
Significance of Acid Washing after Biooxidation of Sulfides in Sequential Biotreatment of Double Refractory Gold Ore from the Syama Mine, Mali



Abstract
:1. Introduction
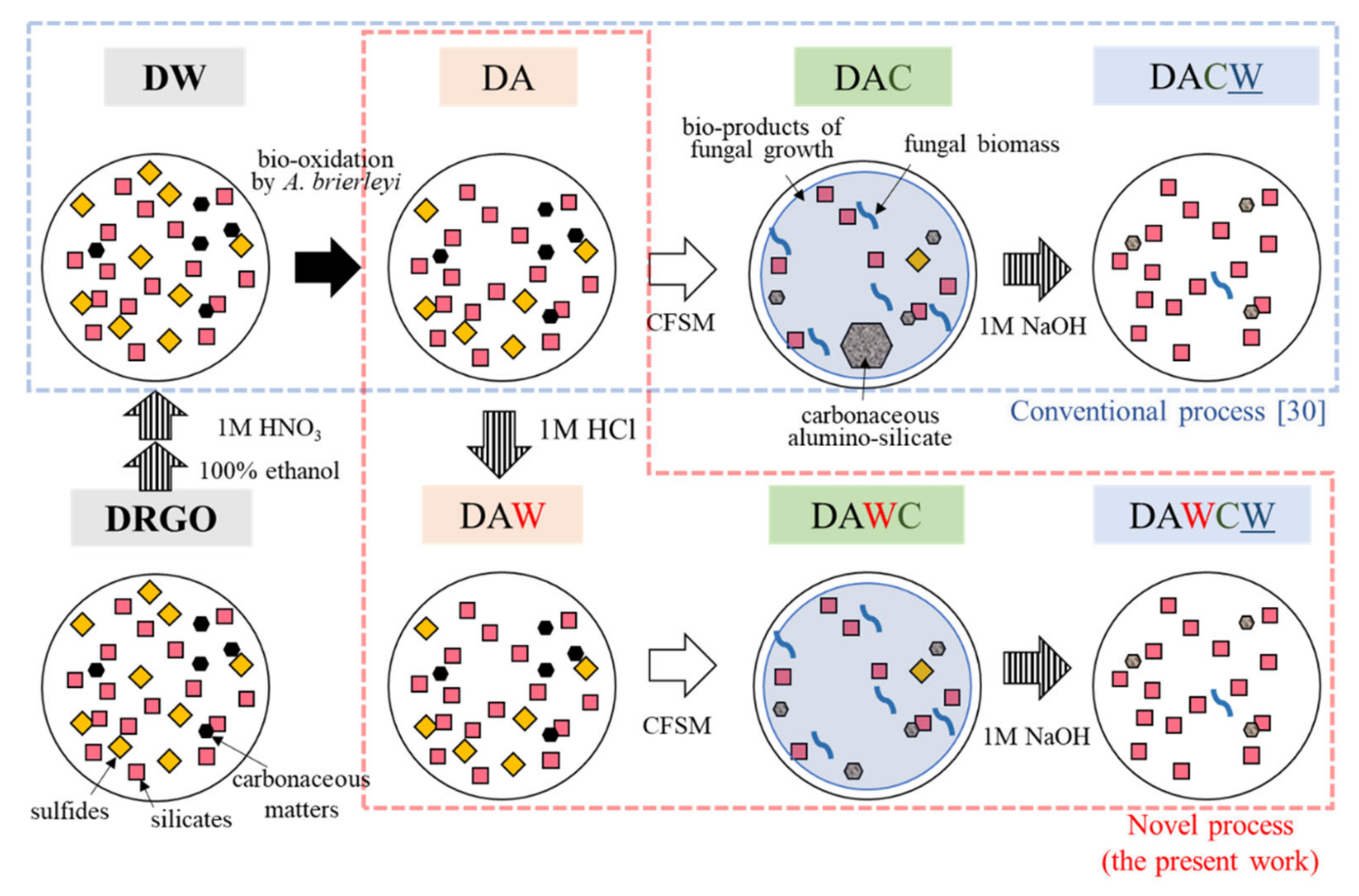
2. Materials and Methods
2.1. DRGO Characterization and Preparation
2.2. Microbiologically Oxidative Dissolution of Sulfides
2.3. Acid Washing of the Residues
2.4. Phanerochaete chrysosporium Cultivation and Enzyme Assay
2.5. Degradation of Carbonaceous Matter by Cell-Free Spent Medium (CFSM) of Phanerochaete chrysosporium
2.6. Alkaline Washing of the Residues
2.7. Gold Recovery
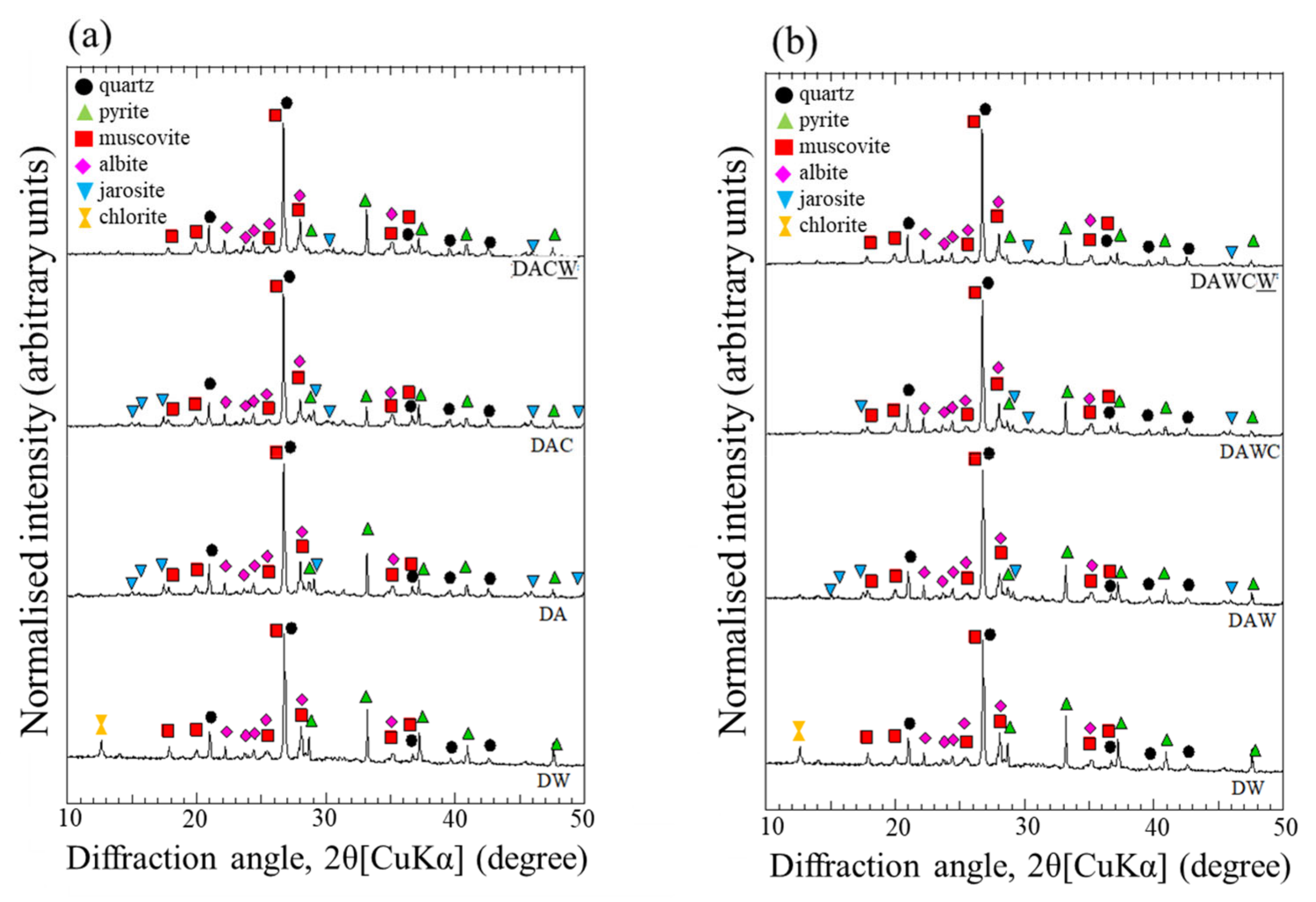
2.8. Characterization of the Solid Residues in Sequential Biotreatment
3. Results and Discussion
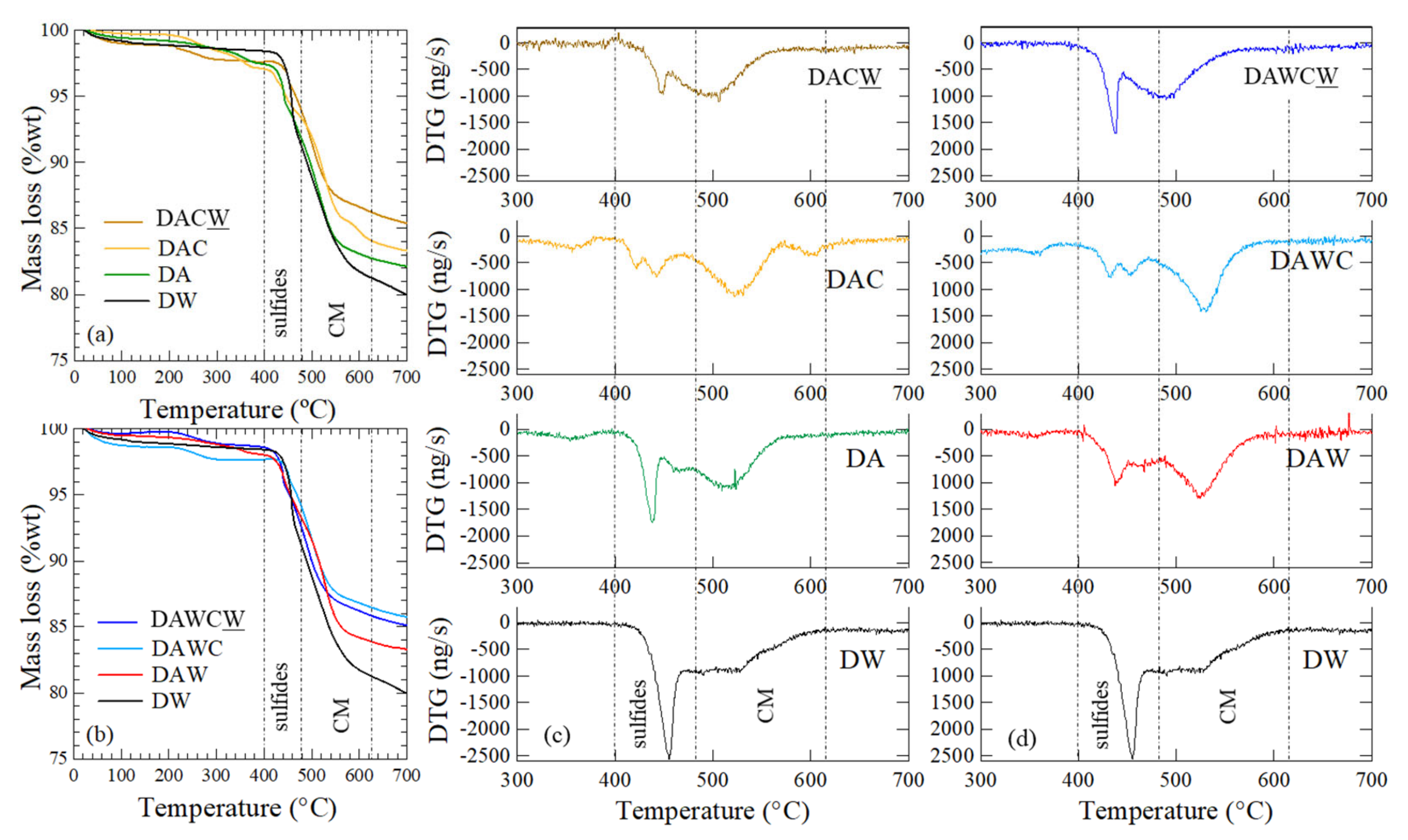
3.1. DW Preparation
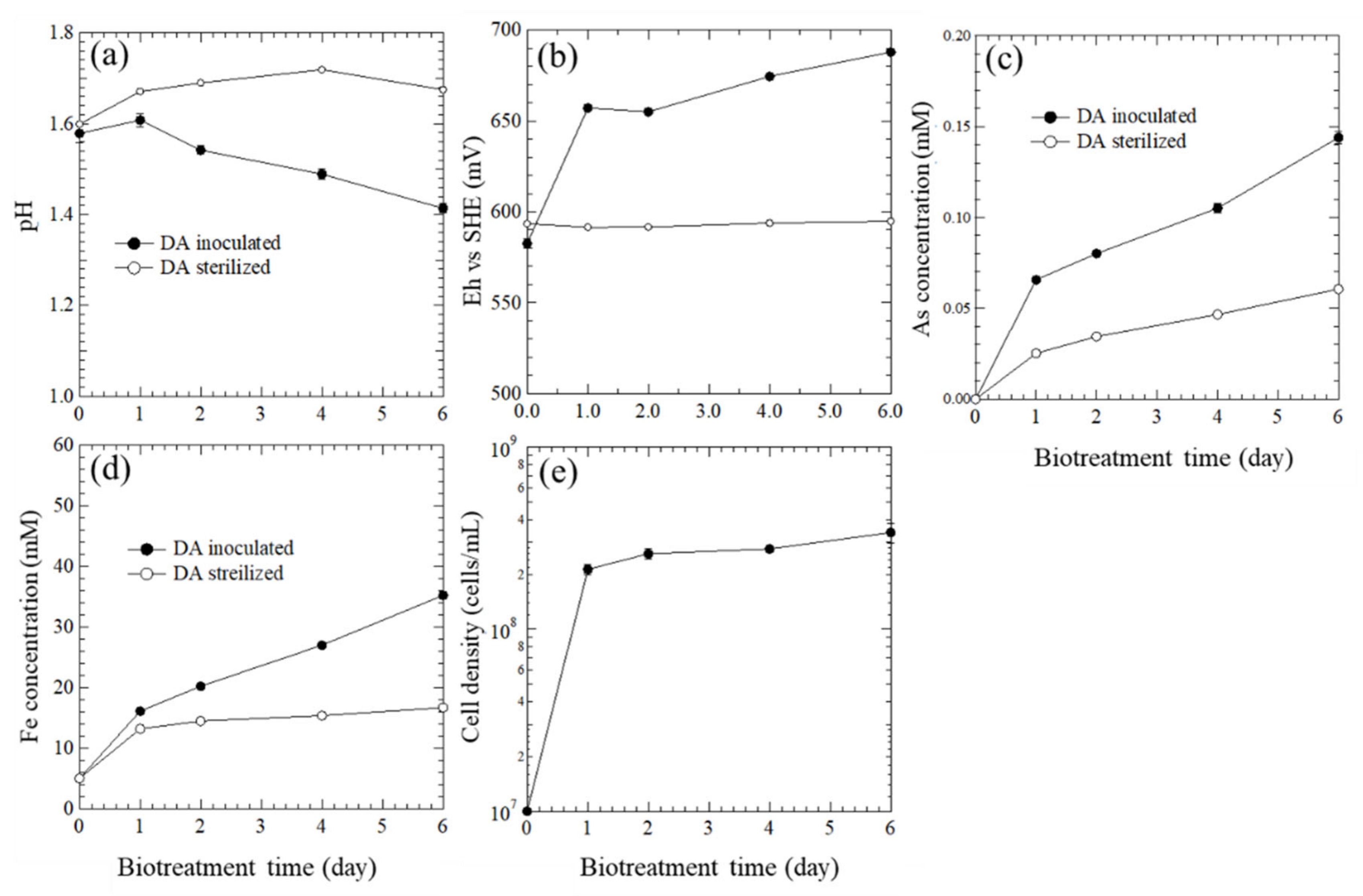
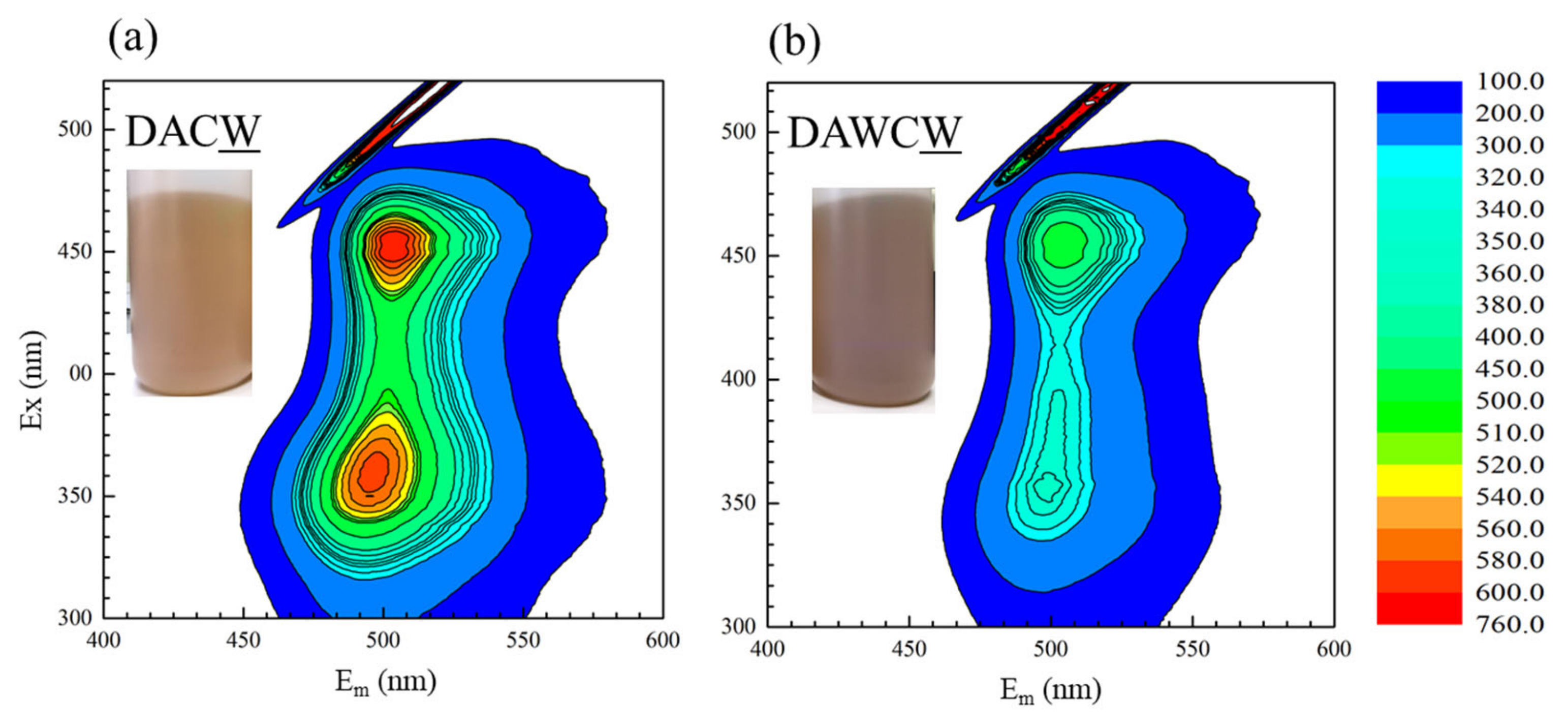
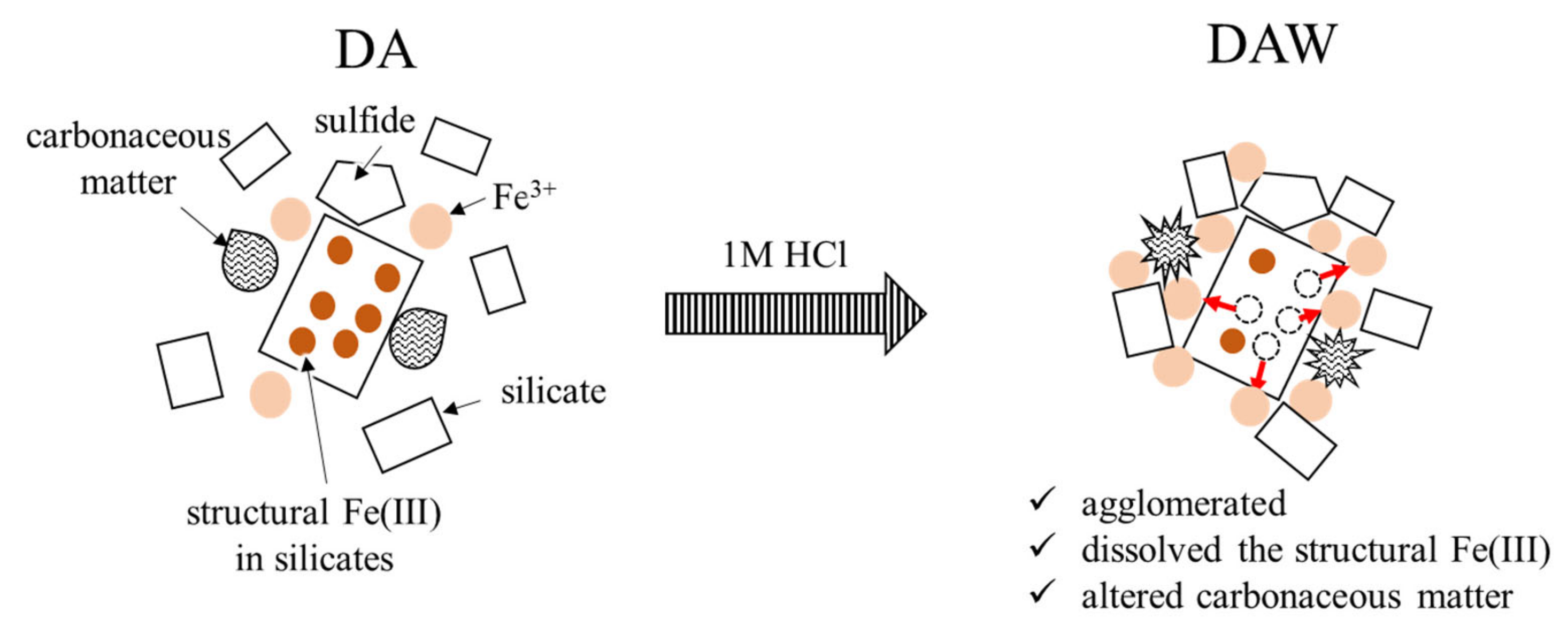
3.2. Biooxidation of Sulfides and HCl Washing
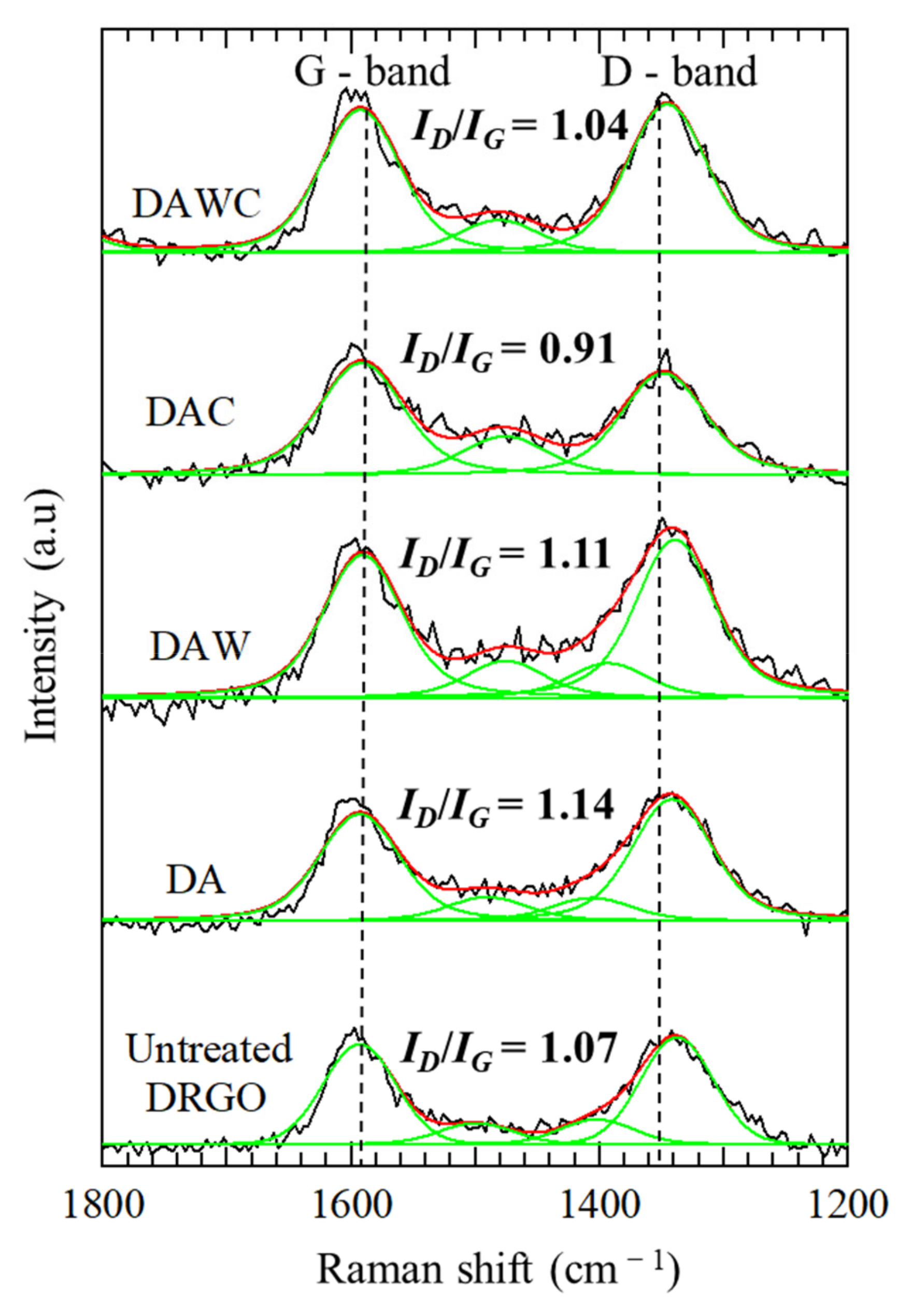
3.3. Degradation of Carbonaceous Matter and NaOH Washing
3.4. Gold Recovery
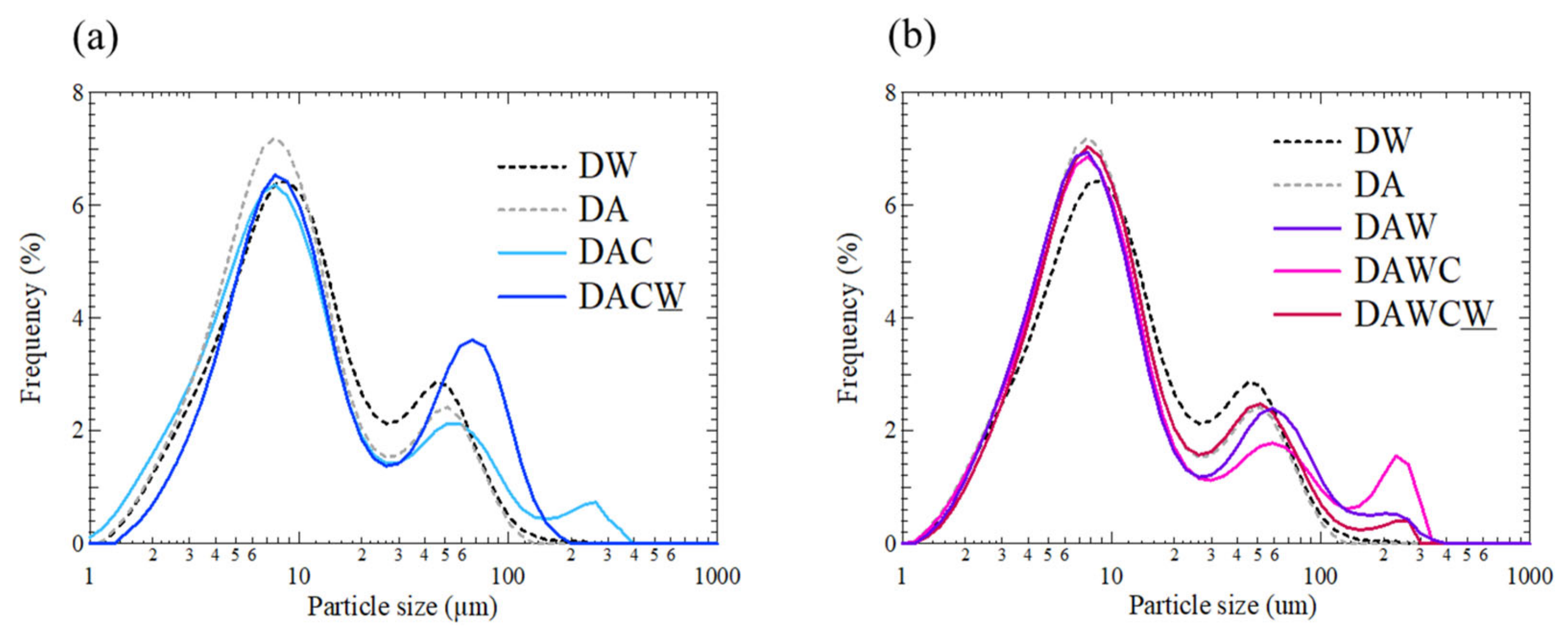
3.5. Particle Size Distribution and Evaluation of HCl Washing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Osseo-Asare, K.; Afenya, P.M.; Abotsi, G.M.K. Carbonaceous matter in gold ores: Isolation, characterization and adsorption behavior in aurocyanide solutions. In Precious Metals: Mining, Extraction and Processing; Metallurgical Society of AIME: Englewood, NJ, USA, 1984; pp. 125–144. [Google Scholar]
- Abotsi, G.M.K.; Osseo-Asare, K. Surface chemistry of carbonaceous gold ores I. Characterization of the carbonaceous matter and adsorption behavior in aurocyanide solution. Int. J. Miner. Process. 1986, 18, 217–236. [Google Scholar] [CrossRef]
