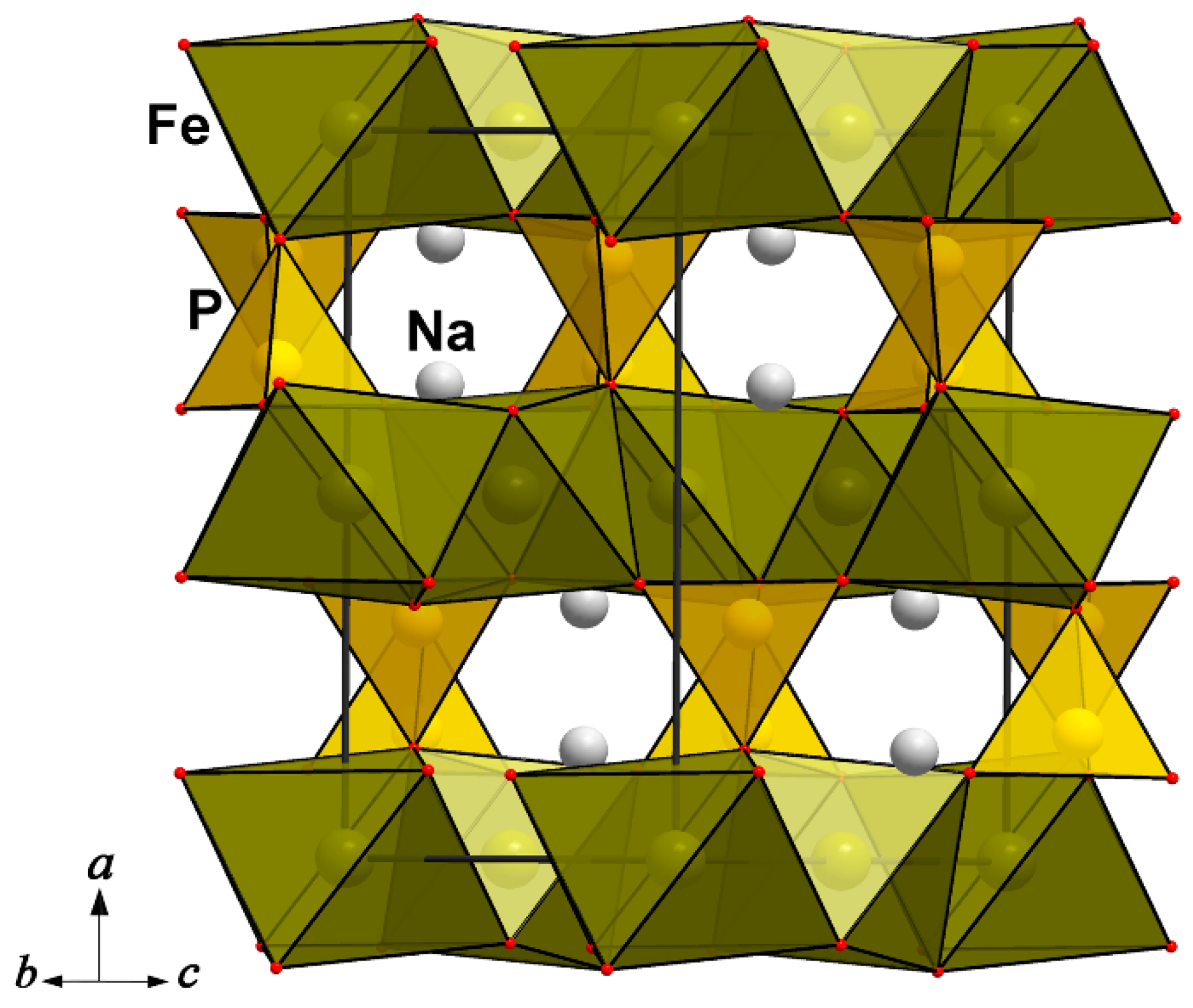
4.1.4. Na-Bearing Triphylite-Lithiophilite Structural Analogous and Their Derivatives as Promising Cathode Materials
4.2. Other Phosphate-Based Series
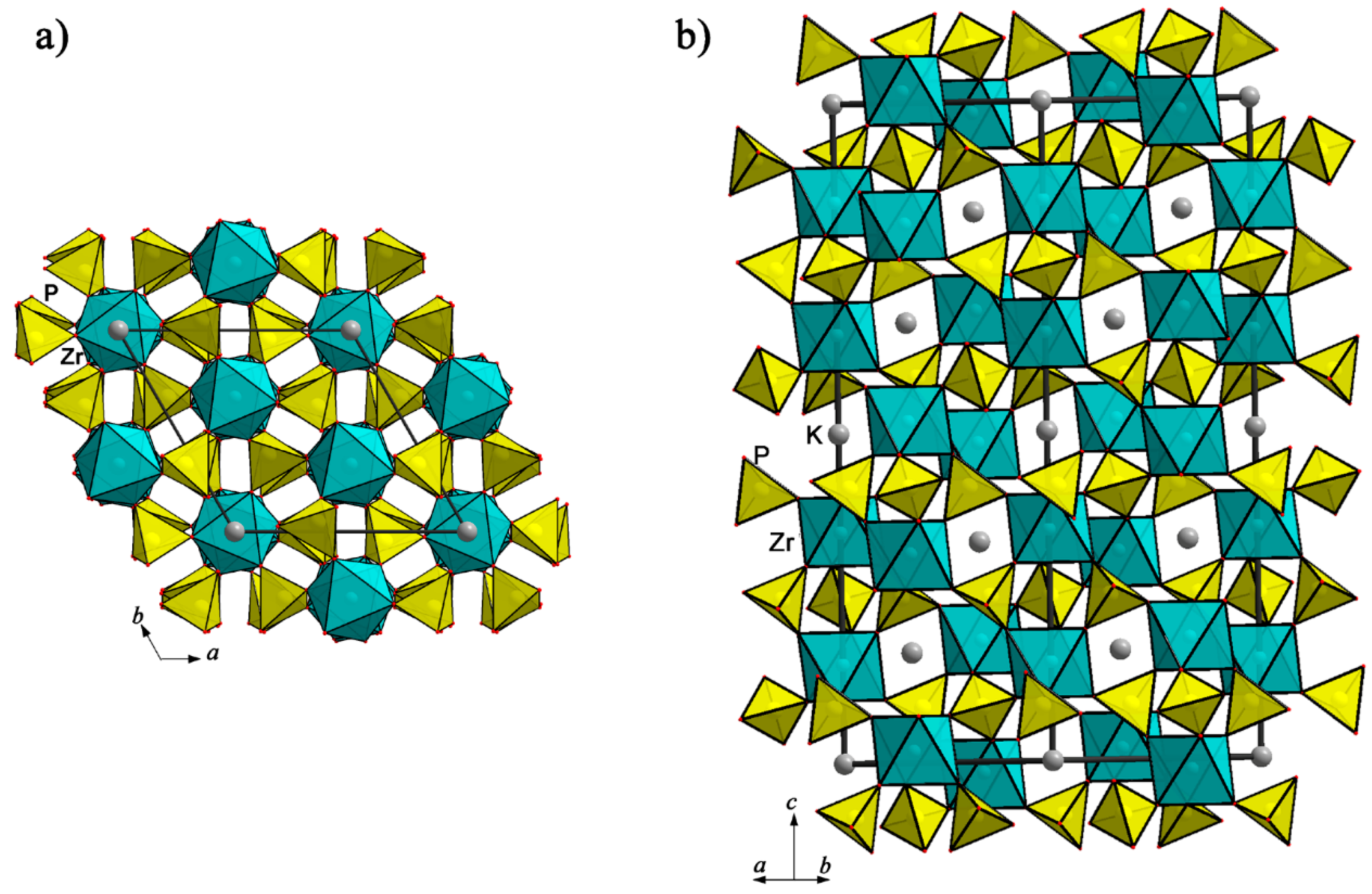
4.2.1. Mineral Kosnarite and Structurally Related Phosphates
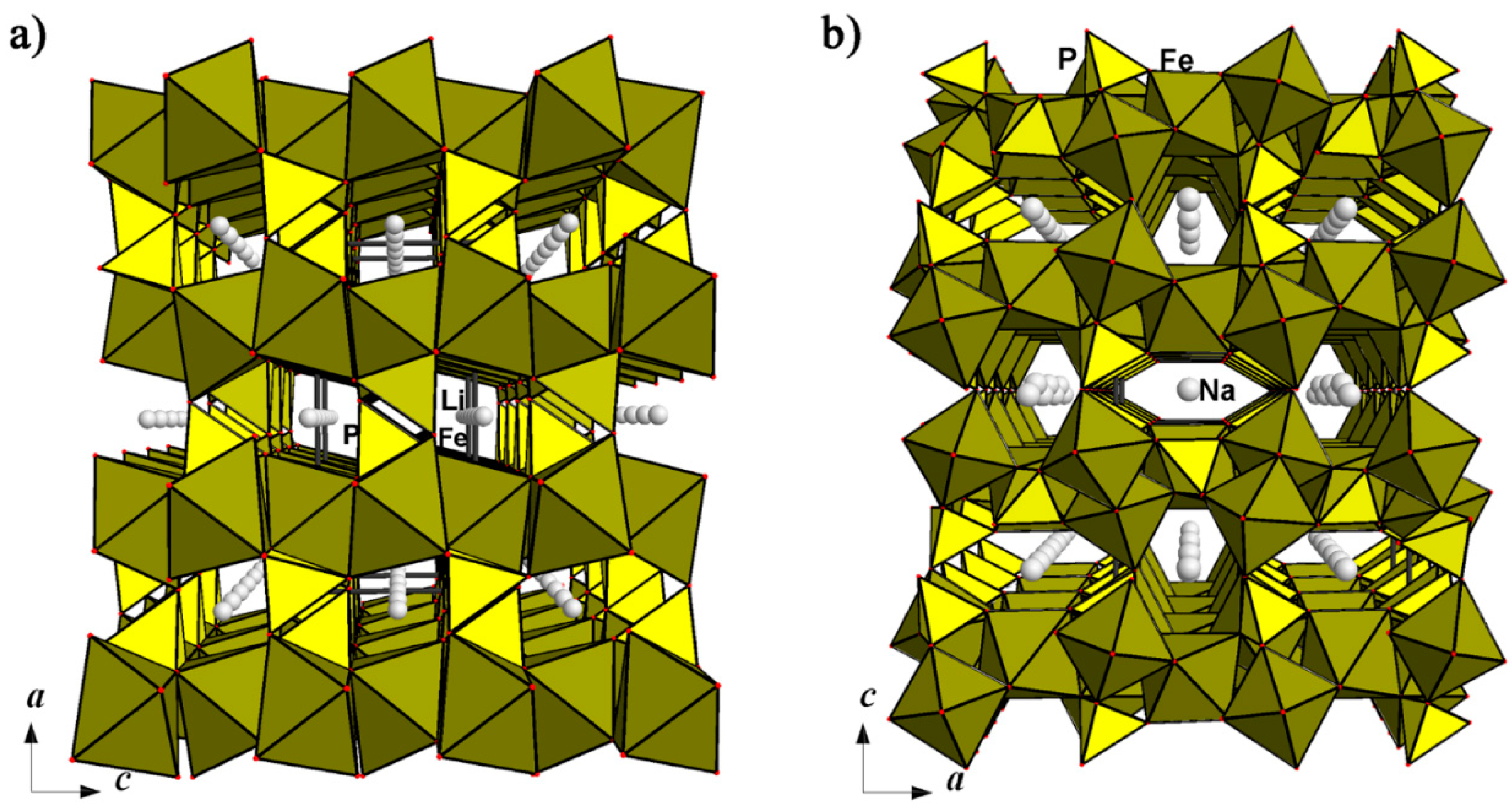
4.2.2. Alluaudite- and Wyllieite-Type Compounds
4.2.3. Minerals with Two Sorts of Complex Anions: Sidorenkite, Na3Mn(PO4)(CO3) and Bonshtedtite, Na3Fe(PO4)(CO3)
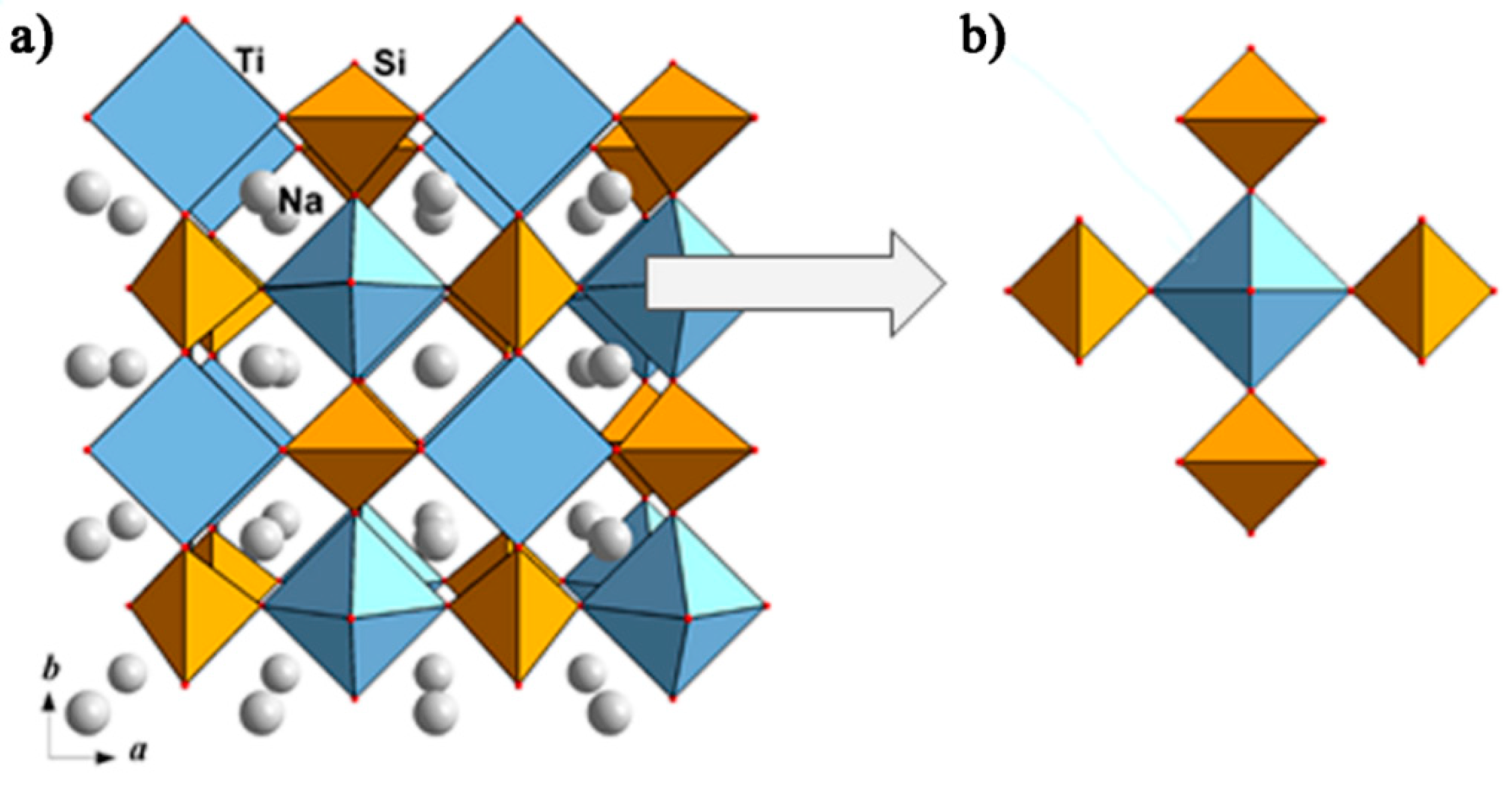
4.2.4. Phosphate Cathode Materials, Structurally Derived from Mineral Natisite, Na2TiSiO5
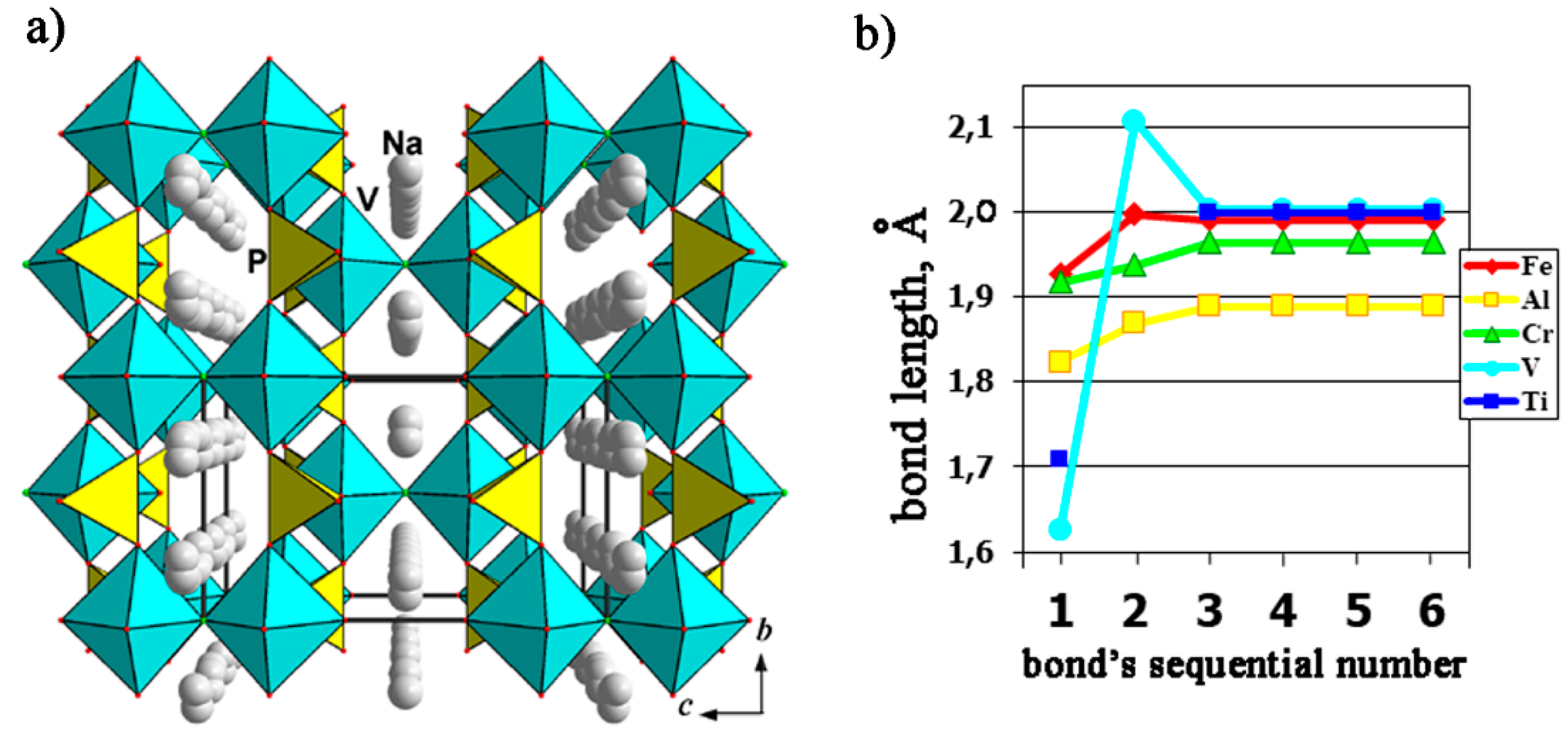
4.2.5. Phosphate Analogues of Katiarsite, KTiOAsO4 and Yurgensonite, K2SnTiO2(AsO4)2, as Successive Electrode Materials for K-Ion Batteries
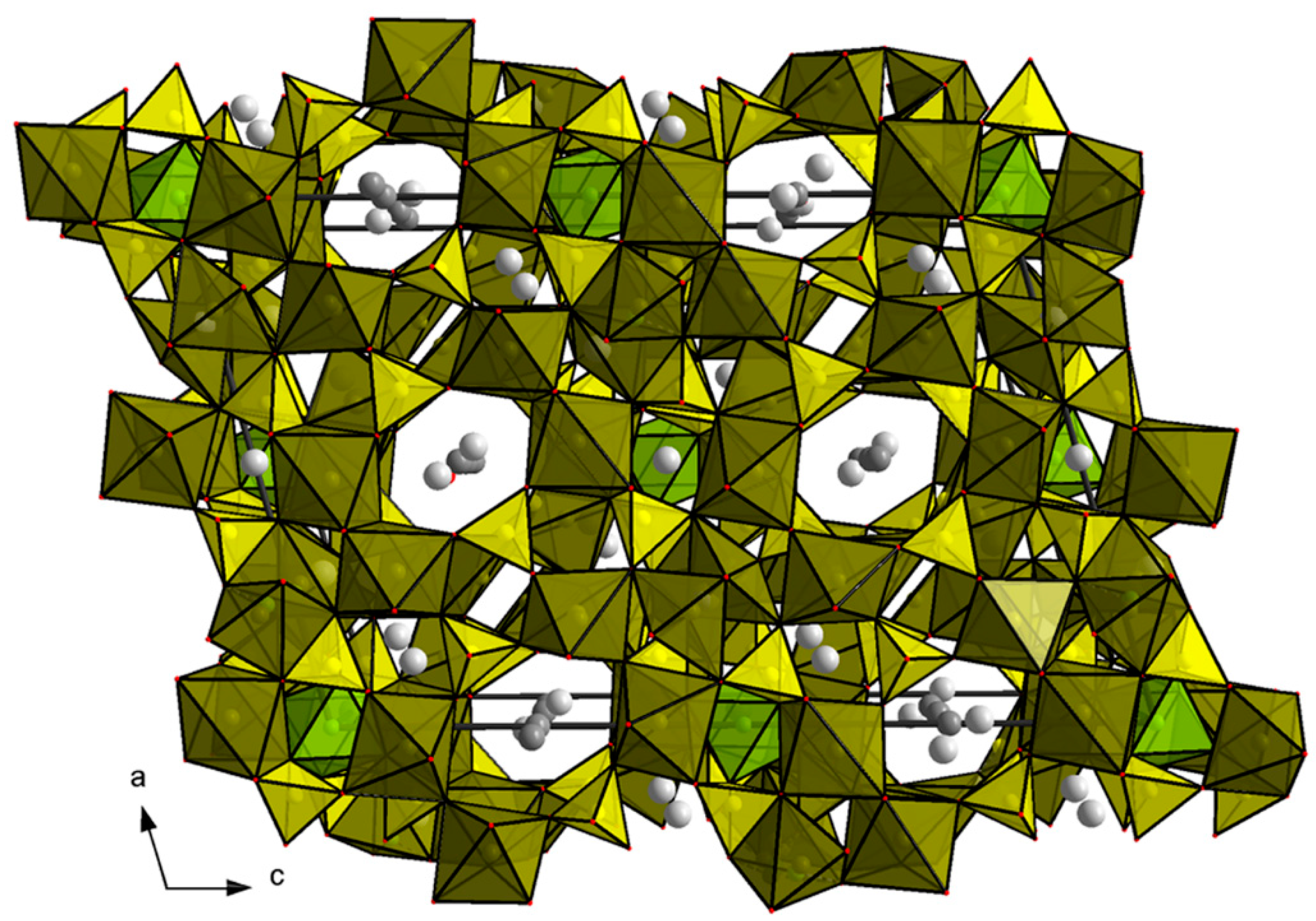
4.2.6. Arrojadite–Type Compounds
5. Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula (Mineral Name) | Unit-Cell Parameters a, b, c, Å and α, β, γ,o | Space Group, V Å3, g/cm3, Z | Ref. |
|---|---|---|---|
| 1. The olivine (Fe,Mg)2(SiO4) Structure Type and Its Derivatives | |||
| Fe End Members | |||
| LiFe(PO4) (triphylite) | a = 10.332(4), b = 6.010(5), c = 4.787(4) | Pnma, 297.25 3.53, 4 | [124] |
| NaFe(PO4) (karenwebberite) | a = 10.4063(6), b = 6.2187(3), c = 4.9469(3) | Pnma, 320.13 3.61, 4 | [53] |
| NaFe(PO4) (marićite) | a = 9.001(8), b = 6.874(3), c = 5.052(4) | Pnma, 312.58 3.69, 4 | [125] |
| Fe3(PO4)2 (sarcopside) | a = 10.450(11), b = 6.033(6), c = 4.782(5) γ = 90.96(8) | P1121/a*, 301.44 3.94, 2 | [126] |
| Fe(PO4) (heterosite) | a = 9.839(1), b = 5.809(3), c = 4,776(1) | Pnma, 272.97 3.67, 4 | [127] |
| NH4Fe(PO4)HF (Fe variety of niahite) | a = 8.958(2), b = 5.649(1), c = 4.811(1) | Pnm21, 243.45 2.58, 2 | [37] |
| Mn End Members | |||
| LiMn(PO4) (lithiophilite) | a = 10.46(3), b = 6. 10(2), c = 4.744(10) | Pnma, 302.7 3.44, 4 | [128] |
| NaMn(PO4) (natrophilite) | a = 10.523(5), b = 6.312(3), c = 4.987(2) | Pnma, 330.48 3.47, 4 | [52] |
| NaMn(PO4) (Mn analogue of marićite) | a = 9.0882(1), b = 6.9041(1), c = 5.1134(1) | Pnma, 320.84 3.58, 4 | [129] |
| Mn(PO4) (purpurite) | a = 9.6237(7), b = 5.9019(3), c = 4.7711(3) | Pnma, 270.99 3.67, 4 | [130] |
| NH4Mn(PO4)(H2O) (niahite centrosymmetric variety) | a = 17.5822(3), b = 5.7310(1), c = 4.9090(1) | Pnma, 494.65 2.50, 4 | [41] |
| 2. The Tavorite, LiFe(PO4)(OH,F) Structure Type | |||
| LiFe(PO4)(OH) (tavorite) | a = 5.3528(6), b=7.2896(7), c = 5.1187(5) α= 109.359(4), β= 97.733(5), γ=106.359(5) | P, 174.98 3.22, 2 | [49] |
| LiV(PO4)F (tavorite) | a = 5.3094(2), b=7.4994(2), c =5.1688(2) α= 112.93 (2), β= 81.66(2), γ=113.12(1) | P, 174.3 3.27, 2 | [9] |
| 3. The Kosnarite, KZr2(PO4)3 (NASICON) Structure Type (Rombohedral Analogues) | |||
| Na3Fe2(PO4)3 | a = 8.7270(2), c = 21.8078(5) | Rc, 1438.38 3.22, 6 | [69] |
| Na3V2(PO4)3 | a = 8.7288(2), c = 21.8042(7) | Rc, 1438.73 3.16, 6 | [68] |
| Li3Fe2(PO4)3 | a = 8.3162(4), c = 22.459(1) | R, 1345.15 3.09, 6 | [69] |
| Na4MnV(PO4)3 | a = 8.96354(2), c = 21.448319(7) | Rc, 1494.82 3.25, 6 | [73] |
| 4. The Alluaudite, Na2Mn2+(Fe3+, Fe2+)2(PO4)3 Structure Type | |||
| Na2Fe2+(Fe2+Fe3+) (PO4)3 (ferroalluaudite) | a = 10.923(1), b = 6.500(1), c = 12.538(2) γ = 98.3(1) | I112/a **, 890.19 3.71, 4 | [79] |
