
Low-Temperature Chlorite Geothermometry and Related Recent Analytical Advances: A Review


Abstract
:1. Introduction
2. Compositional Variability of Chlorites
- Tschermak substitution (TK) as SiIV + (Fe2+,Mg2+)VI = AlIV + AlVI
- di/trioctahedral substitution (DT) as 2(Al3+,Fe3+)VI + □VI = 3(Fe2+,Mg2+)VI
- ferromagnesian substitution (FM) as Mg2+ = Fe2+
- trivalent cation substitution (AF) as Al3+ = Fe3+
3. Concepts of Chlorite Thermometers
4. Chemical Heterogeneity, Achievement of Equilibrium, and Related Analytical Advances
5. Perspectives of New Analytical and Thermometric Developments
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Okuyama-Kusunose, Y.; Itaya, T. Metamorphism of Carbonaceous Material in the Tono Contact Aureole, Kitakami Mountains, Japan. J. Metamorph. Geol. 1987, 5, 121–139. [Google Scholar] [CrossRef]
- Beyssac, O.; Pattison, D.R.M.; Bourdelle, F. Contrasting Degrees of Recrystallization of Carbonaceous Material in the Nelson Aureole, British Columbia and Ballachulish Aureole, Scotland, with Implications for Thermometry Based on Raman Spectroscopy of Carbonaceous Material. J. Metamorph. Geol. 2019, 37, 71–95. [Google Scholar] [CrossRef] [Green Version]
- Beyssac, O.; Rouzaud, J.N.; Goffe, B.; Brunet, F.; Chopin, C. Graphitization in a High-Pressure, Low-Temperature Metamorphic Gradient: A Raman Microspectroscopy and HRTEM Study. Contrib. Mineral. Petrol. 2002, 143, 19–31. [Google Scholar] [CrossRef]
- Lahfid, A.; Beyssac, O.; Deville, E.; Negro, F.; Chopin, C.; Goffe, B. Evolution of the Raman Spectrum of Carbonaceous Material in Low-Grade Metasediments of the Glarus Alps (Switzerland). Terra Nova 2010, 22, 354–360. [Google Scholar] [CrossRef]
- Dubois, M.; Ayt Ougougdal, M.; Meere, P.; Royer, J.-J.; Boiron, M.-C.; Cathelineau, M. Temperature of Paleo- to Modern Self-Sealing within a Continental Rift Basin; the Fluid Inclusion Data (Soultz-Sous-Forets, Rhine Graben, France). Eur. J. Mineral. 1996, 8, 1065–1080. [Google Scholar] [CrossRef]
- Roedder, E. Fluid Inclusions: An Introduction to Studies of All Types of Fluid Inclusions, Gas, Liquid, or Melt, Trapped in Materials from Earth and Space, and Their Application to the Understanding of Geologic Processes; Reviews in Mineralogy; Mineral Soc. of America: Washington, DC, USA, 1984; ISBN 978-0-939950-16-4. [Google Scholar]
- Samson, I. (Ed.) Fluid Inclusions: Analysis and Interpretation; Short Course Series; Mineralogical Association: Ottawa, ON, Canada, 2003; ISBN 978-0-921294-32-0. [Google Scholar]
- Burkhard, M.; Kerrich, R.; Maas, R.; Fyfe, W.S. Stable and Sr-Isotope Evidence for Fluid Advection during Thrusting of the Glarus Nappe (Swiss Alps). Contrib. Mineral. Petrol. 1992, 112, 293–311. [Google Scholar] [CrossRef]
- Vidal, O.; Parra, T.; Trotet, F. A Thermodynamic Model for Fe-Mg Aluminous Chlorite Using Data from Phase Equilibrium Experiments and Natural Pelitic Assemblages in the 100 Degrees to 600 Degrees C, 1 to 25 Kb Range. Am. J. Sci. 2001, 301, 557–592. [Google Scholar] [CrossRef] [Green Version]
