KIMERA: A Kinetic Montecarlo Code for Mineral Dissolution



Abstract
:1. Introduction
2. The Reversible Kinetic Monte Carlo Model for Mineral Dissolution
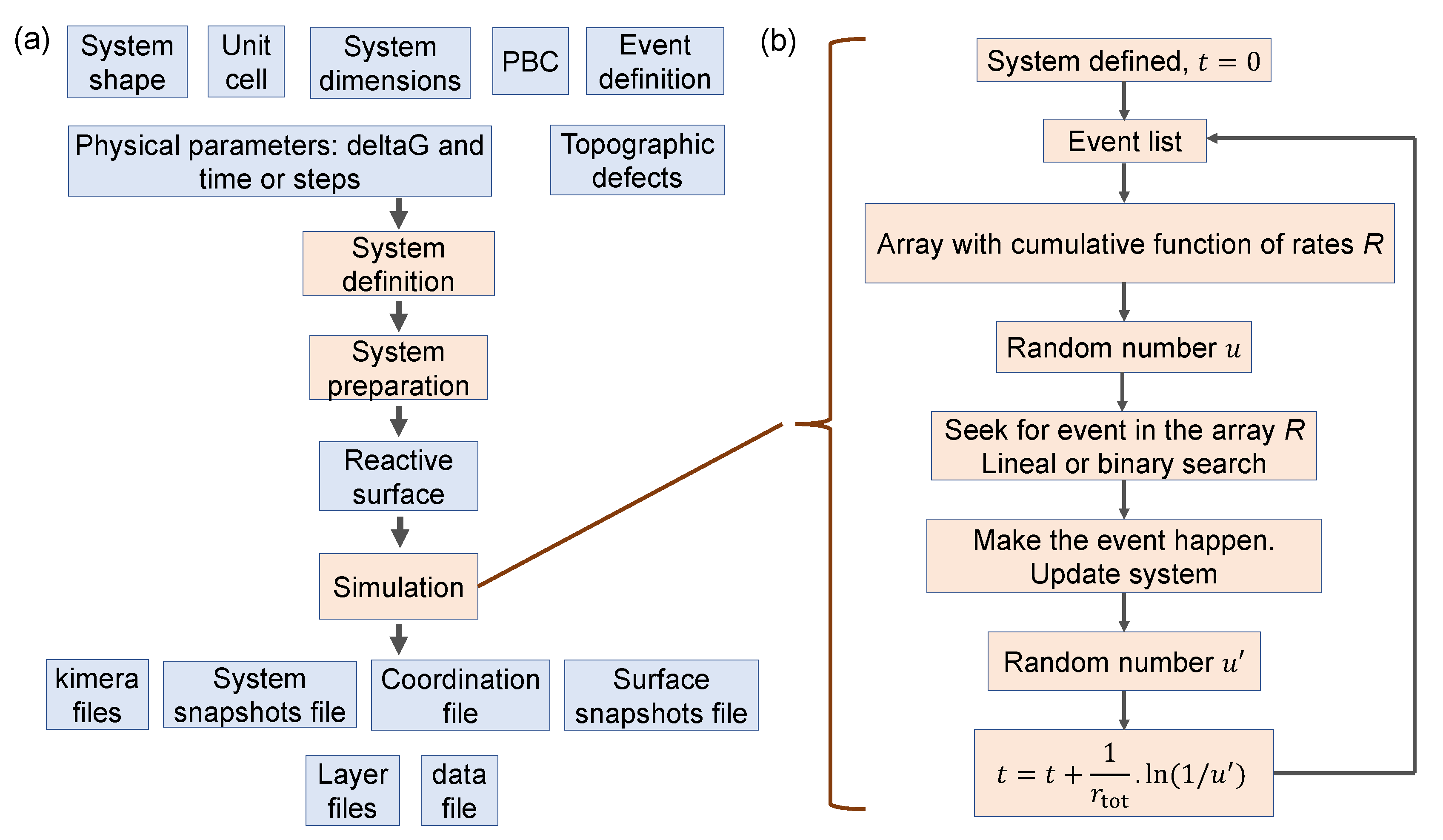
3. The KIMERA Code
3.1. Operation
3.1.1. System Definition
- The mineral structure. The program can either read it from a standard ‘.xyz’ file, or can be defined by commands, or a combination of both. The ‘.xyz’ file [43] is easily obtained by tools such as VESTA [44] from downloadable ‘.cif’ files in mineralogical databases [45]. In principle KIMERA is thought to construct mineral surfaces replicating a small unit cell. Nevertheless, it is also possible to define a complex system within a ‘.xyz’ file and threat it as the whole system box. Coarse grained systems can be also simulated.
- The system dimensions. The program replicates the unit cell in the three spatial directions. Studies of different planes are possible by unit cell transformations with external programs such as VESTA [46]. KIMERA can apply periodic boundary conditions (PBC) in the three spatial directions.
- System shape. The program has commands to create different crystalline shapes of the system into complex surfaces or particles. For the moment the available geometries are cubic, spherical, parallelepiped, ellipsoidal, tick planes, or a combination of them.
- Topographic defects can be defined in the system, such as insoluble regions, dislocations, impurities and vacancies. There are two ways of defining impurities; it is possible to define them in the unit cell indicating their occupancy, or introducing them ex post once the system has been defined.