| (Li,Na)MnFe2(PO4)3 | a = 11.9892(2), b =12.4927(2), c = 6.3859(1) β = 114.639(1) | C2/c, 869.34 3.56, 4 | [82] |
| Na2Fe2(SO4)3 | a = 12.65847(7), b =12.77062(7) c =6.51210(3) β = 115.5391(4) | C2/c, 949.86 3.11, 4 | [80] |
| 5. The Wyllieite, Na2Fe2+2Al(PO4)3 Structure Type | |||
| NaMn2+2.5Mn3+(PO4)3 | a = 6.5291(6), b = 12.653(1), c = 10.952(1) β = 97.18(1) | P21/c, 897.68 3.74, 4 | [86] |
| Ag1.09Mn3.46(AsO4)3 | a = 6.7470(7), b =12.9820(9), c = 11.2970(8) β = 98.85(3) | P21/c, 977.72 4.92 4 | [87] |
| Na1.25Co2+2.187Al1.125(AsO4)3 | a = 6.532(2), b =12.492(2), c = 11.060(2) β = 99.44(2) | P21/c, 890.3 4 | [88] |
| 6. The bradleyite, Na3Mg(CO3)(PO4) Structure Type | |||
| Na3Mn(CO3)(PO4), (sidorenkite) | a = 8.9859(1), b=6.73577(5), c = 5.15998(4) β = 90.123(1) | P21/m, 312.31 2.97, 2 | [94] |
| Na3Fe(CO3)(PO4), (bonshtedtite) | a = 8.9542(1), b=6.61129(5), c = 5.16023(5) β = 89.600(1) | P21/m, 305.47 3.04, 2 | [97] |
| 7. The Natisite, Na2TiSiO5 Homeotypes | |||
| Na3Fe2(OH)2F(PO4)2 | a =9.050(2), c = 10.679(2) | P42/mnm, 874.64 3.22, 8 | [100] |
| Na3Fe2F3(PO4)2 | a =6.399(1), c= 10.679(3) | I4/mmm, 429.11 3.39, 2 | [102] |
| Na3V2O2F(PO4)2 | a =6.3856(2), c = 10.6119(9) | I4/mmm, 431.05 3.17, 2 | [104] |
| 8. The Katiarsite, KTiO(AsO4) (KTP) Structure Type | |||
| KFeF(PO4) | a = 12.885(6), b = 6.370(2), c = 10.656(4) | Pna21, 874.62 3.17, 8 | [131] |
| KVF(PO4) | a = 12.8200(3), b = 6.3952(1), c = 10.6115(2) | Pna21, 869.99 3.11, 8 | [110] |
| KVO(PO4) | a = 12.816(5), b = 6.388(2), c = 10.556(5) | Pna21, 864.21 3.09, 8 | [113] |
| KTiF(PO4) | a = 13.0020(2), b = 6.43420(8), c = 10.7636(2) | Pna21, 900.45 3.01, 8 | [115] |
| 9. The Arrojadite, KNa4Ca(Fe,Mn)14Al(PO4)12(OH,F)2 Structure Type | |||
| KNa5Fe2+14Fe3+(PO4)12(OH)2 (arrojadite) | a = 16.53(2), b = 10.084(9), c = 24.64(3) β = 105.75(7) | C12/c1, 3953 3.69, 4 | [120] |
Author Contributions
Funding
Conflicts of Interest
References
- The Nobel Prize. Available online: https://www.nobelprize.org/prizes/chemistry/2019/press-release/ (accessed on 22 April 2020).
- Kubota, K.; Dahbi, M.; Hosaka, T.; Kumakura, S.; Komaba, S. Towards K-Ion and Na-Ion Batteries as “Beyond Li-Ion”. Chem. Rec. 2018, 18, 459–479. [Google Scholar] [CrossRef]
- Padhi, A.K.; Nanjundaswamy K., S.; Goodenough, J.B. Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. J. Electrochem. Soc. 1997, 144, 1188–1194. [Google Scholar] [CrossRef]
- Phosphates: Geochemical, Geobiological and Materials Importance. In Mineralogical Society of America and Geochemical Society Reviews in Mineralogy and Geochemistry; Kohn, M.J.; Rakovan, J.; Hughes, J.M. (Eds.) Walter de Gruyter GmbH & Co KG: Berlin, Germany, 2002; p. 742. [Google Scholar]
- Goodenough, J.B.; Kim, Y. Challenges for Li Rechargeable Batteries. Chem. Mater. 2010, 22, 587–603. [Google Scholar] [CrossRef]
- Mizushima, K.; Jones, P.C.C.; Wiseman, P.J.; Goodenough, J.B. LixCoO2 (0<x<−1): A new cathode material for batteries of high energy density. Mater. Res. Bull. 1980, 15, 783–789. [Google Scholar]
- Whittingham, M.S. Ultimate Limits to Intercalation Reactions for Lithium Batteries. Chem. Rev. 2014, 114, 11414–11443. [Google Scholar] [CrossRef]
- Masquelier, C.; Croguennec, L. Polyanionic (Phosphates, Silicates, Sulfates) Frameworks as Electrode Materials for Rechargeable Li (or Na) Batteries. Chem. Rev. 2013, 113, 6552–6591. [Google Scholar] [CrossRef] [PubMed]