- Afenya, P.M. Treatment of carbonaceous refractory gold ores. Miner. Eng. 1991, 4, 1043–1055. [Google Scholar] [CrossRef]
- Turner, S.J.; Flindell, P.A.; Hendri, D.; Hardjana, I.; Lauricella, P.F.; Lindsay, R.P.; Marpaung, B.; White, G.P. Sediment-hosted gold mineralisation in the Ratatotok district, North Sulawesi, Indonesia. J. Geochem. Explor. 1994, 50, 317–336. [Google Scholar] [CrossRef]
- Miller, J.D.; Wan, R.Y.; Díaz, X. Preg-robbing gold ores. Dev. Miner. Process. 2005, 15, 937–972. [Google Scholar] [CrossRef]
- Marsden, J.; House, I. The Chemistry of Gold Extraction; SME: Englewood, NJ, USA, 2006; pp. 147–224. [Google Scholar]
- Yang, H.Y.; Liu, Q.; Song, X.L.; Dong, J.K. Research status of carbonaceous matter in carbonaceous gold ores and bio-oxidation pretreatment. Trans. Nonferrous Met. Soc. China 2013, 23, 3405–3411. [Google Scholar] [CrossRef]
- Ofori-Sarpong, G.; Osseo-Asare, K. Preg-robbing of gold from cyanide and non-cyanide complexes: Effect of fungi pretreatment of carbonaceous matter. Int. J. Miner. Process. 2013, 119, 27–33. [Google Scholar] [CrossRef]
- Konadu, K.T.; Mendoza, D.M.; Huddy, R.J.; Harrison, S.T.L.; Kaneta, T.; Sasaki, K. Biological pretreatment of carbonaceous matter in double refractory gold ores: A review and some future considerations. Hydrometallurgy 2020, 196, 105434. [Google Scholar] [CrossRef]
- Hammerschmidt, J.; Guntner, J.; Kerstiens, B. Roasting of gold ore in the circulating fluidized-bed technology. Dev. Miner. Process. 2005, 15, 433–453. [Google Scholar] [CrossRef]
- Thomas, K.G.; Cole, A.P. Roasting Developments—Especially Oxygenated Roasting. In Gold Ore Processing; Ken, T., Murray, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; Volume 3, pp. 403–432. [Google Scholar] [CrossRef]
- La Brooy, S.R.; Linge, H.G.; Walker, G.S. Review of gold extraction from ores. Miner. Eng. 1994, 7, 1213–1241. [Google Scholar] [CrossRef]