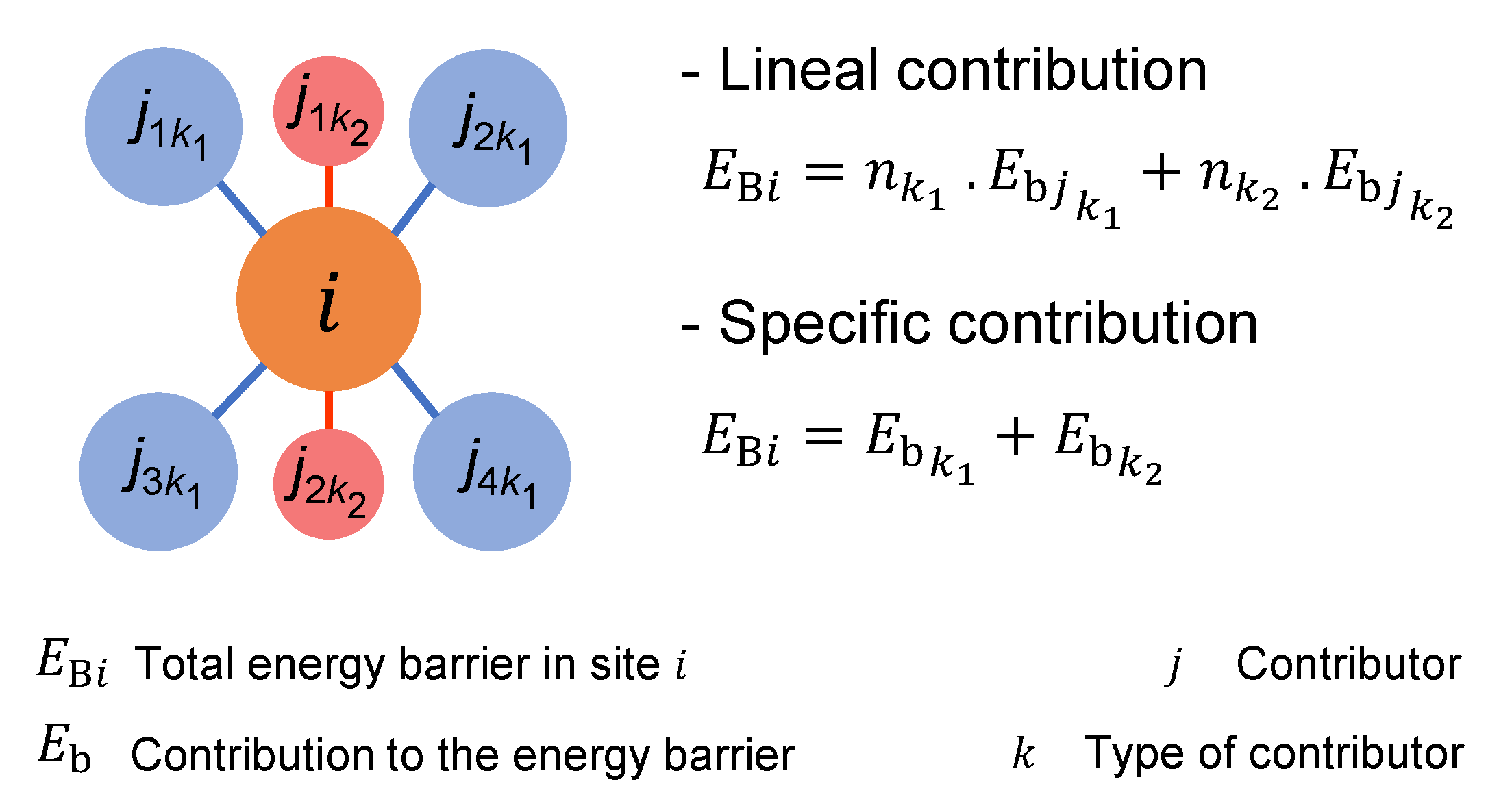
- Event definition. The KMC algorithm simulates the time evolution of a system as a set of possible events. These events take place at a rate that follows an Arrhenius equation (Equation (1)). A recent study demonstrates that the net dissolution of a mineral can be characterized using decoupled reactions of dissolution and precipitation [29]. Hence, we use that KMC scheme, so the fundamental frequency of the Arrhenius equation, f, splits into or , and into or depending if considering a dissolution or a precipitation event respectively. The energy barrier is characteristic of each chemical reaction and its neighbourhood, and can be obtained from the bibliography and/or ab initio calculations [22,23]. Supposing n neighbours of an atom, KIMERA can set and as a linear (Equation (6)) or a specific (Equation (7)) function of each neighbour j [23,47] (see Figure 2):Note that Equation (6) is a specific case of Equation (7). Moreover, since the contribution to the energy barrier can be determined for several types of neighbours, k represents each set of contributors with the same characteristics and and its contribution for dissolution and precipitation energy barrier respectively.With these two ways of defining the energy barrier, two different approaches can be considered to describe the dissolution events:
- 1
- A bond by bond description: Each linking bond breaks sequentially so that when an atom has no bonds left, it is released from the mineral.
- 2
- A site by site description: All bonds reactions are unified in only one event, and each site dissolves with joint probability.
As an additional element, KIMERA supports conditional event definition. Furthermore, it is possible to define the events based on ghost positions in the unit cell without physical meaning and to make a differentiation between atoms of the same type, for example it is possible to split the atoms of silica into Si1, Si2, etc. in the unit cell and then define events for each sub-element. - Target time (s). Predicting the time scale beforehand in a complex system can be tough. There are two options for the simulation to finish. The simplest option is to indicate the number of simulation steps, that is, the number of events to accomplish. The other option is to specify the target time (s) until the simulation is going to run. The user can request the program to do an estimation of it by considering the initial possible events. Given s initial sorted groups of rates corresponding to atom removals with different coordination , the program approximates the total time for the system to dissolve as if all atoms had the same rate value; the previous to the middle one.
- Optional parameters related with the output files. As we will see, output files contain information of the system time evolution like snapshots for visualization or the quantity of dissolved atoms.
3.1.2. System Preparation
3.1.3. Simulation Process
- Initial KIMERA file of the system in its own format (‘.initialkimerabox’). It is designed to save time in calculating neighbourhood, linked and affected atoms. A later simulation which reads this file will not need to do the preparation step.
- Final KIMERA file of the system (‘.finalkimerabox’). When the simulation has finished, or has encountered an error, the system is printed in KIMERA format.
- File with system snapshots (‘.box’) in LAMMPS format [33] for visualization. As this file can contain a lot of data, it may be better to handle the surface file unless for checking reasons.
- File with surface snapshots (‘.surface’) in LAMMPS format. Instead of the whole system, only the atoms on the surface are printed in this file.