- Bailey, S.W. (Ed.) Chlorites: Structures and Crystal Chemistry. In Hydrous Phyllosilicates; De Gruyter: Berlin, Germany; Boston, MA, USA, 1988; ISBN 978-1-5015-0899-8. [Google Scholar]
- Walshe, J.L. A Six-Component Chlorite Solid Solution Model and the Conditions of Chlorite Formation in Hydrothermal and Geothermal Systems. Econ. Geol. 1986, 81, 681–703. [Google Scholar] [CrossRef]
- Cathelineau, M.; Nieva, D. A Chlorite Solid Solution Geothermometer the Los Azufres (Mexico) Geothermal System. Contrib. Mineral. Petrol. 1985, 91, 235–244. [Google Scholar] [CrossRef]
- De Caritat, P.; Hutcheon, I.; Walshe, J. Chlorite Geothermometry—A Review. Clays Clay Miner. 1993, 41, 219–239. [Google Scholar] [CrossRef]
- Essene, E.J.; Peacor, D.R. Clay Mineral Thermometry—A Critical Perspective. Clays Clay Miner. 1995, 43, 540–553. [Google Scholar] [CrossRef]
- Beaufort, D.; Rigault, C.; Billon, S.; Billault, V.; Inoue, A.; Inoue, S.; Patrier, P. Chlorite and Chloritization Processes through Mixed-Layer Mineral Series in Low Temperature Geological Systems—A Review. Clay Miner. 2015, 50, 497–523. [Google Scholar] [CrossRef]
- Masci, L.; Dubacq, B.; Verlaguet, A.; Chopin, C.; De Andrade, V.; Herviou, C. A XANES and EPMA Study of Fe3+ in Chlorite: Importance of Oxychlorite and Implications for Cation Site Distribution and Thermobarometry. Am. Mineral. 2019, 104, 403–417. [Google Scholar] [CrossRef]
- Wiewiora, A.; Weiss, Z. Crystallochemical Classifications of Phyllosilicates Based on the Unified System of Projection of Chemical Composition; II, The Chlorite Group. Clay Miner. 1990, 25, 83–92. [Google Scholar] [CrossRef]
- Meunier, A. Clays; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2005; ISBN 978-3-540-21667-4. [Google Scholar]
- Holland, T.; Baker, J.; Powell, R. Mixing Properties and Activity-Composition Relationships of Chlorites in the System MgO-FeO-Al2O3-SiO2-H2O. Eur. J. Mineral. 1998, 10, 395–406. [Google Scholar] [CrossRef]
- Helgeson, H.C.; Aagaard, P. Activity/Composition Relations among Silicates and Aqueous Solutions; I, Thermodynamics of Intrasite Mixing and Substitutional Order/Disorder in Minerals. Am. J. Sci. 1985, 285, 769–844. [Google Scholar] [CrossRef]
- Stoessell, R.K. Regular Solution Site-Mixing Model for Chlorites. Clays Clay Miner. 1984, 32, 205–212. [Google Scholar] [CrossRef]
- Vidal, O.; Parra, T. Exhumation Paths of High-Pressure Metapelites Obtained from Local Equilibria for Chlorite-Phengite Assemblages. Geol. J. 2000, 35, 139–161. [Google Scholar] [CrossRef]
- Bourdelle, F.; Cathelineau, M. Low-Temperature Chlorite Geothermometry: A Graphical Representation Based on a T-R2+-Si Diagram. Eur. J. Mineral. 2015, 27, 617–626. [Google Scholar] [CrossRef]