- Abotsi, G.M.K.; Osseo-Asare, K. Surface chemistry of carbonaceous gold ores, II. Effects of organic additives on gold adsorption from cyanide solution. Int. J. Miner. Process. 1987, 21, 225–239. [Google Scholar] [CrossRef]
- Kuddus, M.; Joseph, B.; Wasudev Ramteke, P. Production of laccase from newly isolated Pseudomonas putida and its application in bioremediation of synthetic dyes and industrial effluents. Biocatal. Agric. Biotechnol. 2013, 2, 333–338. [Google Scholar] [CrossRef]
- Konadu, K.T.; Sasaki, K.; Ofori-Sarpong, G.; Osseo-Asare, K.; Kaneta, T. Bio-modification of carbonaceous matters in gold ore: Model experiments using powdered activated charcoal and cell-free extracts of Phanerochaete chrysosporium. Adv. Mat. Res. 2015, 1130, 109–113. [Google Scholar] [CrossRef]
- Liu, Q.; Yang, H.Y.; Tong, L.L.; Jin, Z.N.; Sand, W. Fungal degradation of elemental carbon in Carbonaceous gold ore. Hydrometallurgy 2016, 160, 90–97. [Google Scholar] [CrossRef]
- Konadu, K.T.; Sasaki, K.; Kaneta, T.; Ofori-Sarpong, G.; Osseo-Asare, K. Bio-modification of carbonaceous matter in gold ores: Model experiments using powdered activated carbon and cell-free spent medium of Phanerochaete chrysosporium. Hydrometallurgy 2017, 168, 76–83. [Google Scholar] [CrossRef]
- Mendoza, D.M.; Ichinose, H.; Konadu, K.T.; Sasaki, K. Degradation of powder activated carbon by laccase-mediator system: Model experiments for the improvement of gold recovery from carbonaceous gold ore. J. Environ. Chem. Eng. 2021, 9, 106375. [Google Scholar] [CrossRef]
- Ten Have, R.; Teunissen, P.J.M. Oxidative mechanisms involved in lignin degradation by white-rot fungi. Chem. Rev. 2001, 101, 3397–3413. [Google Scholar] [CrossRef] [PubMed]
- Tien, M.; Kirk, T.K. Lignin peroxidase of Phanerochaete chrysosporium. Methods Enzym. 1988, 161, 238–299. [Google Scholar] [CrossRef]