- Data file with the time evolution of the following parameters (‘.data’): The total number of atoms dissolved of each type, its fraction, the surface dispersion, the gyradius (in no PBC systems) as well as all their derivatives.
- Coordination file with the mean coordination to each type of atom along the dissolution process (‘.meandiscoord’). This data is key to calculate correctly value as explained below.
- Layer atom files with the amount of atoms in each layer and each spatial direction (‘.alayer’, ‘.blayer’, ‘.clayer’). For example, the ‘.clayer’ file contains the total number of atoms of the cells in plane ab, layer by layer in c direction.
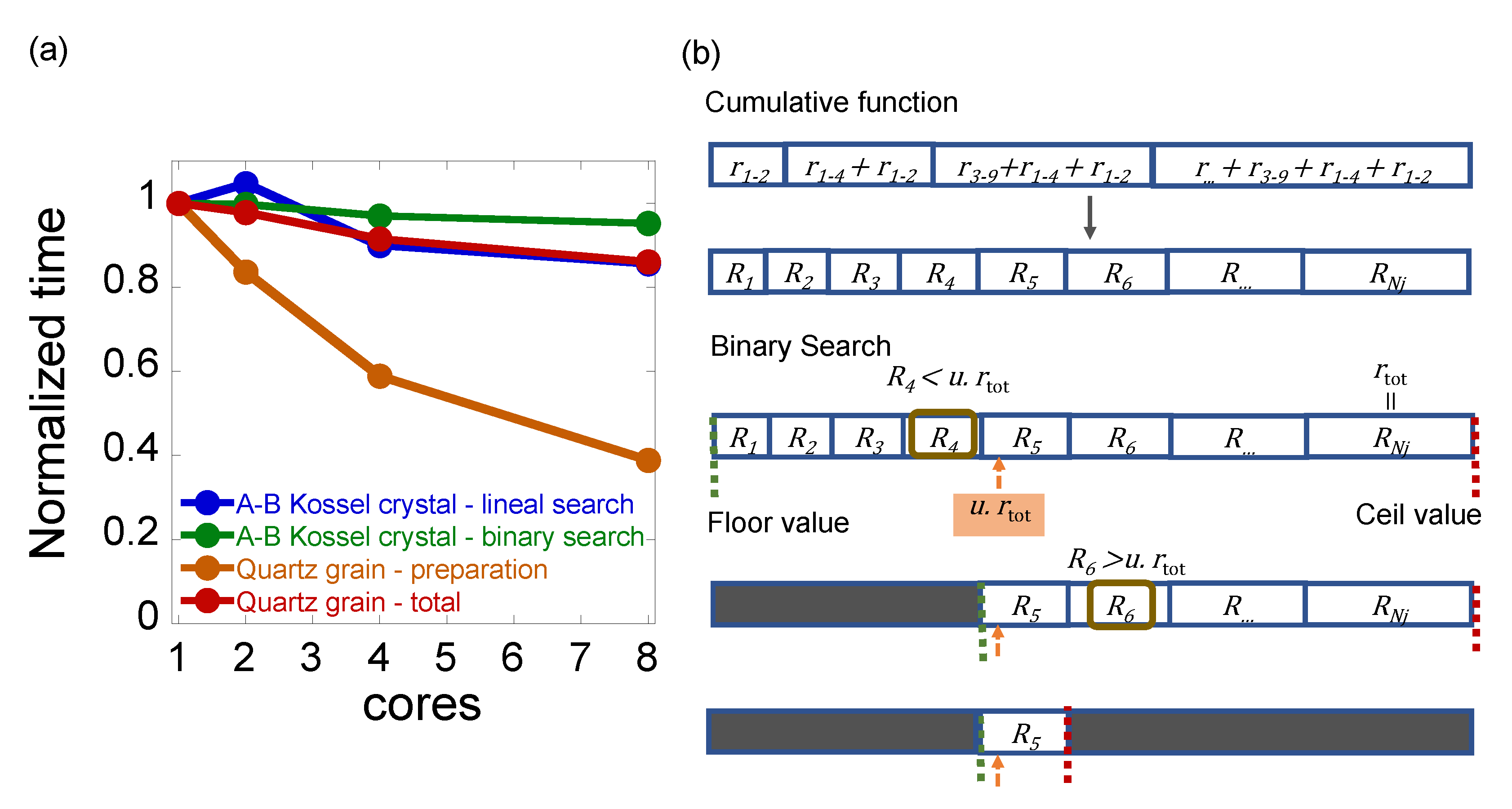
3.2. Parallelization Level
4. Gibbs Free Energy Difference,
5. Examples of KIMERA Capabilities
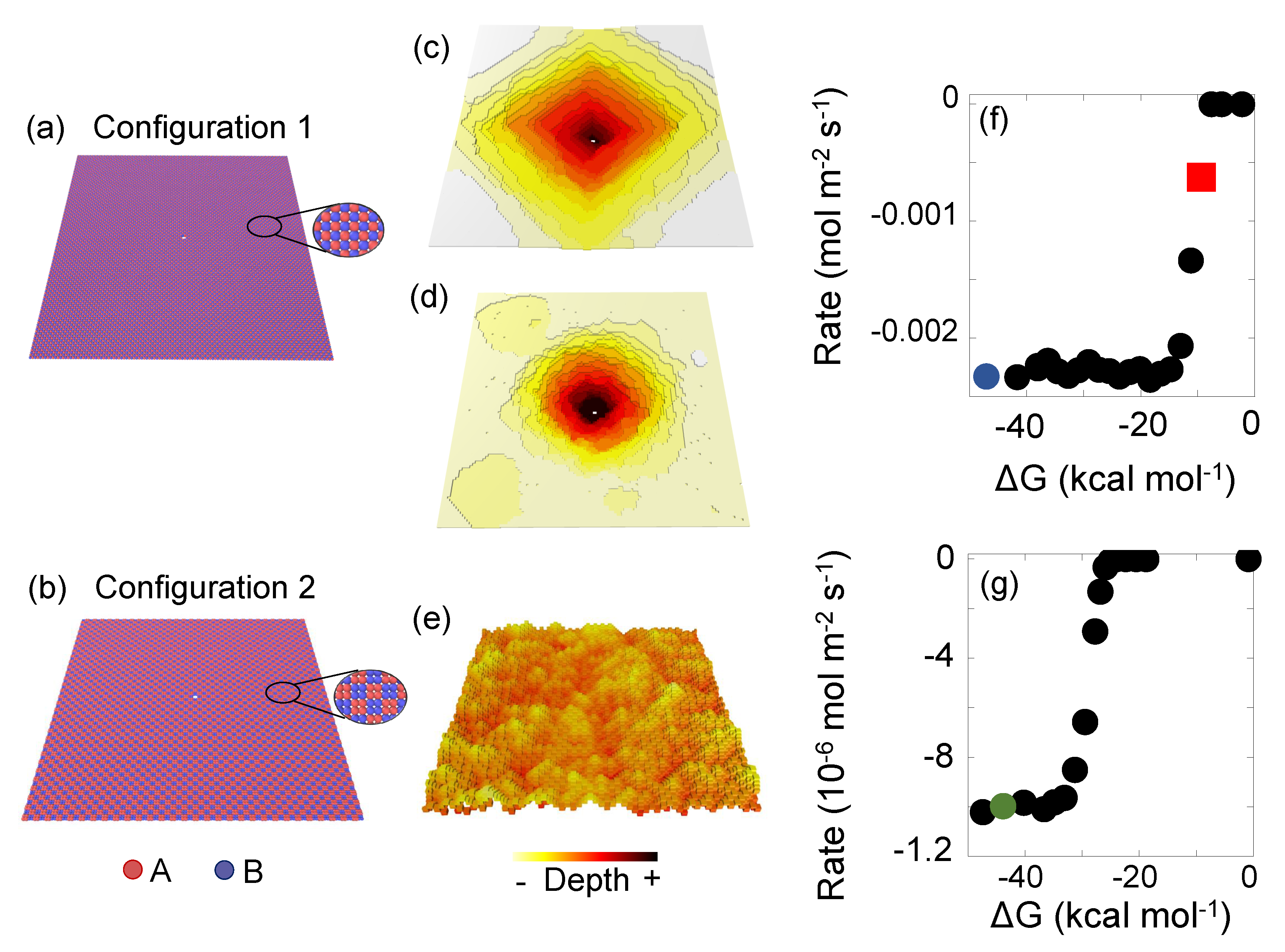
5.1. Model A-B Kossel Crystals
- The system dimensions are indicated; 60 × 60 × 15 unit cells.
- The unit cell parameters. We used Å and . Inside the cell, we define the 8 positions of the atoms in the unit cell that later on is repeated along the system. The positions are: (0,0,0), (2.5,0,0), (0,0,2.5), (2.5,0,2.5), (0,2.5,0), (2.5,2.5,0), (0,2.5,2.5) and (2.5,2.5,2.5) Å. All the positions are initially define as A atoms, and we later will redefine half of them as B type. Note that although the unit cell has Å the distance between atoms is Å, which is a typical distance reported for minerals [54].In ‘configuration 1’, the positions are the same, but half of them are of type B. Specifically, atoms in (0,0,0), (0,0,2.5), (2.5,2.5,0) and (2.5,2.5,2.5) are B type. Since the alternating disposition of the atoms is already taken into account with this definition, no additional commands to modify the system are needed.
- We set periodic boundary conditions along x and y axes.
- Physical parameters for the simulation. The target time s and the local units, which ensures far from equilibrium conditions. In ‘configuration 1’ the time scale is different due to its faster dissolution and s.
- Event definition. We have chosen for this example an energy barrier for A-A atoms of units, for B-B units, which are respectively the higher and lower limit value for most minerals [27]. For the AB interaction the energy barrier is obtained from the Lorentz–Berthelot rule [55], units. The precipitation energy barrier for all the cases is units.For the frequency s−1 s which lies in the range of values for atomic vibration in a mineral (– s−1 at 300 K) [56].Lastly, KIMERA requires the number of neighbours that a bulk atom has to later define the initial reactive surface. For both for A and B atoms, a bulk atom has 3 A type neighbours and 3 B type neighbours. In ‘configuration 1’ the event definition is similar, but the number which defines a bulk atom changes. A bulk A atom has 2 A and 4 B neighbours. A bulk B atom has 4 A and 2 B neighbours.
- Topographic defects. We define the last plane as insoluble and we include one partial dislocation in the center with one third of the system depth. Since there are atoms within the dislocation that have a lower coordination than a bulk atom, the program recognised them as initial reactive surface. Therefore, we remove such condition because it is physically meaningless.