- Hillier, S.; Velde, B. Octahedral Occupancy and the Chemical-Composition of Diagenetic (Low-Temperature) Chlorites. Clay Miner. 1991, 26, 149–168. [Google Scholar] [CrossRef]
- Cathelineau, M. Cation Site Occupancy in Chlorites and Illites as a Function of Temperature. Clay Miner. 1988, 23, 471–485. [Google Scholar] [CrossRef]
- Velde, B.; Medhioub, M. Approach to Chemical Equilibrium in Diagenetic Chlorites. Contrib. Mineral. Petrol. 1988, 98, 122–127. [Google Scholar] [CrossRef]
- Jahren, J.S. Compositional Variations in Diagenetic Chlorites and Illites, and Relationships with Formation-Water Chemistry. Clay Miner. 1989, 24, 157–170. [Google Scholar] [CrossRef]
- Inoue, A.; Meunier, A.; Patrier-Mas, P.; Rigault, C.; Beaufort, D.; Vieillard, P. Application of Chemical Geothermometry to Low-Temperature Trioctahedral Chlorites. Clays Clay Miner. 2009, 57, 371–382. [Google Scholar] [CrossRef]
- Bourdelle, F.; Parra, T.; Beyssac, O.; Chopin, C.; Vidal, O. Clay Minerals as Geo-Thermometer: A Comparative Study Based on High Spatial Resolution Analyses of Illite and Chlorite in Gulf Coast Sandstones (Texas, USA). Am. Mineral. 2013, 98, 914–926. [Google Scholar] [CrossRef]
- McDowell, S.D.; Elders, W.A. Authigenic Layer Silicate Minerals in Borehole Elmore 1, Salton Sea Geothermal Field, California, USA. Contrib. Mineral. Petrol. 1980, 74, 293–310. [Google Scholar] [CrossRef]
- Bevins, R.; Robinson, D.; Rowbotham, G. Compositional Variations in Mafic Phyllosilicates from Regional Low-Grade Metabasites and Application of the Chlorite Geothermometer. J. Metamorph. Geol. 1991, 9, 711–721. [Google Scholar] [CrossRef]
- Jahren, J.; Aagaard, P. Diagenetic Illite-Chlorite Assemblages in Arenites. 1. Chemical Evolution. Clays Clay Miner. 1992, 40, 540–546. [Google Scholar] [CrossRef]
- Rahn, M.; Mullis, J.; Erdelbrock, K.; Frey, M. Very Low-Grade Metamorphism of the Taveyanne Greywacke, Glarus Alps, Switzerland. J. Metamorph. Geol. 1994, 12, 625–641. [Google Scholar] [CrossRef]
- Schmidt, D.; Livi, K.J.T.; Frey, M. Reaction Progress in Chloritic Material: An Electron Microbeam Study of the Taveyanne Greywacke, Switzerland. J. Metamorph. Geol. 1999, 17, 229–241. [Google Scholar] [CrossRef]
- Mas, A.; Guisseau, D.; Mas, P.P.; Beaufort, D.; Genter, A.; Sanjuan, B.; Girard, J.P. Clay Minerals Related to the Hydrothermal Activity of the Bouillante Geothermal Field (Guadeloupe). J. Volcanol. Geotherm. Res. 2006, 158, 380–400. [Google Scholar] [CrossRef]
- Koroknai, B.; Arkai, P.; Horvath, P.; Balogh, K. Anatomy of a Transitional Brittle-Ductile Shear Zone Developed in a Low-T Meta-Andesite Tuff: A Microstructural, Petrological and Geochronological Case Study from the Bukk Mts. (NE Hungary). J. Struct. Geol. 2008, 30, 159–176. [Google Scholar] [CrossRef]
- Beaufort, D.; Patrier, P.; Meunier, A.; Ottaviani, M. Chemical Variations in Assemblages Including Epidote and or Chlorite in the Fossil Hydrothermal System of Saint Martin (Lesser Antilles). J. Volcanol. Geotherm. Res. 1992, 51, 95–114. [Google Scholar] [CrossRef]