- Antipov, E.V.; Khasanova, N.R.; Fedotov, S.S. Perspectives on Li and transition metal fluoride phosphates as cathode materials for a new generation of Li-ion batteries. IUCr J. 2015, 2, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Larcher, D.; Tarascon, J.-M. Towards greener and more sustainable batteries for electrical energy storage. Nat. Chem. 2015, 7, 19–29. [Google Scholar] [CrossRef]
- Kim, H.; Kim, J.C.; Bianchini, M.; Seo, D.-H.; Rodriguez-Garcia, J.; Ceder, G. Recent Progress and Perspective in Electrode Materials for K-Ion Batteries. Adv. Energy Mater. 2018, 8, 1702384. [Google Scholar] [CrossRef] [Green Version]
- London, D.; Kontak, D.J. Granitic Pegmatites: Scientific wonders and economic bonanzas. Elements 2012, 8, 257–261. [Google Scholar] [CrossRef]
- Moore, P.B. Pegmatite phosphates: Descriptive mineralogy and crystal chemistry. Mineral. Rec. 1973, 4, 103–130. [Google Scholar]
- Fisher, D.G. Geochemistry of Phosphorus Containing Minerals. In Environmental Phosphorus Handbook; Griffith, E., Beeton, A., Spenser, J., Eds.; Wiley: New York, NY, USA, 1973. [Google Scholar]
- Černý, P.; Ercit, S. The classification of granitic pegmatites revisited. Can. Mineral. 2005, 43, 2005–2026. [Google Scholar] [CrossRef] [Green Version]
- Keller, P.; Fransolet, A.-M.; Fontan, F. Triphylite–lithiophilite and triplite–zwieselite in granitic pegmatites: Their textures and genetic relationships. N. Jahrb. Mineral. Abh. 1994, 168, 127–145. [Google Scholar]
- Moore, P.B. Crystallochemical Aspects of the Phosphate Minerals. In Phosphate Minerals; Nriagu, J.O., Moore, P.B., Eds.; Springer: Berlin/Heidelberg, Germany, 1984; pp. 155–170. [Google Scholar]
- Moore, P.B. The primary pegmatite phosphate minerals. The mineralogy of pegmatites, Вrown, G.Е., Jr. Аmer. Miner. 1982, 67, 180–189. [Google Scholar]
- Mücke, A. The paragenesis of the phosphate minerals of the Hagendorf pegmatite—A general view. Chem. Erde. 1981, 40, 217–237. [Google Scholar]
- Sandomirskii, P.A.; Belov, N.V. Crystal Chemistry of Mixed Anionic Radicals; Nauka: Moscow, Russia, 1984. (In Russian) [Google Scholar]
- Yakubovich, O.V.; Urusov, V.S. The genetic crystal chemistry of pegmatite phosphates. Mosc. Univ. Geol. Bull. 1996, 51, 18–40. [Google Scholar]
- Yakubovich, O.V. Phosphates with amphoteric oxo-complexes: From structural features to genetic conclusions. Z. Krist. 2008, 223, 126–131. [Google Scholar] [CrossRef]
- Yakubovich, O.V. Phosphates with Amphoteric Oxocomplexes: Crystal Chemical Features and Expected Physical Properties In Minerals as Advanced Materials I; Krivovichev, S., Ed.; Springer: Berlin/Heidelberg, Germany, 2008; p. 255. [Google Scholar]
- Shigley, J.E.; Brown, G.E. Lithiophilite formation in granitic pegmatites: A reconnaissance experimental study of phosphate crystallization from hydrous aluminosilicate melts. Am. Mineral. 1986, 71, 356–366. [Google Scholar]
- Eventoff, W.; Martin, R.; Peacor, D.R. The crystal structure of heterosite. Am. Mineral. 1972, 57, 45–51. [Google Scholar]
- Iakubovich, O.V.; Simonov, M.A.; Belov, N.V. Crystal structure of synthetic triphyline LiFe[PO4]. Dokl. Akad. Nauk SSSR 1977, 235, 93–95. [Google Scholar]