- Wariishi, H.; Valli, K.; Gold, M.H. In vitro depolymerization of lignin by Manganese Peroxidase of Phanerochaete chrysosporium. Biochem. Biophys. Res. Commun. 1991, 176, 269–275. [Google Scholar] [CrossRef]
- Ofori-Sarpong, G.; Osseo-Asare, K.; Tien, M. Mycohydrometallurgy: Biotransformation of double refractory gold ores by the fungus, Phanerochaete chrysosporium. Hydrometallurgy 2013, 137, 38–44. [Google Scholar] [CrossRef]
- Konadu, K.T.; Harrison, S.T.L.; Osseo-Asare, K.; Sasaki, K. Transformation of the carbonaceous matter in double refractory gold ore by crude lignin peroxidase released from the white-rot fungus. Int. Biodeterior. Biodegrad. 2019, 143, 104735. [Google Scholar] [CrossRef]
- Ciftci, H.; Akcil, A. Effect of biooxidation conditions on cyanide consumption and gold recovery from a refractory gold concentrate. Hydrometallurgy 2010, 104, 142–149. [Google Scholar] [CrossRef]
- Asamoah, R.K. Specific refractory gold flotation and bio-oxidation products: Research overview. Minerals 2021, 11, 93. [Google Scholar] [CrossRef]
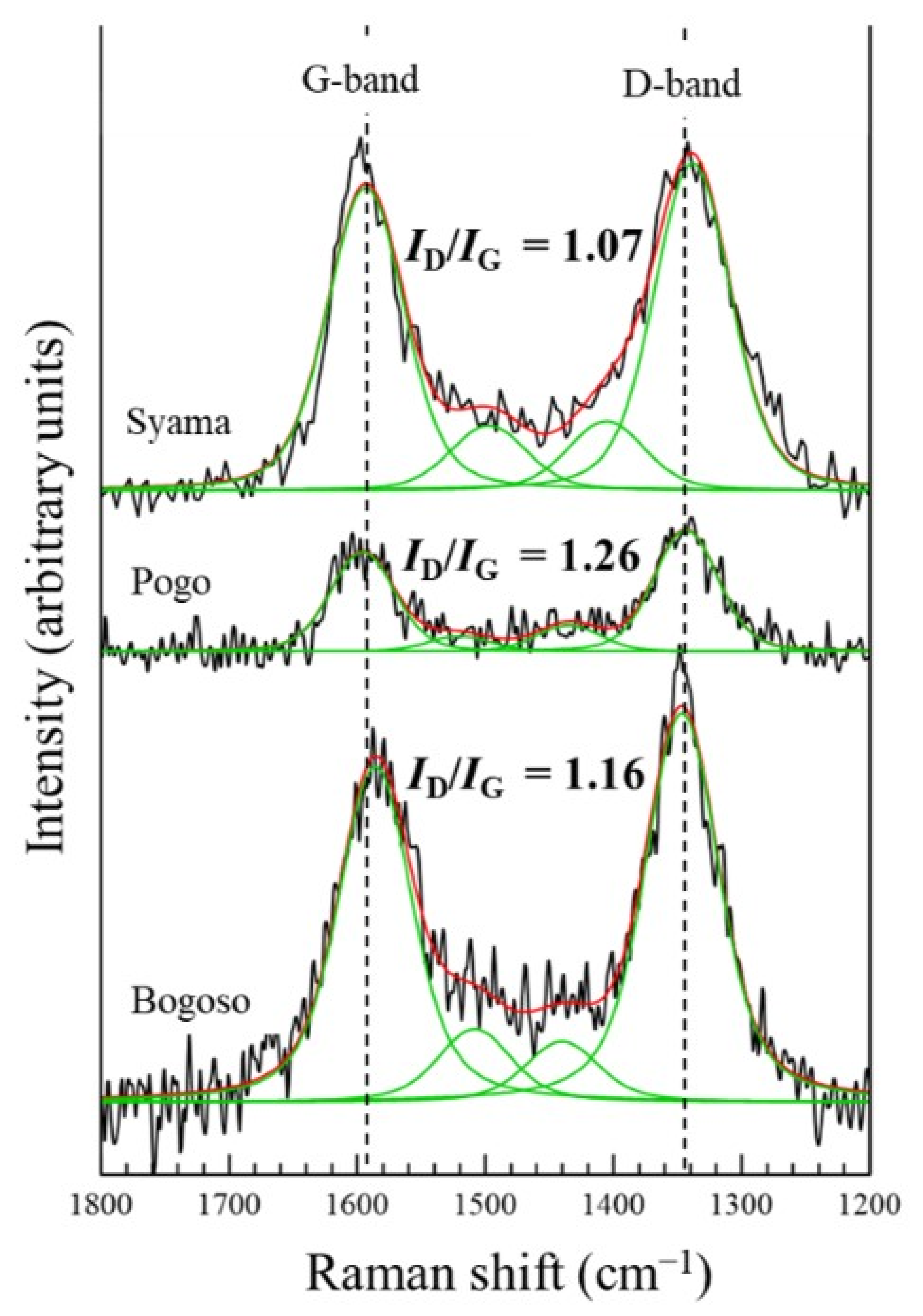
- Ferrari, A.C. Raman spectroscopy of graphene and graphite: Disorder, electron-phonon coupling, doping and nonadiabatic effects. Solid State Commun. 2007, 143, 47–57. [Google Scholar] [CrossRef]
- Mendoza, D.M.; Konadu, K.T.; Aoki, Y.; Kameya, M.; Sasaki, K. Carbonaceous matter degradation by fungal enzyme treatment to improve Ag recovery from an Au-Ag-bearing concentrate. Miner. Eng. 2021, 163, 106768. [Google Scholar] [CrossRef]
- Dimov, S.S.; Hart, B.R. Applications of microbeam analytical techniques in gold deportment studies and characterization of losses during the gold recovery process. Surf. Interface Anal. 2017, 49, 1404–1415. [Google Scholar] [CrossRef]