- Xie, X.G.; Byerly, G.R.; Ferrell, R.E. IIb Trioctahedral Chlorite from the Barberton Greenstone Belt: Crystal Structure and Rock Composition Constraints with Implications to Geothermometry. Contrib. Mineral. Petrol. 1997, 126, 275–291. [Google Scholar] [CrossRef]
- Lopez-Munguira, A.; Nieto, F.; Morata, D. Chlorite Composition and Geothermometry: A Comparative HRTEM/AEM-EMPA-XRD Study of Cambrian Basic Lavas from the Ossa Morena Zone, SW Spain. Clay Miner. 2002, 37, 267–281. [Google Scholar] [CrossRef] [Green Version]
- Potel, S. Very Low-Grade Metamorphic Study in the Pre-Late Cretaceous Terranes of New Caledonia (Southwest Pacific Ocean). Isl. Arc 2007, 16, 291–305. [Google Scholar] [CrossRef]
- Percival, J.B.; Kodama, H. Sudoite from Cigar Lake, Saskatchewan. Can. Mineral. 1989, 27, 633–641. [Google Scholar]
- Hutcheon, I. Clay Carbonate Reactions in the Venture Area, Scotian Shelf, Nova Scotia, Canada. Geochem. Soc. 1990, 2, 199–212. [Google Scholar]
- Bourdelle, F.; Beyssac, O.; Parra, T.; Chopin, C. Nanoscale Chemical Zoning of Chlorite and Implications for Low-Temperature Thermometry: Application to the Glarus Alps (Switzerland). Lithos 2018, 314, 551–561. [Google Scholar] [CrossRef]
- Hutcheon, I.; Oldershaw, A.; Ghent, E.D. Diagenesis of Cretaceous Sandstones of the Kootenay Formation at Elk Valley (Southeastern British Columbia) and Mt Allan (Southwestern Alberta). Geochim. Cosmochim. Acta 1980, 44, 1425–1435. [Google Scholar] [CrossRef]
- Zang, W.; Fyfe, W. Chloritization of the Hydrothermally Altered Bedrock at the Igarape-Bahia Gold Deposit, Carajas, Brazil. Miner. Depos. 1995, 30, 30–38. [Google Scholar] [CrossRef]
- Waldie, C.J.; Jowett, E.C.; Swinden, H.S. The Crescent Lake Copper Deposit, Central Newfoundland: Deep Levels of a Volcanogenic Hydrothermal System? Atl. Geol. 1991, 27. [Google Scholar] [CrossRef] [Green Version]
- Kranidiotis, P.; MacLean, W.H. Systematics of Chlorite Alteration at the Phelps Dodge Massive Sulfide Deposit, Matagami, Quebec. Econ. Geol. 1987, 82, 1898–1911. [Google Scholar] [CrossRef]
- Jowett, E.C. Fitting Iron and Magnesium into the Hydrothermal Chlorite Geothermometer. In Proceedings of the GAC/MAC/SEG Joint Annual Meeting, Toronto, ON, Canada, 27–29 May 1991. [Google Scholar]
- Vidal, O.; Parra, T.; Vieillard, P. Thermodynamic Properties of the Tschermak Solid Solution in Fe-Chlorite: Application to Natural Examples and Possible Role of Oxidation. Am. Mineral. 2005, 90, 347–358. [Google Scholar] [CrossRef]
- Vidal, O.; De Andrade, V.; Lewin, E.; Munoz, M.; Parra, T.; Pascarelli, S. P-T-Deformation-Fe3+/Fe2+ Mapping at the Thin Section Scale and Comparison with XANES Mapping: Application to a Garnet-Bearing Metapelite from the Sambagawa Metamorphic Belt (Japan). J. Metamorph. Geol. 2006, 24, 669–683. [Google Scholar] [CrossRef]