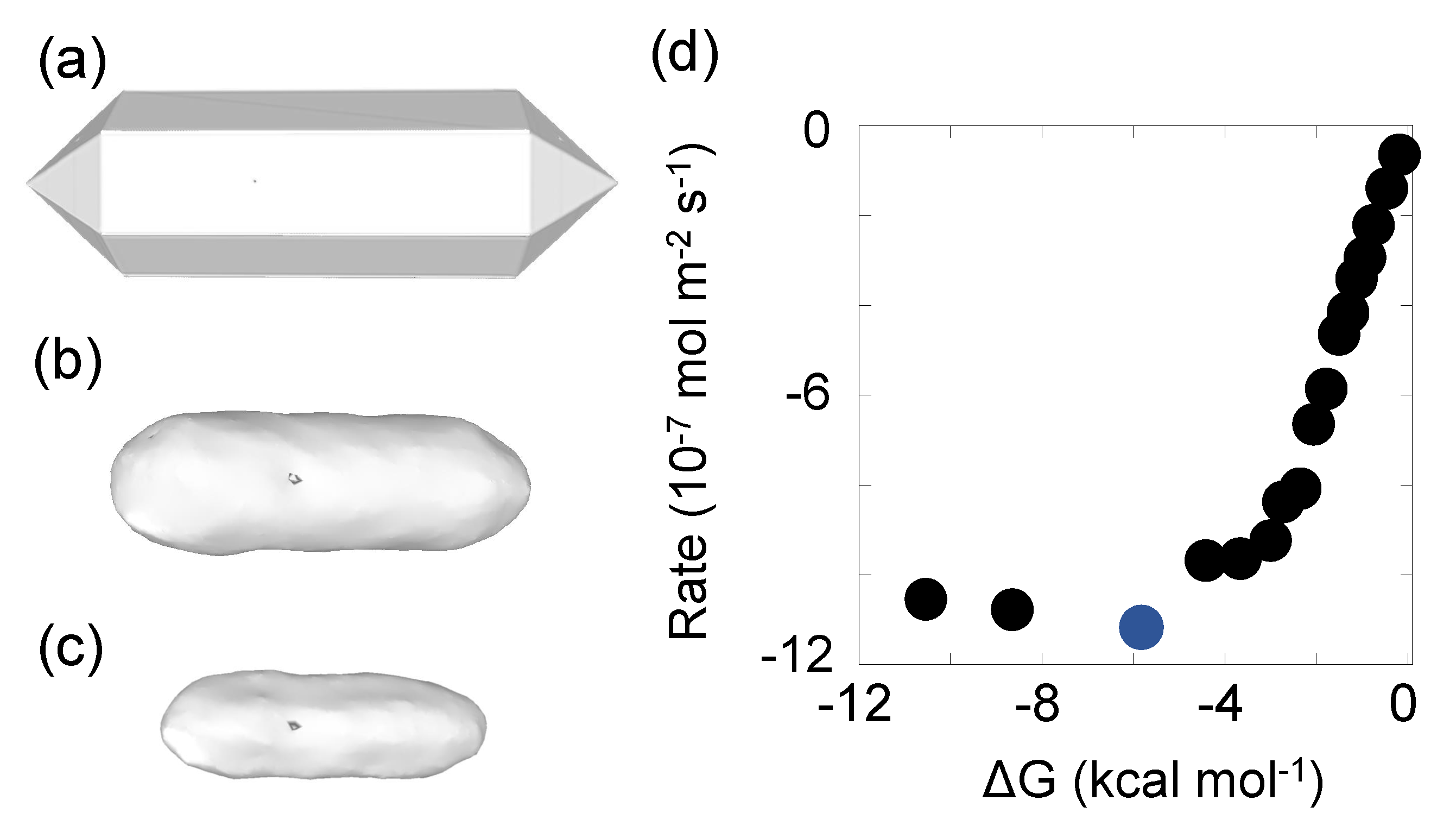
5.2. Quartz Model I: Dissolution of an Ellipsoidal Grain
- System dimensions. A box in which we will define the ellipsoid is created with 50 × 40 × 30 unit cells.
- The unit cell parameters. For -quartz , , and . Inside the cell, we load a ‘.xyz’ file containing the positions, which has been converted from a ‘.cif’ file downloaded from The American Mineralogist crystal structure database [45]. Oxygen atoms can be removed for performance purposes since they are not explicitly taken into account for the quartz dissolution reaction in this case. The dissolution of a SiO2 is considered in a single step with a joint probability (Equation (6)). This can be interpreted as a coarse grain of a SiO2 unit in each Si site.
- Physical parameters. The target time s and the local units.
- Topographic defects. An ellipsoid with radius in the three axes, Å, Å and Å is defined as the simulation system. A dislocation along the x axis is placed in the middle.
- Event definition. The energy barrier with first neighboring silicon atom is units and with second . Precipitation energies of and are used. All the four first silicon neighbours are at 3.09832 Å. If an atom is surrounded by the four first neighbours, it is considered to be in bulk. 12 second silicon neighbours are at 5.01 Å. Finally, the fundamental frequencies values are s−1 [57].
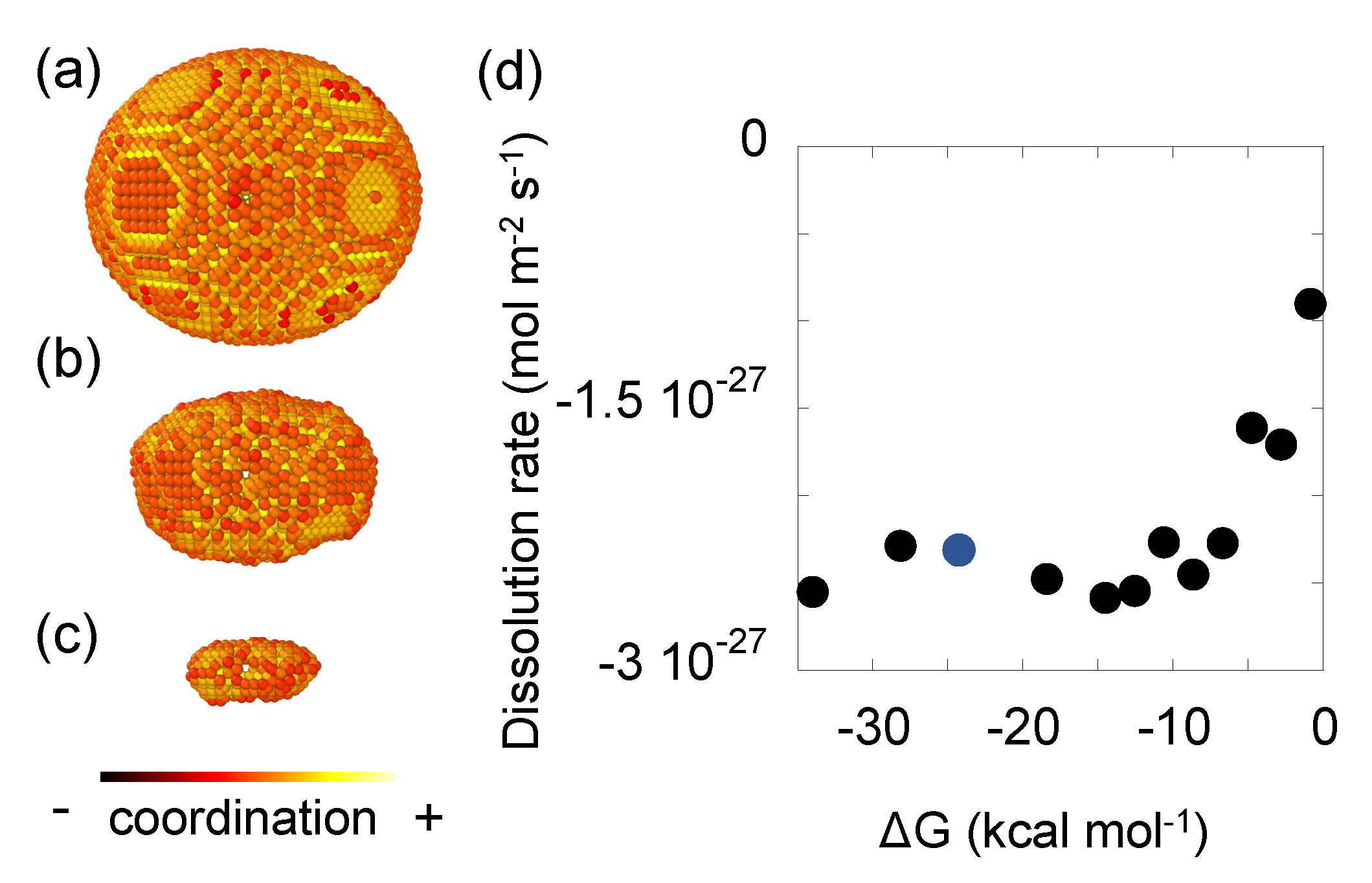
5.3. Quartz Model II: Dissolution of a Wulff Shape Particle
- System dimensions. A box in which the wulff shape fit in is created with 16 × 16 × 47 units cells
- Same unit cell parameters as the previous example. , , and . The ‘.xyz’ file is also called, but this time the oxygen atoms do play an important role and they cannot be removed.
- This time instead of target time, we define a target step = 62,700 steps, which is approximately the total amount of silicon and oxygen atoms forming the particle.
- Topographic defects. The wulff shape is sculpted from the system by defining planes in which the atoms are removed. The equations of these planes are taken from GEODEBRA3D tool [60] which was used to visualise the system beforehand. Besides, two dislocations are defined and inner atoms removed from the initial surface. One dislocation is placed transversally in the center of the {100} plane, and another one perpendicular to the previous and longitudinally to the wulff shape
- Event definition. The and for the linking oxygen bond is directly related with the number of oxygen atoms within Å. 6 surrounding oxygen atoms indicates that the considered one is in a bulk position and therefore it is not reactive. Besides, the silicon atom must be automatically released if all of its four surrounding oxygen atoms have reacted. Finally, the fundamental frequency values s−1 are indicated [61]. The value sets very far from equilibrium conditions.
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Breemen, N.; Buurman, P. Soil Formation; Springer: Berlin/Heidelberg, Germany, 2002. [Google Scholar]
- Kalbitz, K.; Solinger, S.; Park, J.H.; Michalzik, B.; Matzner, E. Controls on the dynamics of dissolved organic matter in soils: A review. Soil Sci. 2000, 165, 277–304. [Google Scholar] [CrossRef]
- Krouse, H.R.; Viau, C.A.; Eliuk, L.S.; Ueda, A.; Halas, S. Chemical and isotopic evidence of thermochemical sulphate reduction by light hydrocarbon gases in deep carbonate reservoirs. Nature 1988, 333, 415. [Google Scholar] [CrossRef]
- Canals, M.; Meunier, J.D. A model for porosity reduction in quartzite reservoirs by quartz cementation. Geochim. Cosmochim. Acta 1995, 59, 699–709. [Google Scholar] [CrossRef]