- Sonibare, O.O.; Haeger, T.; Foley, S.F. Structural characterization of Nigerian coals by X-ray diffraction, Raman and FTIR spectroscopy. Energy 2010, 35, 5347–5353. [Google Scholar] [CrossRef]
- Konadu, K.T.; Huddy, R.J.; Harrison, S.T.L.; Osseo-Asare, K.; Sasaki, K. Sequential pretreatment of double refractory gold ore (DRGO) with a thermophilic iron oxidizing archaeon and fungal crude enzymes. Miner. Eng. 2019, 138, 86–94. [Google Scholar] [CrossRef]
- Zeng, G.M.; Zhao, M.H.; Huang, D.L.; Lai, C.; Huang, C.; Wei, Z.; Cheng, M. Purification and biochemical characterization of two extracellular peroxidases from Phanerochaete chrysosporium responsible for lignin biodegradation. Int. Biodeterior. Biodegrad. 2013, 85, 166–172. [Google Scholar] [CrossRef]
- West, T.O.; McBride, A.C. The contribution of agricultural lime to carbon dioxide emissions in the United States: Dissolution, transport, and net emissions. Agric. Ecosyst. Environ. 2005, 108, 145–154. [Google Scholar] [CrossRef]
- Ofori-Sarpong, G.; Adam, A.-S.; Komla Asamoah, R.; Kwasi Amankwah, R. Characterisation of biooxidation feed and products for improved understanding of biooxidation and gold extraction performance. Int. J. Miner. Process. Extr. Metall. 2020, 5, 20. [Google Scholar] [CrossRef]
- Sasaki, K. Raman study of the microbially mediated dissolution of pyrite by Thiobacillus ferrooxidans. Can. Mineral. 1997, 35, 999–1008. [Google Scholar]
- Smith, M.M.; Wolery, T.J.; Carroll, S.A. Kinetics of chlorite dissolution at elevated temperatures and CO2 conditions. Chem. Geol. 2013, 347, 1–8. [Google Scholar] [CrossRef]
- Kuwahara, M.; Glenn, J.K.; Morgan, M.A.; Gold, M.H. Separation and characterization of two extracelluar H2O2-dependent oxidases from ligninolytic cultures of Phanerochaete chrysosporium. FEBS Lett. 1984, 169, 247–250. [Google Scholar] [CrossRef] [Green Version]