- Lanari, P.; Wagner, T.; Vidal, O. A Thermodynamic Model for Di-Trioctahedral Chlorite from Experimental and Natural Data in the System MgO-FeO-Al2O3- SiO2-H2O: Applications to P-T Sections and Geothermometry. Contrib. Mineral. Petrol. 2014, 167, 968. [Google Scholar] [CrossRef] [Green Version]
- Parra, T.; Vidal, O.; Theye, T. Experimental Data on the Tschermak Substitution in Fe-Chlorite. Am. Mineral. 2005, 90, 359–370. [Google Scholar] [CrossRef]
- Vidal, O.; Lanari, P.; Munoz, M.; Bourdelle, F.; De Andrade, V. Deciphering Temperature, Pressure and Oxygen-Activity Conditions of Chlorite Formation. Clay Miner. 2016, 51, 615–633. [Google Scholar] [CrossRef] [Green Version]
- Trincal, V.; Lanari, P. Al-Free Di-Trioctahedral Substitution in Chlorite and a Ferri-Sudoite End-Member. Clay Miner. 2016, 51, 675–689. [Google Scholar] [CrossRef]
- Bourdelle, F.; Parra, T.; Chopin, C.; Beyssac, O. A New Chlorite Geothermometer for Diagenetic to Low-Grade Metamorphic Conditions. Contrib. Mineral. Petrol. 2013, 165, 723–735. [Google Scholar] [CrossRef]
- Aja, S. Excess Functions of Chlorite Solid Solutions and Neoformation of Fe-Chlorites: Some Implications of Recent Thermochemical Measurements. Am. Mineral. 2019, 104, 232–243. [Google Scholar] [CrossRef]
- Inoue, A.; Kurokawa, K.; Hatta, T. Application of Chlorite Geothermometry to Hydrothermal Alteration in Toyoha Geothermal System, Southwestern Hokkaido, Japan. Resour. Geol. 2010, 60, 52–70. [Google Scholar] [CrossRef]
- Harbi, H.M.; Surour, A.A.; Davidson, G.J. Genesis of Neoproterozoic Au-Bearing Volcanogenic Sulfides and Quartz Veins in the Ar Rjum Goldfield, Saudi Arabia. Ore Geol. Rev. 2014, 58, 110–125. [Google Scholar] [CrossRef]
- Mamadou, M.M.; Cathelineau, M.; Bourdelle, F.; Boiron, M.-C.; Elmaleh, A.; Brouand, M. Hot Fluid Flows Around a Major Fault Identified by Paleothermometric Studies (Tim Mersoi Basin, Niger). J. Sediment. Res. 2016, 86, 914–928. [Google Scholar] [CrossRef]
- Perez-Caceres, I.; Martinez Poyatos, D.J.; Vidal, O.; Beyssac, O.; Nieto, F.; Simancas, J.F.; Azor, A.; Bourdelle, F. Deciphering the Metamorphic Evolution of the Pulo Do Lobo Metasedimentary Domain (SW Iberian Variscides). Solid Earth 2020, 11, 469–488. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, M.; Nieto, F.; Morata, D.; Droguett, B.; Carrillo-Rosua, F.J.; Morales, S. Evolution of Clay Mineral Assemblages in the Tinguiririca Geothermal Field, Andean Cordillera of Central Chile: An XRD and HRTEM-AEM Study. J. Volcanol. Geotherm. Res. 2014, 282, 43–59. [Google Scholar] [CrossRef] [Green Version]
- Dolores Rodriguez-Ruiz, M.; Abad, I.; Bentabol, M.J. Permo-Triassic Clastic Rocks from the Ghomaride Complex and Federico Units (Rif Cordillera, N Morocco): An Example of Diagenetic-Metamorphic Transition. Minerals 2019, 9, 738. [Google Scholar] [CrossRef] [Green Version]