- Lal, R. Carbon sequestration. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 815–830. [Google Scholar] [CrossRef] [PubMed]
- Lal, R. Soil Carbon Sequestration Impacts on Global Climate Change and Food Security. Science 2004, 304, 1623–1627. [Google Scholar] [CrossRef] [Green Version]
- Blain, S.; Quéguiner, B.; Armand, L.; Belviso, S.; Bombled, B.; Bopp, L.; Bowie, A.; Brunet, C.; Brussaard, C.; Carlotti, F.; et al. Effect of natural iron fertilization on carbon sequestration in the Southern Ocean. Nature 2007, 446, 1070. [Google Scholar] [CrossRef]
- Giammar, D.E.; Bruant, R.G., Jr.; Peters, C.A. Forsterite dissolution and magnesite precipitation at conditions relevant for deep saline aquifer storage and sequestration of carbon dioxide. Chem. Geol. 2005, 217, 257–276. [Google Scholar] [CrossRef]
- Juilland, P.; Gallucci, E. Morpho-topological investigation of the mechanisms and kinetic regimes of alite dissolution. Cem. Concr. Res. 2015, 76, 180–191. [Google Scholar] [CrossRef]
- Juilland, P.; Gallucci, E.; Flatt, R.; Scrivener, K. Dissolution theory applied to the induction period in alite hydration. Cem. Concr. Res. 2010, 40, 831–844. [Google Scholar] [CrossRef]
- Lasaga, A.C.; Blum, A.E. Surface chemistry, etch pits and mineral-water reactions. Geochim. Cosmochim. Acta 1986, 50, 2363–2379. [Google Scholar] [CrossRef]
- Brand, A.S.; Bullard, J.W. Dissolution kinetics of cubic tricalcium aluminate measured by digital holographic microscopy. Langmuir 2017, 33, 9645–9656. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.S.; Feng, P.; Bullard, J.W. Calcite dissolution rate spectra measured by in situ digital holographic microscopy. Geochim. Cosmochim. Acta 2017, 213, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Brand, A.S.; Chen, L.; Bullard, J.W. In situ nanoscale observations of gypsum dissolution by digital holographic microscopy. Chem. Geol. 2017, 460, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Cama, J.; Ganor, J.; Ayora, C.; Lasaga, C.A. Smectite dissolution kinetics at 80 °C and pH 8.8. Geochim. Cosmochim. Acta 2000, 64, 2701–2717. [Google Scholar] [CrossRef]
- Dove, P.M.; Han, N.; De Yoreo, J.J. Mechanisms of classical crystal growth theory explain quartz and silicate dissolution behavior. Proc. Natl. Acad. Sci. USA 2005, 102, 15357–15362. [Google Scholar] [CrossRef] [Green Version]
- Shvab, I.; Brochard, L.; Manzano, H.; Masoero, E. Precipitation mechanisms of mesoporous nanoparticle aggregates: Off-lattice, coarse-grained, kinetic simulations. Cryst. Growth Des. 2017, 17, 1316–1327. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Manzano, H.; Guo, Y.; Lopez-Arbeloa, I.; Shen, X. Hydration mechanism of reactive and passive dicalcium silicate polymorphs from molecular simulations. J. Phys. Chem. C 2015, 119, 19869–19875. [Google Scholar] [CrossRef]
- Wang, Q.; Manzano, H.; López-Arbeloa, I.; Shen, X. Water adsorption on the β-dicalcium silicate surface from DFT simulations. Minerals 2018, 8, 386. [Google Scholar] [CrossRef] [Green Version]
- Manzano, H.; Gartzia-Rivero, L.; Bañuelos, J.; López-Arbeloa, I.N. Ultraviolet–visible dual absorption by single BODIPY dye confined in LTL zeolite nanochannels. J. Phys. Chem. C 2013, 117, 13331–13336. [Google Scholar] [CrossRef]
- de Assis, T.A.; Reis, F.D.A. Dissolution of minerals with rough surfaces. Geochim. Cosmochim. Acta 2018, 228, 27–41. [Google Scholar] [CrossRef]
- Kurganskaya, I.; Luttge, A. Kinetic Monte Carlo approach to study carbonate dissolution. J. Phys. Chem. C 2016, 120, 6482–6492. [Google Scholar] [CrossRef]