- Ravet, N.; Chouinard, Y.; Magnan, J.F.; Besner, S.; Gauthier, M.; Armand, M. Electroactivity of natural and synthetic triphilyte. J. Power Sources 2001, 97–98, 503–507. [Google Scholar] [CrossRef]
- Huang, H.; Yin, S.C.; Nazar, L.F. Approachim theoretical capacity of LiFePO4 at room temperature at high rates. Electrochem. Solid State Lett. 2001, 4, A170–A172. [Google Scholar] [CrossRef]
- Dominko, R.; Gaberšček, M.; Drofenik, J.; Bele, M.; Pejovnik, S. A Novel Coating Technology for Preparation of Cathodes in Li-Ion Batteries. Electrochem. Solid State Lett. 2001, 4, A187–A190. [Google Scholar] [CrossRef]
- Chung, S.-J.; Bloking, J.T.; Chiang, Y.M. Electronically conductive phospho-olivines as lithium storage electrodes. Nat. Mat. 2002, 1, 123–128. [Google Scholar] [CrossRef]
- Morgan, D.; Van der Ven, A.; Ceder, G. Li Conductivity in LixMPO4 (M =Mn, Fe, Co, Ni) Olivine Materials. Electrochem. Solid-State Lett. 2004, 7, A30–A32. [Google Scholar] [CrossRef]
- Islam, M.S.; Driscoll, D.J.; Fisher, C.A.; Slater, P.R. Atomic-Scale Investigation of Defects, Dopants, and Lithium Transport in the LiFePO4 Olivine-Type Battery Material. Chem. Mater. 2005, 17, 5085–5092. [Google Scholar] [CrossRef]
- Nishimura, S.; Kobayashi, G.; Ohoyama, K.; Kanno, K.; Yashima, M.; Yamada, A. Experimental visualization of lithium diffusion in LixFePO4. Nat. Mater. 2008, 7, 707–711. [Google Scholar] [CrossRef]
- Qin, X.; Wang, X.; Xiang, H.; Xie, J.; Li, J.; Zhou, Y. Mechanism for Hydrothermal Synthesis of LiFePO4 Platelets as Cathode Material for Lithium-Ion Batteries. J. Phys. Chem. C 2010, 114, 16806–16812. [Google Scholar] [CrossRef]
- Sharikov, F.Y.; Drozhzhin, O.A.; Sumanov, V.D.; Baranov, A.N.; Abakumov, A.M.; Antipov, E.V. Exploring the Peculiarities of LiFePO4 Hydrothermal Synthesis Using In Situ Calvet Calorimetry. Cryst. Growth Des. 2018, 18, 879–882. [Google Scholar] [CrossRef]
- Ravnsbæk, D.B.; Xiang, K.; Xing, W.; Borkiewicz, O.J.; Wiaderek, K.M.; Gione, P.; Chapman, K.W.; Chupas, P.J.; Chiang, Y.-M. Extended Solid Solutions and Coherent Transformations in Nanoscale Olivine Cathodes. Nano Lett. 2014, 14, 1484–1491. [Google Scholar]
- Yakubovich, O.V.; Karimova, O.V.; Melnikov, O.K.; Urusov, V.S. Crystal structure of (NH4)(FePO4)HF-niahite synthetic variant. Dokl. Akad. Nauk SSSR 1995, 342, 40–44. [Google Scholar]
- Yakubovich, O.V.; Karimova, O.V.; Dimitrova, O.V.; Mass, A.W. Layer structure of (NH4)CoPO4 H2O. Acta Cryst. C 1999, 55, 151–153. [Google Scholar] [CrossRef]
- Bridge, P.J.; Robinson, B.W. Niahite—a new mineral from Malaysia. Mineral. Mag. 1983, 47, 79–80. [Google Scholar] [CrossRef]
- Livage, J. Chimie douce: From shake-and-bake processing to wet chemistry. New J. Chem. 2001, 25, 1. [Google Scholar] [CrossRef]
- Kiriukhina, G.V.; Yakubovich, O.V.; Dimitrova, O.V. Crystal structure of a new polymorphic modification of niahite, NH4MnPO4 H2O. Cryst. Rep. 2015, 60, 198–203. [Google Scholar] [CrossRef]
- Wu, C.; Xie, J.; Cao, G.; Zhao, X.; Zhang, S. Ordered LiMPO4 (M = Fe, Mn) nanorods synthesized from NH4MPO4·H2O microplates by stress involved ion exchange for Li-ion batteries. Cryst. Eng. Comm. 2014, 16, 2239–2245. [Google Scholar] [CrossRef]
- Bramnik, N.; Ehrenberg, H. Precursor-based synthesis and electrochemical performance of LiMnPO4. J. Alloys Compd. 2008, 464, 259–264. [Google Scholar] [CrossRef]