- Fakoussa, R.M.; Hofrichter, M. Biotechnology and microbiology of coal degradation. Appl. Microbiol. Biotechnol. 1999, 52, 25–40. [Google Scholar] [CrossRef]
- Sasaki, K.; Tsunekawa, M.; Hasebe, K.; Konno, H. Effect of anionic ligands on the reactivity of pyrite with Fe(III) ions in acid solutions. Colloids Surf. A 1995, 101, 39–49. [Google Scholar] [CrossRef]
- Gao, J.; Liang, C.; Shen, G.; Lv, J.; Wu, H. Spectral characteristics of dissolved organic matter in various agricultural soils throughout China. Chemosphere 2017, 176, 108–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.Y.; Cabri, L.J. Gold process mineralogy: Objectives, techniques, and applications. JOM 2004, 56, 49–52. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Elements (wt%), and Their Relative Intensities (RI) Normalized to Si Contents | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C | O | Na | Mg | Al | Si | P | S | Cl | K | Ca | Ti | Cr | Mn | Fe | Co | Ni | Cu | Zn | As | Sr | Zr | ||
| DRGO | wt% | 6.83 | 41.75 | 1.00 | 1.67 | 7.45 | 15.69 | 0.07 | 6.13 | n.d. | 1.98 | 3.11 | 0.55 | 0.05 | 0.09 | 13.38 | n.d. | 0.03 | 0.10 | 0.03 | 0.08 | 0.02 | 0.01 |
| RI | 0.435 | 2.661 | 0.064 | 0.106 | 0.475 | 1.000 | 0.004 | 0.391 | n.d. | 0.126 | 0.198 | 0.035 | 0.003 | 0.006 | 0.853 | n.d. | 0.002 | 0.006 | 0.002 | 0.005 | 0.001 | 0.001 | |
| DW | wt% | 6.09 | 39.49 | 1.16 | 0.83 | 8.37 | 18.28 | 0.03 | 8.35 | n.d. | 2.43 | 0.24 | 0.56 | 0.04 | n.d. | 13.80 | 0.04 | 0.05 | 0.10 | 0.02 | 0.10 | 0.01 | 0.02 |
| RI | 0.333 | 2.160 | 0.063 | 0.045 | 0.458 | 1.000 | 0.002 | 0.457 | n.d. | 0.133 | 0.013 | 0.030 | 0.002 | n.d. | 0.755 | 0.002 | 0.003 | 0.006 | 0.001 | 0.005 | 0.000 | 0.001 | |
| DA | wt% | 6.33 | 44.97 | 1.12 | 0.43 | 8.99 | 20.73 | 0.11 | 3.65 | n.d. | 3.55 | n.d. | 0.85 | 0.04. | n.d. | 9.04 | n.d. | 0.02 | 0.04 | n.d. | 0.08 | 0.01 | 0.05 |