- Kurganskaya, I.; Luttge, A. Kinetic Monte Carlo Simulations of Silicate Dissolution: Model Complexity and Parametrization. J. Phys. Chem. C 2013, 117, 24894–24906. [Google Scholar] [CrossRef]
- Rohlfs, R.D.; Fischer, C.; Kurganskaya, I.; Luttge, A. Crystal dissolution kinetics studied by a combination of Monte Carlo and Voronoi methods. Minerals 2018, 8, 133. [Google Scholar] [CrossRef] [Green Version]
- Briese, L.; Arvidson, R.S.; Luttge, A. The effect of crystal size variation on the rate of dissolution—A kinetic Monte Carlo study. Geochim. Cosmochim. Acta 2017, 212, 167–175. [Google Scholar] [CrossRef]
- Lasaga, A.C.; Luttge, A. Variation of crystal dissolution rate based on a dissolution stepwave model. Science 2001, 291, 2400–2404. [Google Scholar] [CrossRef]
- Meakin, P.; Rosso, K.M. Simple kinetic Monte Carlo models for dissolution pitting induced by crystal defects. J. Chem. Phys. 2008, 129. [Google Scholar] [CrossRef]
- Fischer, C.; Luttge, A. Pulsating dissolution of crystalline matter. Proc. Natl. Acad. Sci. USA 2018, 115, 897–902. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.; Manzano, H.; Dolado, J.S. Mechanisms and Dynamics of Mineral Dissolution: A New Kinetic Monte Carlo Model. Adv. Theory Simul. 2019, 2, 1900114. [Google Scholar] [CrossRef] [Green Version]
- Artacho, E.; Anglada, E.; Diéguez, O.; Gale, J.D.; García, A.; Junquera, J.; Martin, R.M.; Ordejón, P.; Pruneda, J.M.; Sánchez-Portal, D.; et al. The SIESTA method; developments and applicability. J. Phys. Condens. Matter 2008, 20, 064208. [Google Scholar] [CrossRef]
- Gale, J.D. GULP: A computer program for the symmetry-adapted simulation of solids. J. Chem. Soc. Faraday Trans. 1997, 93, 629–637. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics; Technical Report; Sandia National Labs.: Albuquerque, NM, USA, 1993.
- Plimpton, S.; Thompson, A.; Slepoy, A. Stochastic Parallel PARticle Kinetic Simulator; Technical Report; Sandia National Laboratories: Albuquerque, NM, USA, 2008.
- Jørgensen, M.; Grönbeck, H. MonteCoffee: A programmable kinetic Monte Carlo framework. J. Chem. Phys. 2018, 149, 114101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.J.; Matera, S.; Reuter, K. Kmos: A lattice kinetic Monte Carlo framework. Comput. Phys. Commun. 2014, 185, 2138–2150. [Google Scholar] [CrossRef] [Green Version]
- Holm, E.A.; Hoffmann, T.D.; Rollett, A.D.; Roberts, C.G. Particle-Assisted Abnormal Grain Growth; IOP Conference Series: Materials Science and Engineering; IOP Publishing: Risø, Denmark, 2015; Volume 89, p. 012005. [Google Scholar]
- Rodgers, T.M.; Madison, J.D.; Tikare, V.; Maguire, M.C. Predicting mesoscale microstructural evolution in electron beam welding. JOM 2016, 68, 1419–1426. [Google Scholar] [CrossRef] [Green Version]
- Jansen, A.P.J. An Introduction to Kinetic Monte Carlo Simulations of Surface Reactions; Springer: Berlin/Heidelberg, Germany, 2012; Volume 856. [Google Scholar]
- Bortz, A.B.; Kalos, M.H.; Lebowitz, J.L. A new algorithm for Monte Carlo simulation of Ising spin systems. J. Comput. Phys. 1975, 17, 10–18. [Google Scholar] [CrossRef]
- Dybeck, E.C.; Plaisance, C.P.; Neurock, M. Generalized temporal acceleration scheme for kinetic monte carlo simulations of surface catalytic processes by scaling the rates of fast reactions. J. Chem. Theory Comput. 2017, 13, 1525–1538. [Google Scholar] [CrossRef]
- Tian, X.; Bik, A.; Girkar, M.; Grey, P.; Saito, H.; Su, E. Intel® OpenMP C++/Fortran Compiler for Hyper-Threading Technology: Implementation and Performance. Intel Technol. J. 2002, 6, 36–46. [Google Scholar]
- Unofficial XYZ File Format Specification. Available online: http://en.wikipedia.org/wiki/XYZ_file_format (accessed on 1 October 2019).