- Alyoshin, V.A.; Pleshakov, E.A.; Ehrenberg, H.; Michailova, D. Platelike LiMPO4 (M=Fe, Mn, Co, Ni) for Possible Application in Rechargeable Li Ion Batteries: Beyond Nanosize. J. Phys. Chem. C 2014, 118, 17426–17435. [Google Scholar] [CrossRef]
- Yakubovich, O.V.; Bairakov, V.V.; Simonov, M.A. Crystal structure of simferite Li(Mg,Fe3+,Mn3+)2[PO4]2. Soviet Physics Doklady. 1989, 34, 669–671. [Google Scholar]
- Omenya, F.; Miller, J.K.; Fang, J.; Wen, B.; Zhang, R.; Wang, Q.; Chernova, N.A.; Whittingham, M.S. Single-Phase Lithiation and Delithiation of Simferite Compounds Li(Mg,Mn,Fe)PO4. Chem. Mater. 2014, 26, 6206–6212. [Google Scholar] [CrossRef]
- Moore, P.B. Crystal chemistry of the basic iron phosphates. Am. Mineral. 1970, 55, 135–169. [Google Scholar]
- Yakubovich, O.V.; Urusov, V.S. The structure and electron density of Fe2+-bearing tavorite in relation to the genetic crystal chemistry of secondary phosphates of lithium pegmatites. Geochem. Intern. 1997, 35, 630–638. [Google Scholar]
- Marx, N.; Croguennec, L.; Carlier, D.; Wattiaux, A.; Le Cras, F.; Suard, E.; Delmas, C. The structure of tavorite LiFePO4(OH) from diffraction and GGA + U studies and its preliminary electrochemical characterization. Dalton Trans. 2010, 39, 5108–5116. [Google Scholar] [CrossRef]
- Barker, J.; Gover, R.K.B.; Burns, P.; Bryan, A. LiVPO4F: A new active material for safe lithium-ion batteries. Electrochem. Solid-State Lett. 2005, 8, A285–A287. [Google Scholar] [CrossRef]
- Vignola, P.; Hatert, F.; Fransolet, A.-M.; Medenbach, O.; Diella, V.; Andò, S. Karenwebberite, Na(Fe2+,Mn2+)PO4, a new member of the triphylite group from the Malpensata pegmatite, Lecco Province, Italy. Am. Mineral. 2013, 98, 767–772. [Google Scholar] [CrossRef]
- Moore, P.B. Natrophilite, NaMn(PO4), has ordered cations. Am. Mineral. 1972, 57, 1333–1344. [Google Scholar]
- Moreau, P.; Guyomard, D.; Gaubicher, J.; Boucher, F. Structure and Stability of Sodium Intercalated Phases in Olivine FePO4. Chem. Mater. 2010, 22, 4126–4128. [Google Scholar] [CrossRef]
- Yabuuchi, N.; Kubota, K.; Dahbi, M.; Komaba, S. Research Development on Sodium-Ion Batteries. Chem. Rev. 2014, 114, 11636–11682. [Google Scholar] [CrossRef]
- Casas-Cabanas, M.; Roddatis, V.V.; Saurel, D.; Kubiak, P.; Carretero-Gonzalez, J.; Palomares, V.; Serras, P.; Rojo, T. Crystal chemistry of Na insertion/deinsertion in FePO4–NaFePO4. J. Mater. Chem. 2012, 22, 1742–17423. [Google Scholar] [CrossRef]
- Le Page, Y.; Donnay, G. The crystal structure of the new mineral marićite, NaFePO4. Can. Mineral. 1977, 15, 518–521. [Google Scholar]
- Yakubovich, O.V.; Urusov, V.S. Electron density distribution in lithiophosphatite Li3PO4: Crystallochemical features of orthophosphates with hexagonal close packing. Cryst. Rep. 1997, 42, 261–268. [Google Scholar]
- Corlett, M.I.; Armbruster, T. Structural relations between marićite and natrophilite in the system NaFePO4-NaMnPO4. GAC-MAC Join Annu. Meet. 1979, 4, 1979. [Google Scholar]
- Kim, J.; Seo, D.-H.; Kim, H.; Park, I.; Yoo, Y.-K.; Jung, S.-K.; Park, Y.-U.; Goddard, W.A.; Kang, K. Unexpected discovery of low-cost maricite NaFePO4 as a high-performance electrode for Na-ion batteries. Energy Environ. Sci. 2015, 8, 540–545. [Google Scholar] [CrossRef] [Green Version]