| RI | 0.306 | 2.169 | 0.054 | 0.021 | 0.434 | 1.000 | 0.005 | 0.176 | n.d. | 0.171 | n.d. | 0.041 | 0.002 | n.d. | 0.436 | n.d. | 0.001 | 0.002 | n.d. | 0.004 | 0.001 | 0.002 | |
| DAW | wt% | 7.53 | 43.87 | 1.21 | 0.37 | 8.73 | 20.72 | 0.05 | 4.92 | n.d. | 3.03 | 0.02 | 0.83 | 0.05 | n.d. | 8.54 | n.d. | 0.02 | 0.03 | 0.01 | 0.05 | 0.01 | 0.02 |
| RI | 0.363 | 2.118 | 0.058 | 0.018 | 0.422 | 1.000 | 0.002 | 0.238 | n.d. | 0.147 | 0.001 | 0.040 | 0.002 | n.d. | 0.412 | n.d. | 0.001 | 0.001 | 0.001 | 0.002 | 0.000 | 0.001 | |
| DAC | wt% | 8.30 | 44.80 | 1.22 | 0.32 | 8.21 | 20.40 | 0.19 | 4.18 | n.d. | 3.21 | 0.04 | 0.76 | 0.05 | n.d. | 8.16 | n.d. | n.d. | 0.03 | 0.03 | 0.05 | 0.01 | 0.01 |
| RI | 0.407 | 2.196 | 0.060 | 0.016 | 0.403 | 1.000 | 0.009 | 0.205 | n.d. | 0.158 | 0.002 | 0.037 | 0.002 | n.d. | 0.400 | n.d. | n.d. | 0.002 | 0.001 | 0.003 | 0.001 | 0.001 | |
| DAWC | wt% | 8.38 | 43.30 | 1.19 | 0.38 | 9.19 | 21.71 | 0.12 | 3.86 | n.d. | 3.36 | 0.04 | 0.77 | 0.05 | n.d. | 7.50 | n.d. | 0.01 | 0.03 | 0.03 | 0.04 | 0.01 | 0.02 |
| RI | 0.386 | 1.994 | 0.055 | 0.018 | 0.423 | 1.000 | 0.006 | 0.178 | n.d. | 0.155 | 0.002 | 0.035 | 0.002 | n.d. | 0.345 | n.d. | 0.001 | 0.001 | 0.001 | 0.002 | 0.001 | 0.001 | |
| DACW | wt% | 8.53 | 44.12 | 1.51 | 0.40 | 9.06 | 21.34 | 0.02 | 2.55 | 0.06 | 3.05 | 0.04 | 0.78 | 0.06 | n.d. | 8.36 | n.d. | n.d. | 0.03 | 0.03 | 0.04 | 0.01 | 0.02 |
| RI | 0.400 | 2.068 | 0.071 | 0.019 | 0.425 | 1.000 | 0.001 | 0.119 | 0.003 | 0.143 | 0.002 | 0.037 | 0.003 | n.d. | 0.392 | n.d. | n.d. | 0.001 | 0.001 | 0.002 | 0.001 | 0.001 | |
| DAWCW | wt% | 8.85 | 43.34 | 1.57 | 0.33 | 9.07 | 21.81 | 0.02 | 3.35 | 0.07 | 2.98 | 0.03 | 0.82 | n.d. | n.d. | 7.66 | n.d. | n.d. | 0.03 | 0.02 | 0.03 | n.d. | 0.02 |