- Momma, K.; Izumi, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Downs, R.T.; Hall-Wallace, M. The American Mineralogist crystal structure database. Am. Mineral. 2003, 88, 247–250. [Google Scholar]
- Unit Cell Rotation with VESTA. Available online: https://ma.issp.u-tokyo.ac.jp/en/app-post/1788 (accessed on 15 March 2020).
- Kohli, C.; Ives, M. Computer simulation of crystal dissolution morphology. J. Cryst. Growth 1972, 16, 123–130. [Google Scholar] [CrossRef]
- Lasaga, A.C.; Lüttge, A. Mineralogical approaches to fundamental crystal dissolution kinetics. Am. Mineral. 2004, 89, 527–540. [Google Scholar] [CrossRef]
- Williams, L.F., Jr. A modification to the half-interval search (binary search) method. In Proceedings of the 14th Annual Southeast Regional Conference, Birmingham, AL, USA, 22–24 April 1976; pp. 95–101. [Google Scholar]
- Hill, M.D.; Marty, M.R. Amdahl’s law in the multicore era. Computer 2008, 41, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Dukhin, S.S.; Kretzschmar, G.; Miller, R. Dynamics of Adsorption at Liquid Interfaces: Theory, Experiment, Application; Elsevier: Amsterdam, The Netherlands, 1995. [Google Scholar]
- Oura, K.; Katayama, M.; Saranin, A.; Lifshits, V.; Zotov, A. Surface Science; Springer: Berlin/Heidelberg, Germany, 2003; Volume 602, pp. 229–233. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Model. Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Gibbs, G.V.; Downs, R.T.; Cox, D.F.; Ross, N.L.; Prewitt, C.T.; Rosso, K.M.; Lippmann, T.; Kirfel, A. Bonded interactions and the crystal chemistry of minerals: A review. Z. Krist. Cryst. Mater. 2008, 223, 1–40. [Google Scholar] [CrossRef]
- Berthelot, D. Sur le mélange des gaz. Compt. Rendus 1898, 126, 1703–1855. [Google Scholar]
- Lasaga, A.C. Kinetic Theory in the Earth Sciences; Princeton University Press: Princeton, NJ, USA, 2014. [Google Scholar]
- Pelmenschikov, A.; Leszczynski, J.; Pettersson, L.G. Mechanism of dissolution of neutral silica surfaces: Including effect of self-healing. J. Phys. Chem. A 2001, 105, 9528–9532. [Google Scholar] [CrossRef]
- Martin, P.; Gaitero, J.J.; Dolado, J.S.; Manzano, H. A Kinetic Monte Carlo Model for Quartz Dissolution. 2020; in preparation. [Google Scholar]
- Wendler, F.; Okamoto, A.; Blum, P. Phase-field modeling of epitaxial growth of polycrystalline quartz veins in hydrothermal experiments. Geofluids 2016, 16, 211–230. [Google Scholar] [CrossRef] [Green Version]
- Geodebra3d. Available online: https://www.geogebra.org/3d?lang=en (accessed on 15 March 2020).
- Casey, W.H.; Lasaga, A.C.; Gibbs, G. Mechanisms of silica dissolution as inferred from the kinetic isotope effect. Geochim. Cosmochim. Acta 1990, 54, 3369–3378. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A-A | A-B | B-B | B-A | ||
|---|---|---|---|---|---|
| Configuration 1 | 1.0 | 1.33 | 1.0 | 2.66 | |
| Configuration 2 | 1.5 | 0.423 | 1.5 | 2.576 |
| Si-Si-3.09832 | Si-Si-5.0100 | Si-Si-5.66774 | Si-Si-4.42416 | |
|---|---|---|---|---|
| 1.95 | 1.92 | 1.92 | 1.95 |
| Bond | Surrounding Oxygen Atoms | ||
|---|---|---|---|
| Q1-Q1 | - | - | 0 |
| Q1-Q2 | 75 | 60 | 1 |
| Q1-Q3 | 85 | 70 | 2 |
| Q1-Q4 | 95 | 80 | 3 |
| Q2-Q2 | 85 | 70 | 2 |
| Q2-Q3 | 95 | 80 | 3 |
| Q2-Q4 | 105 | 105 | 4 |
| Q3-Q3 | 105 | 105 | 4 |
| Q3-Q4 | 135 | 135 | 5 |
| Q4-Q4 | - | - | 6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, P.; Gaitero, J.J.; Dolado, J.S.; Manzano, H. KIMERA: A Kinetic Montecarlo Code for Mineral Dissolution. Minerals 2020, 10, 825. https://doi.org/10.3390/min10090825
Martin P, Gaitero JJ, Dolado JS, Manzano H. KIMERA: A Kinetic Montecarlo Code for Mineral Dissolution. Minerals. 2020; 10(9):825. https://doi.org/10.3390/min10090825
Chicago/Turabian StyleMartin, Pablo, Juan J. Gaitero, Jorge S. Dolado, and Hegoi Manzano. 2020. "KIMERA: A Kinetic Montecarlo Code for Mineral Dissolution" Minerals 10, no. 9: 825. https://doi.org/10.3390/min10090825
APA StyleMartin, P., Gaitero, J. J., Dolado, J. S., & Manzano, H. (2020). KIMERA: A Kinetic Montecarlo Code for Mineral Dissolution. Minerals, 10(9), 825. https://doi.org/10.3390/min10090825