- Dowey, P.J.; Hodgson, D.M.; Worden, R.H. Pre-Requisites, Processes, and Prediction of Chlorite Grain Coatings in Petroleum Reservoirs: A Review of Subsurface Examples. Mar. Pet. Geol. 2012, 32, 63–75. [Google Scholar] [CrossRef]
- Mosser-Ruck, R.; Pignatelli, I.; Bourdelle, F.; Abdelmoula, M.; Barres, O.; Guillaume, D.; Charpentier, D.; Rousset, D.; Cathelineau, M.; Michau, N. Contribution of Long-Term Hydrothermal Experiments for Understanding the Smectite-to-Chlorite Conversion in Geological Environments. Contrib. Mineral. Petrol. 2016, 171, 97. [Google Scholar] [CrossRef]
- Jiang, W.; Peacor, D.; Buseck, P. Chlorite Geothermometry—Contamination and Apparent Octahedral Vacancies. Clays Clay Miner. 1994, 42, 593–605. [Google Scholar] [CrossRef]
- Hillier, S.; Velde, B. Chlorite Interstratified with a 7-a Mineral—An Example from Offshore Norway and Possible Implications for the Interpretation of the Composition of Diagenetic Chlorites. Clay Miner. 1992, 27, 475–486. [Google Scholar] [CrossRef]
- Curtis, C.D.; Ireland, B.J.; Whiteman, J.A.; Mulvaney, R.; Whittle, C.K. Authigenic Chlorites: Problems with Chemical Analysis and Structural Formula Calculations. Clay Miner. 1984, 19, 471–481. [Google Scholar] [CrossRef]
- Putnis, A. Mineral Replacement Reactions. In Thermodynamics and Kinetics of Water-Rock Interaction; Oelkers, E.H., Schott, J., Eds.; De Gruyter: Berlin, Germany, 2009; Volume 70, pp. 87–124. [Google Scholar]
- Putnis, C.V.; Geisler, T.; Schmid-Beurmann, P.; Stephan, T.; Giampaolo, C. An Experimental Study of the Replacement of Leucite by Analcime. Am. Mineral. 2007, 92, 19–26. [Google Scholar] [CrossRef]
- Trincal, V.; Lanari, P.; Buatier, M.; Lacroix, B.; Charpentier, D.; Labaume, P.; Munoz, M. Temperature Micro-Mapping in Oscillatory-Zoned Chlorite: Application to Study of a Green-Schist Facies Fault Zone in the Pyrenean Axial Zone (Spain). Am. Mineral. 2015, 100, 2468–2483. [Google Scholar] [CrossRef]
- Ganne, J.; De Andrade, V.; Weinberg, R.F.; Vidal, O.; Dubacq, B.; Kagambega, N.; Naba, S.; Baratoux, L.; Jessell, M.; Allibon, J. Modern-Style Plate Subduction Preserved in the Palaeoproterozoic West African Craton. Nat. Geosci. 2012, 5, 60–65. [Google Scholar] [CrossRef]
- Vidal, O.; Baldeyrou, A.; Beaufort, D.; Fritz, B.; Geoffroy, N.; Lanson, B. Experimental Study of the Stability and Phase Relations of Clays at High Temperature in a Thermal Gradient. Clays Clay Miner. 2012, 60, 200–225. [Google Scholar] [CrossRef]
- Jahren, J. Evidence of Ostwald Ripening Related Recrystallization of Diagenetic Chlorites from Reservoir Rocks Offshore Norway. Clay Miner. 1991, 26, 169–178. [Google Scholar] [CrossRef]
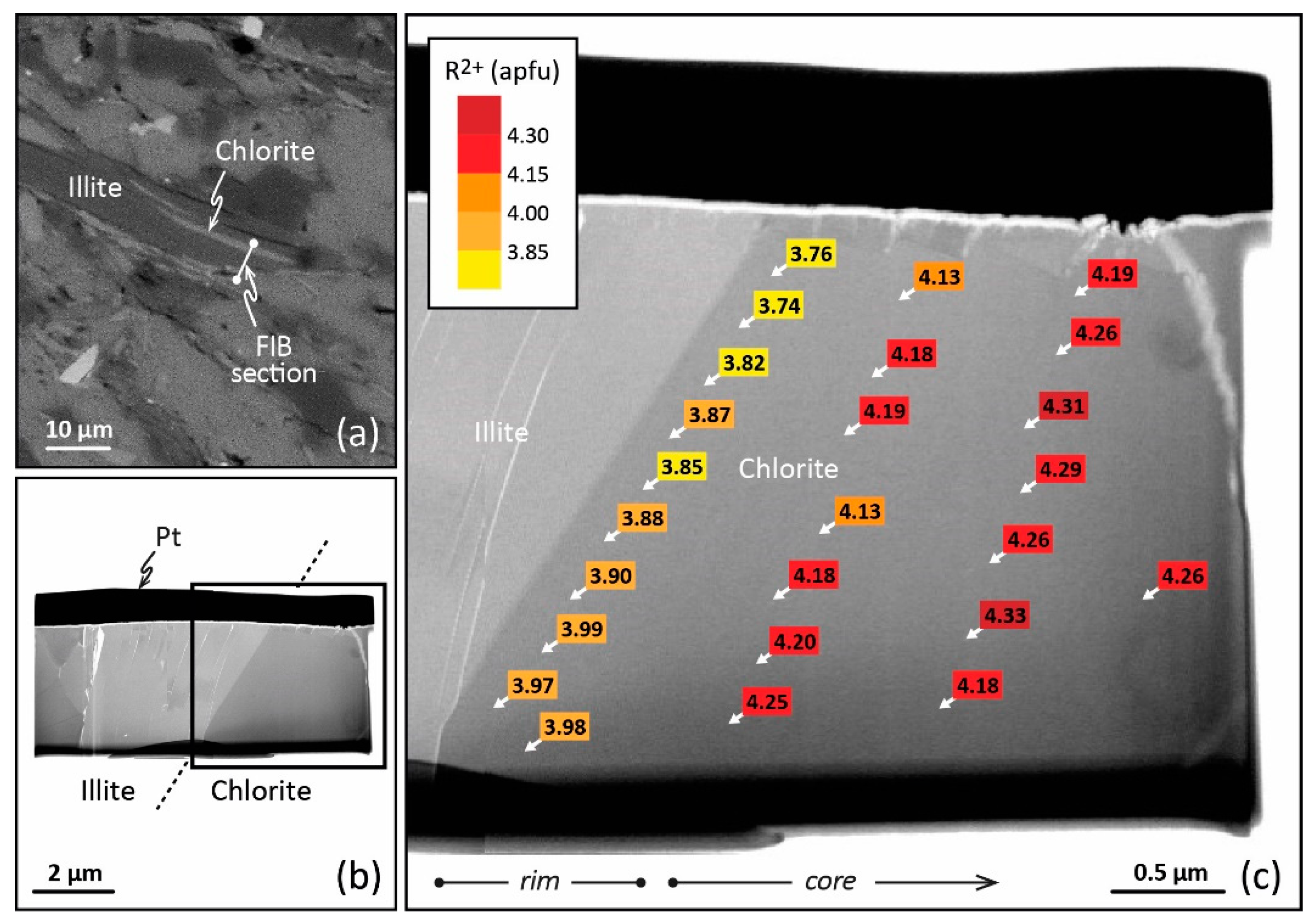
- Bourdelle, F.; Parra, T.; Beyssac, O.; Chopin, C.; Moreau, F. Ultrathin Section Preparation of Phyllosilicates by Focused Ion Beam Milling for Quantitative Analysis by TEM-EDX. Appl. Clay Sci. 2012, 59–60, 121–130. [Google Scholar] [CrossRef]