| RI | 0.406 | 1.987 | 0.072 | 0.015 | 0.416 | 1.000 | 0.001 | 0.154 | 0.003 | 0.137 | 0.001 | 0.037 | n.d. | n.d. | 0.351 | n.d. | n.d. | 0.001 | 0.001 | 0.002 | n.d. | 0.001 | |
| Sample | C (%) | H (%) | N (%) |
|---|---|---|---|
| DRGO | 5.27 | 0.30 | 0.08 |
| DW | 4.16 | 0.27 | 0.07 |
| DA | 6.07 | 0.44 | 0.19 |
| DAC | 7.40 | 0.57 | 0.21 |
| DACW | 7.22 | 0.50 | 0.14 |
| DAW | 6.53 | 0.40 | 0.18 |
| DAWC | 6.84 | 0.46 | 0.17 |
| DAWCW | 6.87 | 0.42 | 0.13 |
| Sample | Recovery (%) |
|---|---|
| DRGO | 30 ± 6.8 |
| DW | 41.5 ± 0.3 |
| DA | 62.6 ± 3.0 |
| DAC | 64.4 ± 9.2 |
| DACW | 72.5 ± 0.9 |
| DAW | 66.4 ± 1.7 |
| DAWC | 84.9 ± 0.7 |
| DAWCW | 71.3 ± 5.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cindy; Sakai, R.; Mendoza, D.M.; Konadu, K.T.; Sasaki, K. Significance of Acid Washing after Biooxidation of Sulfides in Sequential Biotreatment of Double Refractory Gold Ore from the Syama Mine, Mali. Minerals 2021, 11, 1316. https://doi.org/10.3390/min11121316
Cindy, Sakai R, Mendoza DM, Konadu KT, Sasaki K. Significance of Acid Washing after Biooxidation of Sulfides in Sequential Biotreatment of Double Refractory Gold Ore from the Syama Mine, Mali. Minerals. 2021; 11(12):1316. https://doi.org/10.3390/min11121316
Chicago/Turabian StyleCindy, Ryotaro Sakai, Diego M. Mendoza, Kojo T. Konadu, and Keiko Sasaki. 2021. "Significance of Acid Washing after Biooxidation of Sulfides in Sequential Biotreatment of Double Refractory Gold Ore from the Syama Mine, Mali" Minerals 11, no. 12: 1316. https://doi.org/10.3390/min11121316
APA StyleCindy, Sakai, R., Mendoza, D. M., Konadu, K. T., & Sasaki, K. (2021). Significance of Acid Washing after Biooxidation of Sulfides in Sequential Biotreatment of Double Refractory Gold Ore from the Syama Mine, Mali. Minerals, 11(12), 1316. https://doi.org/10.3390/min11121316