- Moore, P.B. Sarcopside: Its atomic arrangement. Am. Mineral. 1972, 57, 24–35. [Google Scholar]
- Omenya, F.; Chernova, N.A.; Zhang, R.; Fang, J.; Huang, Y.; Cohen, F.; Dobrzynski, N.; Senanayake, S.; Xu, W.; Whittingham, M.S. Why Substitution Enhances the Reactivity of LiFePO4. Chem. Mater. 2013, 25, 85–89. [Google Scholar] [CrossRef]
- Janssen, Y.; Santhanagopalan, D.; Qian, D.; Chi, M.; Wang, X.; Hoffmann, C.; Meng, Y.S.; Khalifah, P. Reciprocal Salt Flux Growth of LiFePO4 Single Crystals with Controlled Defect Concentrations. Chem. Mater. 2013, 25, 4574–4584. [Google Scholar] [CrossRef]
- Crouzet, C.; Recham, N.; Brunet, F.; Findling, N. A novel route for FePO4 olivine synthesis from sarcopside oxidation. Solid State Sci. 2016, 62, 29–33. [Google Scholar] [CrossRef]
- Bnownfield, M.-E.; Foord, E.E.; Sutley, S.J.; Botinelly, T. Kosnarite, KZr2(PO4)3, a new mineral from Mount Mica and Black Mountain, Oxford County, Maine. Am. Mineral. 1993, 78, 653–656. [Google Scholar]
- Šljukić, M.; Matković, B.; Prodić, B.; Anderson, D. The crystal structure of KZr2(PO4)3. Z. Krist. 1969, 130, 148–161. [Google Scholar] [CrossRef]
- D’Yvoire, F.; Pintard-Screpel, M.; Bretey, E.; De la Roche`re, M. Phase transitions and ionic conduction in 3D skeleton phosphates A3M2(PO4)3: A = Li, Na, Ag, K; M = Cr, Fe. Solid State Ionics 1983, 9–10, 851–857. [Google Scholar] [CrossRef]
- Gaubicher, J.; Wurm, C.; Goward, G.; Masquelier, C.; Nazar, L.F. Rhombohedral Form of Li3V2(PO4)3 as a Cathode in Li-Ion Batteries. Chem. Mater. 2000, 12, 3240–3242. [Google Scholar] [CrossRef]
- Zatovsky, I.V. NASICON-type Na3V2(PO4)3. Acta Cryst. 2010, E66, i12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masquelier, C.; Wurm, C.; Rodriguez-Carvajal, J.; Gaubicher, J.; Nazar, L.F. A Powder Neutron Diffraction Investigation of the Two Rhombohedral NASICON Analogues: γ-Na3Fe2(PO4)3 and Li3Fe2(PO4)3. Chem. Mater. 2000, 12, 522–532. [Google Scholar] [CrossRef]
- Chen, S.; Wu, C.; Shen, L.; Zhu, C.; Huang, Y.; Xi, K.; Maier, J.; Yu, Y. Challenges and Perspectives for NASICON-Type Electrode Materials for Advanced Sodium-Ion Batteries. Adv. Mater. 2017, 29, 170043. [Google Scholar] [CrossRef]
- Jian, Z.; Han, W.; Liang, Y.; Lan, Y.; Fang, Z.; Hu, Y.-S.; Yao, Y. Carbon-coated rhombohedral Li3V2(PO4)3 as both cathode and anode materials for lithium-ion batteries: Electrochemical performance and lithium storage mechanism. J. Mater. Chem. A 2014, 2, 20231–20236. [Google Scholar] [CrossRef]
- Zhou, W.; Xue, L.; Lü, X.; Gao, H.; Li, Y.; Xin, S.; Fu, G.; Cui, Z.; Zhu, Y.; Goodenough, J.B. NaxMV(PO4)3 (M = Mn, Fe, Ni) Structure and Properties for Sodium Extraction. Nano Lett. 2016, 16, 7836–7841. [Google Scholar] [CrossRef]
- Chen, F.; Kovrugin, V.M.; David, R.; Mentré, O.; Fauth, F.; Chotard, J.-N.; Masquelier, C. A NASICON-Type Positive Electrode for Na Batteries with High Energy Density: Na4MnV(PO4)3. Small Methods 2018, 3, 1800218. [Google Scholar] [CrossRef]
- Zakharkin, M.V.; Drozhzhin, O.A.; Tereshchenko, I.V.; Chernyshov, D.; Abakumov, A.M.; Antipov, E.V.; Stevenson, K.J. Enhancing Na+ Extraction Limit through High Voltage Activation of the NASICON-Type Na4MnV(PO4)3 Cathode. Appl. Energy Mater. 2018, 1, 5842–5846. [Google Scholar] [CrossRef]
- Mindat.org. Available online: https://www.mindat.org/min-142.html (accessed on 22 April 2020).