- Wirth, R. Focused Ion Beam (FIB): A Novel Technology for Advanced Application of Micro- and Nanoanalysis in Geosciences and Applied Mineralogy. Eur. J. Mineral. 2004, 16, 863–876. [Google Scholar] [CrossRef] [Green Version]
- Munoz, M.; De Andrade, V.; Vidal, O.; Lewin, E.; Pascarelli, S.; Susini, J. Redox and Speciation Micromapping Using Dispersive X-Ray Absorption Spectroscopy: Application to Iron Chlorite Mineral of a Metamorphic Rock Thin Section. Geochem. Geophys. Geosystems 2006, 7, Q11020. [Google Scholar] [CrossRef]
- Rat, J.; Mouthereau, F.; Brichau, S.; Cremades, A.; Bernet, M.; Balvay, M.; Ganne, J.; Lahfid, A.; Gautheron, C. Tectonothermal Evolution of the Cameros Basin: Implications for Tectonics of North Iberia. Tectonics 2019, 38, 440–469. [Google Scholar] [CrossRef]
- Grosch, E.G.; Vidal, O.; Abu-Alam, T.; McLoughlin, N. P-T Constraints on the Metamorphic Evolution of the Paleoarchean Kromberg Type-Section, Barberton Greenstone Belt, South Africa. J. Petrol. 2012, 53, 513–545. [Google Scholar] [CrossRef] [Green Version]
- Grosch, E.G.; McLoughlin, N.; Lanari, P.; Erambert, M.; Vidal, O. Microscale Mapping of Alteration Conditions and Potential Biosignatures in Basaltic-Ultramafic Rocks on Early Earth and Beyond. Astrobiology 2014, 14, 216–228. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Inoue, S.; Utada, M. Application of Chlorite Thermometry to Estimation of Formation Temperature and Redox Conditions. Clay Miner. 2018, 53, 143–158. [Google Scholar] [CrossRef]
- Bourdelle, F.; Benzerara, K.; Beyssac, O.; Cosmidis, J.; Neuville, D.R.; Brown, G.E.; Paineau, E. Quantification of the Ferric/Ferrous Iron Ratio in Silicates by Scanning Transmission X-Ray Microscopy at the Fe L-2,L-3 Edges. Contrib. Mineral. Petrol. 2013, 166, 423–434. [Google Scholar] [CrossRef]
- Sussenberger, A.; Pospiech, S.; Schmidt, S.T. [MnO Vertical Bar SiO2, Al2O3, FeO, MgO] Balanced Log-Ratio in Chlorites: A Tool for Chemo-Stratigraphic Mapping and Proxy for the Depositional Environment. Clay Miner. 2018, 53, 351–375. [Google Scholar] [CrossRef]
- Bobos, I.; Noronha, F.; Mateus, A. Fe-, Fe,Mn- and Fe,Mg-Chlorite: A Genetic Linkage to W, (Cu, Mo) Mineralization in the Magmatic-Hydrothermal System at Borralha, Northern Portugal. Mineral. Mag. 2018, 82, S259–S279. [Google Scholar] [CrossRef] [Green Version]
- Smith, W.C.; Bannister, F.A.; Hey, M.H. Pennantite, a New Manganese-Rich Chlorite from Benallt Mine, Rhiw, Carnarvonshire. Mineral. Mag. 1946, 27, 217–220. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
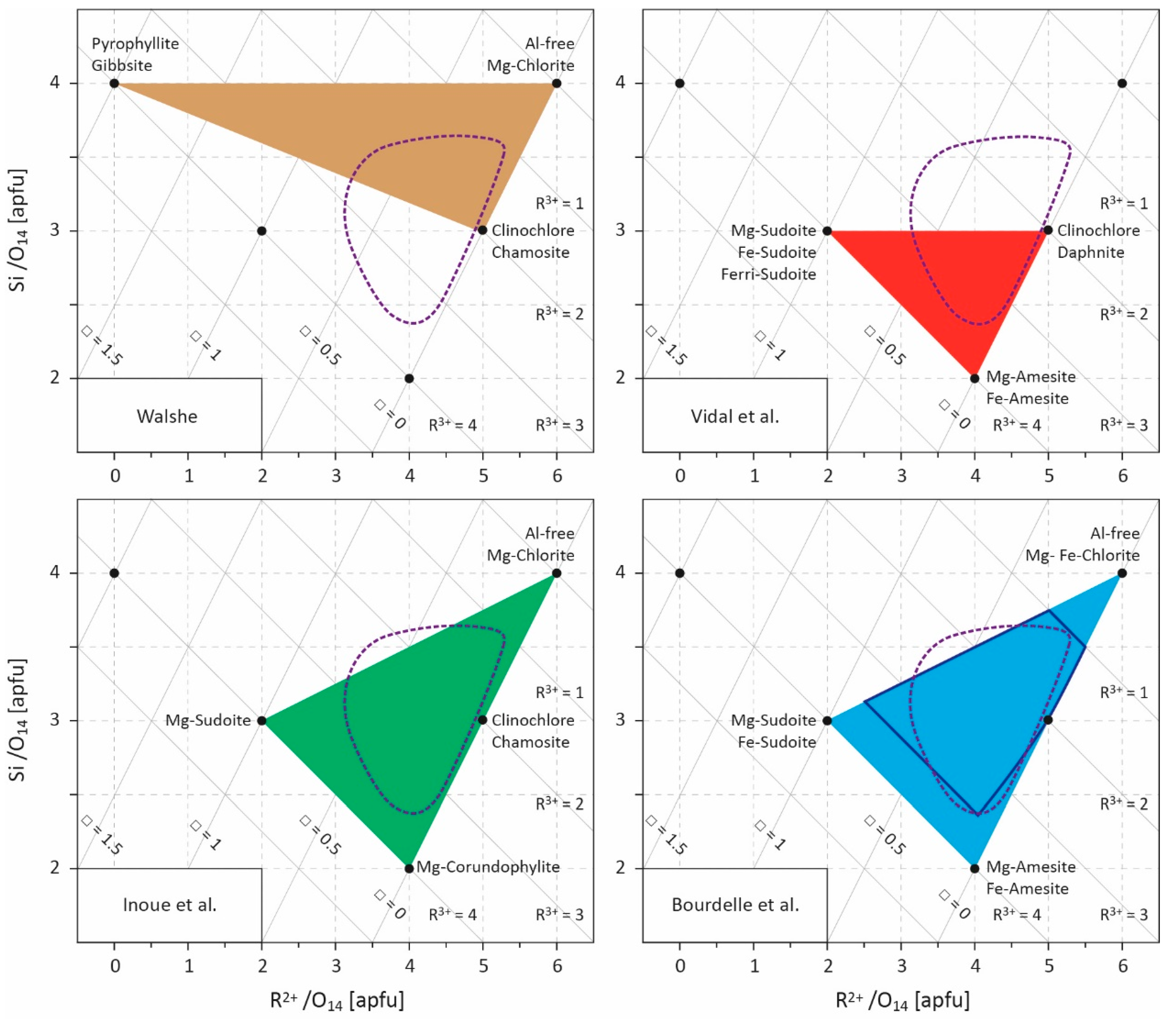
| Ref. | Year | Type | Equation (Oxygen Basis (14 or 28) in Brackets)/Equilibrium 1,2,3 | Elements/End-Members |
|---|---|---|---|---|
| Cathelineau and Nieva [12] | 1985 | empirical equation | T = 212.3AlIV + 17.5 (14) | AlIV content |
| Kranidiotis and McLean [47] | 1987 | empirical equation | T = 106(AlIV + 0.7XFe) + 18 (28) | AlIV, Fe, Mg contents |