- Moore, P.B. Crystal chemistry of the alluaudite structure type: Contribution to the paragenesis of pegmatite phosphate giant crystals. Am. Mineral. 1971, 56, 1955–1975. [Google Scholar]
- Hatert, F. The crystal chemistry of lithium in the alluaudite structure: A study of the (Na1-xLix)1.5Mn1.5Fe1.5(PO4)3 solid solution (x=0 to 1). Mineral. Petrol. 2004, 81, 205–217. [Google Scholar] [CrossRef]
- Hatert, F.; Ottolini, L.; Schmid-Beurmann, P. Experimental investigation of the alluaudite+triphlyite assemblage, and development of the Na-in-triphylite geothermometer: Application to natural pegmatite phosphates. Contrib. Mineral. Petrol. 2011, 161, 531–546. [Google Scholar] [CrossRef]
- Iakubovich, O.V.; Simonov, M.A.; Egorov-Tismenko, I.K.; Belov, N.V. Crystal structure of Na2(Fe0.53+Fe0.52+)2Fe2+[PO4]3-synthetic variety of alluaudite. Dokl. Akad. Nauk SSSR. 1977, 236, 1123–1126. [Google Scholar]
- Barpanda, P.; Oyama, G.; Nishimura, S.I.; Chung, S.C.; Yamada, A. A 3.8-V earth- abundant sodium battery electrode. Nat. Commun. 2014, 5, 4358. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Yang, K.; Song, L.; Feng, H.-J.; Gao, J. An alluaudite-type sodium-ion battery cathode candidate Na2Mn2V(PO4)3: Crystal growth, preparation, structure and electrochemical properties. J. Alloys Compd. 2019, 783, 409–415. [Google Scholar] [CrossRef]
- Tard, K.; Carlier, D.; Croguennec, L.; Wattiaux, A.; Ben Amara, M.; Delmas, C. Structural study of the Li(0.5)Na(0.5)MnFe2(PO4)3 and Li(0.75)Na(0.25)MnFe2(PO4)3 alluaudite phases and their electrochemical properties as positive electrodes in lithium batteries. Inorg. Chem. 2010, 49, 10378–10389. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.B.; Ito, J. Wyllieite, Na2Fe2+2Al[PO4]3, a new species. Mineral. Rec. 1973, 4, 131–136. [Google Scholar]
- Moore, P.B.; Ito, J. Alluaudites, wyllieites, arrojadites: Crystal chemistry and nomenclature. Mineral. Mag. 1979, 6, 227–235. [Google Scholar] [CrossRef]
- Yang, H.; Downs, R.T.; Gu, X.; Xie, X.; Kobsch, A. Fupingqiuite, IMA 2016-087. CNMNC Newsletter No. 35, February 2017, 211. Mineral. Mag. 2017, 81, 209–213. [Google Scholar]
- Yakubovich, O.V.; Massa, W.; Gavrilenko, P.; Dimitrova, O. The crystal structure of new synthetic member in the wyllieite group: Na1.265Mn2+2.690Mn3+0.785(PO4)3. Eur. J. Mineral. 2005, 17, 741–747. [Google Scholar] [CrossRef]
- Frigui, W.; Zid, M.F.; Driss, A. Wyllieite-type Ag1.09Mn3.46(AsO4)3. Acta Cryst. 2012, E68, i40–i41. [Google Scholar] [CrossRef]
- Marzouki, R.; Ben Smida, Y.; Avdeev, M.; Alghamdi, M.M.; Zid, M.F. Synthesis, structure and Na+ migration pathways of new Wylleite-type Na1.25Co2.187Al1.125(AsO4)3. Mater. Res. Express 2019, 6, 126313. [Google Scholar] [CrossRef]
- Khomyakov, A.P.; Semenov, E.I.; Kazakova, M.E.; Shumyatskaya, N.G. Sidorenkite, Na3Mn(PO4)(CO3), a new mineral. Zapiski VMO 1979, 108, 56–59. (In Russian) [Google Scholar] [CrossRef]
- Khomyakov, A.P.; Aleksandrov, V.V.; Krasnova, N.I.; Ermilov, V.V.; Smolyaninova, V.V. Bonshtedtite, Na3Fe(PO4)(CO3), a new mineral. Zapiski VMO 1982, 111, 486–490. (In Russian) [Google Scholar] [CrossRef]
- Kurova, T.A.; Shumyatskaya, N.G.; Voronkov, A.A.; Pyatenko, Y.A. Crystal structure of sidorenkite Na3Mn(PO4)(CO3). Dokl. Akad. Nauk. SSSR 1980, 251, 605–607. [Google Scholar]
- Krivovichev, S.V.; Chernyatieva, A.P.; Britvin, S.N.; Yakovenchuk, V.N.; Krivovichev, V.G. Refinement of the crystal structure of bonshtedtite, Na3Fe(PO4)(CO3). Geol. Ore Depos. 2013, 55, 669–675. [Google Scholar] [CrossRef]
- Hautier, G.; Jain, A.; Chen, H.; Moore, C.; Ong, S.P.; Ceder, G. Novel mixed polyanions lithium-ion battery cathode materials predicted by high-throughput ab initio computations. J. Mater. Chem. 2011, 21, 17147–17153. [Google Scholar] [CrossRef]
- Chen, H.; Hao, Q.; Zivkovic, O.; Hautier, G.; Du, L.-S.; Tang, Y.; Hu, Y.-Y.; Ma, X.; Grey, C.P.; Ceder, G. Sidorenkite (Na3MnPO4CO3): A New Intercalation Cathode Material for Na-Ion Batteries. Chem. Mater. 2013, 25, 2777–2787. [Google Scholar] [CrossRef]
- Wang, C.; Sawicki, M.; Emani, S.; Liu, C.; Shaw, L.L. Na3MnCO3PO4–A High Capacity, Multi-Electron Transfer Redox Cathode Material for Sodium Ion Batteries. Electrochim. Acta 2015, 161, 322–328. [Google Scholar] [CrossRef]
- Hassanzadeh, N.; Sadrnezhaad, S.K.; Chen, G. In-situ hydrothermal synthesis of Na3MnCO3PO4/rGO hybrid as a cathode for Na-ion battery. Electrochim. Acta 2016, 208, 188–194. [Google Scholar] [CrossRef]
- Huang, W.; Zhou, J.; Li, B.; Ma, J.; Tao, S.; Xia, D.; Chu, W.; Wu, Z. Detailed investigation of Na2.24FePO4CO3 as a cathode material for Na-ion batteries. Sci. Rep. 2014, 4, 4188. [Google Scholar] [CrossRef]
- Men’shikov, Y.P.; Pakhomovskii, Y.A.; Goiko, E.A.; Bussen, I.V.; Mer´kov, A.N. A natural tetragonal titanosilicate of sodium, natisite. Zapiski VMO 1975, 104, 314–317. (In Russian) [Google Scholar]
- Nyman, H.; O’Keeffe, M.; Bovin, J.O. Sodium titanium silicate, Na2TiSiO5. Acta Cryst. 1978, B34, 905–906. [Google Scholar] [CrossRef]
- Yakubovich, O.V.; Simonov, M.A.; Melnikov, O.K. The mixed Fe-P framework in the crystal structure of Na3Fe23+[PO4]2(OH)2F. Kristallografiya 1984, 29, 484–488. [Google Scholar]
- Slobodyanik, N.S.; Nagornyi, P.C.; Kornienko, Z.I.; Kapshuk, A.A.; Mitkevich, V.V. Preparation and crystal structure of Na3Cr2F3(PO4)2. Russ. J. Inorg. Chem. 1989, 34, 1413–1414. [Google Scholar]
- Yakubovich, O.V.; Melnikov, O.K.; Urusov, V.S. The framework of mixed nature in Na3{Fe2F3[PO4]2} crystal structure-a new homeotype in the series of natisite. Dokl. Akad. Nauk SSSR 1995, 342, 615–620. [Google Scholar]
- Yakubovich, O.V.; Melnikov, O.K. Anionic framework of a mixed type in Na3{Al2F3[PO4]2} crystalline structure. Kristallografiya 1996, 41, 663–668. [Google Scholar]
- Massa, W.; Yakubovich, O.V.; Dimitrova, O.V. Crystal structure of a new sodium vanadyl(IV) fluoride phosphate Na3{V2O2F[PO4]2}. Solid State Sci. 2002, 4, 495–501. [Google Scholar] [CrossRef]
- Serras, P.; Palomares, V.; Goni, A.; Gil de Muro, I.; Kubiak, P.; Lezama, L.; Rojo, T. High voltage cathode materials for Na-ion batteries of general formula Na3V2O2x(PO4)2F3−2x. J. Mater. Chem. 2012, 22, 22301–22308. [Google Scholar] [CrossRef]
- Park, Y.U.; Seo, D.H.; Kwon, H.S.; Kim, B.; Kim, J.; Kim, H.; Kim, I.; Yoo, H.-I.; Kang, K. A new high-energy cathode for a Na-ion battery with ultrahigh stability. J. Am. Chem. Soc. 2013, 135, 13870–13878. [Google Scholar] [CrossRef]