| Cathelineau [25] | 1988 | empirical equation | T = 321.98AlIV − 61.92 (14) | AlIV content |
| Jowett [48] | 1991 | empirical equation | T = 319(AlIV + 0.1XFe) − 69 (14) | AlIV, Fe, Mg contents |
| Hillier and Velde [24] | 1991 | empirical equation | T = 249.56AlIV − 320.28 (28) | AlIV content |
| Zang and Fyfe [45] | 1995 | empirical equation | T = 106.2(AlIV − 0.88[XFe − 0.34]) + 17.5 (28) | AlIV, Fe, Mg contents |
| Xie et al. [38] | 1997 | empirical equation | T = 321.98(AlIV + 0.133[0.31 − XFe (<0.31)]) − 61.92 (14) | AlIV, Fe, Mg contents |
| Vidal et al. [9] | 2001 | thermodynamic model | 2 clinochlore + 3 Mg-sudoite = 4 Mg-amesite + 7 quartz + 4 H2O | clinochlore daphnite Mg-amesite Mg-sudoite |
| Vidal et al. [49,50] | 2005 2006 | thermodynamic model | + 5 Fe-amesite + 4 clinochlore = 5 Mg-amesite + 4 daphnite 4 16 daphnite + 15 Mg-sudoite = 20 Fe-amesite + 6 clinochlore + 35 quartz + 20 H2O 4 daphnite + 6 Mg-sudoite = 5 Fe-amesite + 3 Mg-amesite + 14 quartz + 8 H2O | + Fe-amesite |
| Lanari et al. [51] | 2014 | thermodynamic model | + 2 daphnite + 3 Fe-sudoite = 4 Fe-amesite + 7 quartz + 4 H2O Mg-amseite + 2 Fe-sudoite = Fe-amesite + 2 Mg-sudoite 2 clinochlore + 5 Fe-sudoite = 2 daphnite + 5 Mg-sudoite | + Fe-sudoite |
| Vidal et al. [53] | 2016 | thermodynamic model | + 14 equilibrium implying quartz, O2, and ferri-sudoite | + Ferri-sudoite |
| Walshe [11] | 1986 | semi-empirical model | T = 1626/(6.542 + 0.33(log K) − 273 5 Al-free Mg-chlorite + 3 pyrophyllite-gibbsite = 6 clinochlore + 14 quartz + 8 H2O | Al-free Mg-chlorite clinochlore chamosite (daphnite) pyrophyllite-gibbsite |
| Inoue et al. [28] | 2009 | semi-empirical model | T = 1/(0.00293 − 0.000513(log K) + 0.00003904(log K)²) − 273 Al-free Mg-chlorite + 3 Mg-sudoite = 3 Mg-amesite + 7 quartz + 4H2O | Al-free Mg-chlorite Mg-corundophilite (amesite) Mg-sudoitechamosite (daphnite) |
| Bourdelle et al. [55] | 2013 | semi-empirical model | T = −9400/(log K − 23.40) − 273 Al-free Mg-chlorite + 3 Mg-sudoite = 3 Mg-amesite + 7 quartz + 4H2O | Al-free Fe-, Mg-chlorite Fe-, Mg-amesite Fe-, Mg-sudoite |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourdelle, F. Low-Temperature Chlorite Geothermometry and Related Recent Analytical Advances: A Review. Minerals 2021, 11, 130. https://doi.org/10.3390/min11020130
Bourdelle F. Low-Temperature Chlorite Geothermometry and Related Recent Analytical Advances: A Review. Minerals. 2021; 11(2):130. https://doi.org/10.3390/min11020130
Chicago/Turabian StyleBourdelle, Franck. 2021. "Low-Temperature Chlorite Geothermometry and Related Recent Analytical Advances: A Review" Minerals 11, no. 2: 130. https://doi.org/10.3390/min11020130
APA StyleBourdelle, F. (2021). Low-Temperature Chlorite Geothermometry and Related Recent Analytical Advances: A Review. Minerals, 11(2), 130. https://doi.org/10.3390/min11020130