- Broux, T.; Bamine, T.; Fauth, F.; Simonelli, L.; Olszewski, W.; Marini, C.; Meneétrier, M.; Carlier, D.; Masquelier, C.; Croguennec, L. Strong impact of the oxygen content in Na3V2(PO4)2F3-yOy (0 ≤ y ≤0.5) on its structural and electrochemical properties. Chem. Mater. 2016, 28, 7683–7692. [Google Scholar] [CrossRef] [Green Version]
- Pekov, I.V.; Yapaskurt, V.O.; Britvin, S.N.; Zubkova, N.V.; Vigasina, M.F.; Sidorov, E.G. New arsenate minerals from the Arsenatnaya fumarole, Tolbachik volcano, Kamchatka, Russia. V. Katiarsite, KTiO(AsO4). Miner. Mag. 2016, 80, 639–646. [Google Scholar] [CrossRef]
- Pekov, I.V.; Zubkova, N.V.; Agakhanov, A.A.; Yapaskurt, V.O.; Belakovskiy, D.I.; Vigasina, M.F.; Britvin, S.N.; Turchkova, A.G.; Sidorov, E.G.; Pushcharovsky, D.Y. Yurgensonite, IMA 2019-059. CNMNC Newsletter No. 52. Mineral. Mag. 2019, 83. [Google Scholar] [CrossRef] [Green Version]
- Recham, N.; Gwenaelle, R.; Sougrati, M.T.; Chotard, J.-N.; Frayret, C.; Mariyappan, S.; Melot, B.C.; Jumas, J.-C.; Tarascon, J.-M. Preparation and Characterization of a Stable FeSO4F-Based Framework for Alkali Ion Insertion Electrodes. Chem. Mater. 2012, 24, 4363–4370. [Google Scholar] [CrossRef]
- Fedotov, S.S.; Khasanova, N.R.; Samarin, A.S.; Drozhzhin, O.A.; Batuk, D.; Karakulina, O.M.; Hadermann, J.; Abakumov, A.M.; Antipov, E.V. AVPO4F (A = Li, K): A 4 V Cathode Material for High-Power Rechargeable Batteries. Chem. Mater. 2016, 28, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Nikitina, V.A.; Fedotov, S.S.; Vassiliev, S.Y.; Samarin, A.S.; Khasanova, N.R.; Antipov, E.V. Transport and Kinetic Aspects of Alkali Metal Ions Intercalation into AVPO4F Framework. J. Electrochem. Soc. 2017, 164, A6373–A6380. [Google Scholar] [CrossRef]
- Chihara, K.; Katogi, A.; Kubota, K.; Komaba, S. KVPO4F and KVOPO4 toward 4 volt-class potassium-ion batteries. Chem. Commun. 2017, 53, 5208–5211. [Google Scholar] [CrossRef]
- Kim, H.; Seo, D.-H.; Bianchini, M.; Clément, R.J.; Kim, H.; Kim, J.C.; Tian, Y.; Shi, T.; Yoon, W.-S.; Ceder, C. A new strategy for high-voltage cathodes for K-ion batteries: Stoichiometric KVPO4F. Adv. Energy Mater. 2018, 8, 1801591. [Google Scholar] [CrossRef] [Green Version]
- Fedotov, S.S.; Luchinin, N.D.; Aksyonov, D.A.; Morozov, A.V.; Ryazantsev, S.V.; Gaboardi, M.; Plaisier, J.R.; Stevenson, K.J.; Abakumov, A.M.; Antipov, E.V. Titanium-based potassium-ion battery positive electrode with extraordinarily high redox potential. Nat. Commun. 2020, 11, 1484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Headden, W.P. A phosphate near triphylite from the Black Hills. Amer. J. Sci. 1891, 41, 416–417. [Google Scholar]
- Krutik, V.M.; Puscharovskii, D.Y.; Pobedimskaya, E.A.; Belov, N.V. Crystal structure of arrojadite. Soy. Phys.-Crystallogr. 1979, 24, 425–429. [Google Scholar]
- Moore, P.B.; Araki, T.; Merlino, S.; Mellini, M.; Zanazzi, P.F. The arrojadite-dickinson series, KNa4Ca(Fe,Mn)2+14Al(OH)2(PO4)12: Crystal structure and crystal chemistry. Am. Mineral. 1981, 66, 1034–1049. [Google Scholar]
- Kallfaß, C.; Hoch, C.; Schier, H.; Simon, A.; Schubert, H. The transition metal-rich orthophosphate arrojadite with special structural features. Z. Naturforsch. B 2010, 65, 1427–1433. [Google Scholar] [CrossRef]
- Yakubovich, O.V.; Matvienko, E.N.; Simonov, M.A.; Mel’nikov, O.K. Crystal structure of synthetic Fe3+-arrojadite with ideal formula of K2Na5Fe2+14Fe3+[PO4]12(OH)2. Mosc. Univ. Geol. Bull. 1986, 41, 35–47. [Google Scholar]
- Hoch, C. Vom Mineral lernen. Nachr. Chem. 2012, 60, 1181–1184. [Google Scholar] [CrossRef]
- Fedotov, S.S.; Aksyonov, D.A.; Samarin, A.S.; Karakulina, O.M.; Hadermann, J.; Stevenson, K.J.; Khasanova, N.R.; Abakumov, A.M.; Antipov, E.V. Tuning the Crystal Structure of A2CoPO4F (A = Li, Na) Fluoride-Phosphates: A New Layered Polymorph of LiNaCoPO4F. Eur. J. Inorg. Chem. 2019, 4365–4372. [Google Scholar] [CrossRef]
- Yakubovich, O.V.; Mass, W.; Kireev, V.V.; Urusov, V.S. Cation distribution in the crystal structure of Li2.32Fe0.563+[PO4]F-a synthetic structural analog of grandidierite. Dokl. Phys. 2006, 51, 474–480. [Google Scholar] [CrossRef]
- Yakubovich, O.V.; Belokoneva, E.L.; Tsirelson, V.G.; Urusov, V.S. Electron density distribution in synthetic triphylite LiFePO4. Mosc. Univ. Geol. Bull. 1990, 45, 93–99. [Google Scholar]
- Yakubovich, O.V.; Belokoneva, E.L.; Tsirelson, V.G.; Urusov, V.S. Electron density distribution and chemical bond in maricite NaFePO4. Mosc. Univ. Geol. Bull. 1992, 47, 46–56. [Google Scholar]
- Yakubovich, O.V.; Belokoneva, E.L.; Tsirelson, V.G.; Urusov, V.S. Distribution pattern of electronic density in Fe-sarcopside crystals Fe3(PO4)2. Mosc. Univ. Geol. Bull. 1991, 46, 77–84. [Google Scholar]
- Delacourt, C.; Rodríguez-Carvajal, J.; Schmitt, B.; Tarascon, J.-M.; Masquelier, C. Crystal chemistry of the olivine-type LixFePO4 system (0 ≤ x ≤ 1) between 25 and 370 °C. Solid State Sci. 2005, 7, 1506–1516. [Google Scholar] [CrossRef]
- Geller, S.; Durand, J.L. Refinement of the structure of LiMnPO4. Acta Cryst. 1960, 13, 325–331. [Google Scholar] [CrossRef] [Green Version]
- Moring, J.; Kostiner, E. The crystal structure of NaMnPO4. J. Solid State Chem. 1986, 61, 379–383. [Google Scholar] [CrossRef]
- Mishima, Y.; Hojo, T.; Nishio, T.; Sadamura, H.; Oyama, N.; Moriyoshi, C.; Kuroiwa, Y. MEM charge density study of olivine LiMPO4 and MPO4 (M = Mn, Fe) as cathode materials for lithium-ion batteries. J. Phys. Chem. C 2013, 117, 2608–2615. [Google Scholar] [CrossRef]
- Matvienko, E.N.; Iakubovich, O.V.; Simonov, M.A.; Belov, N.V. Crystal structure of K, Fe3+-orthophosphate, KFe[PO4]F. Dokl. Akad. Nauk SSSR 1979, 246, 875–878. [Google Scholar]















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yakubovich, O.; Khasanova, N.; Antipov, E. Mineral-Inspired Materials: Synthetic Phosphate Analogues for Battery Applications. Minerals 2020, 10, 524. https://doi.org/10.3390/min10060524
Yakubovich O, Khasanova N, Antipov E. Mineral-Inspired Materials: Synthetic Phosphate Analogues for Battery Applications. Minerals. 2020; 10(6):524. https://doi.org/10.3390/min10060524
Chicago/Turabian StyleYakubovich, Olga, Nellie Khasanova, and Evgeny Antipov. 2020. "Mineral-Inspired Materials: Synthetic Phosphate Analogues for Battery Applications" Minerals 10, no. 6: 524. https://doi.org/10.3390/min10060524
APA StyleYakubovich, O., Khasanova, N., & Antipov, E. (2020). Mineral-Inspired Materials: Synthetic Phosphate Analogues for Battery Applications. Minerals, 10(6), 524. https://doi.org/10.3390/min10060524