1. Introduction
Dissolution of minerals and salts is one of the most important reactions in chemistry. Because it is possibly the oldest known class of reactions, dissolution is known by different names in a wide range of fields—dissociation, digestion, etching, corrosion, leaching, weathering, and decay are some examples. Despite the obvious importance of this reaction, there is no generally accepted theory of the mechanism of dissolution that accounts for various experimental observations. Recently, the author has proposed a novel theory based on the ionic charging of the surface [
1,
2,
3,
4,
5,
6]. This theory has been shown to describe the kinetic observations for a diverse range of minerals, including quartz and silica (SiO
2) [
7], salt (NaCl) [
8], forsterite (Mg
2SiO
4) and the olivine group [
9], feldspars (KAlSi
3O
8, NaAlSi
3O
8, CaAl
2Si
2O
8) [
10,
11], oxide minerals [
3] and silicate minerals [
2]. The purpose of this paper is to provide an exposition of this theory with specific focus on the underlying physical model, showing that the model describes the principal features of dissolution reactions.
Kinetic models proposed for dissolution reactions have generally been unsuccessful in three major areas: (i) the orders of reaction with respect to the concentrations of reactant in solution, mostly importantly H+ and OH− ions; (ii) the acceleration or deceleration in the initial rate of reaction; (iii) the development of surface charge during dissolution or crystallization.
The first of these principal difficulties in understanding the mechanism of dissolution is that the kinetics exhibit fractional orders of reaction. Frequently, they are close to one-half. Considering that dissolution reactions are chemically quite simple, for example, , the fact that the rate is proportional to the concentration of H+ to the power of 0.5 indicates that these simple reactions are not elementary reactions. As the minimum stoichiometric number in an elementary reaction is one, it is difficult to derive values for orders of reaction below one, such as those found in many dissolution reactions, using multistep mechanisms. That the order of reaction is fractional points to a depth to dissolution reactions that is not easily explained.
The second principal difficulty in the understanding of the mechanism of dissolution is the observation that the initial rate of dissolution is sometimes accelerating and at other times decelerating. In other words, the initial rate per unit area under some conditions is initially fast and then slows to a steady value. Conversely, under other conditions, the rate of the reaction starts slowly, and accelerates to a constant value [
12]. This points to a change in a property of the surface that is initially away from and then moves towards a stationary value. None of the current models of the dissolution clearly account for this phenomenon.
The third principal difficulty in the understanding of the mechanism of dissolution is the observation that the surfaces of salts and minerals are electrically charged. For minerals such as oxides and clays, this property is well-known, and mechanisms, such as H+ adsorption for oxides and ion-exchange for clays, have been proposed to account for this charge.
More importantly, what is generally not recognized in the dissolution literature is that the surfaces of salts in aqueous solution are also electrically charged [
8,
13]. The explanation of charge on the surface of salts is not possible by an explanation of proton absorption, surface complexation or ion exchange because there are no foreign ions in solution. This means that there is a mechanism for the formation of surface charge on solids that is not dependent on mechanisms of adsorption of foreign ions.
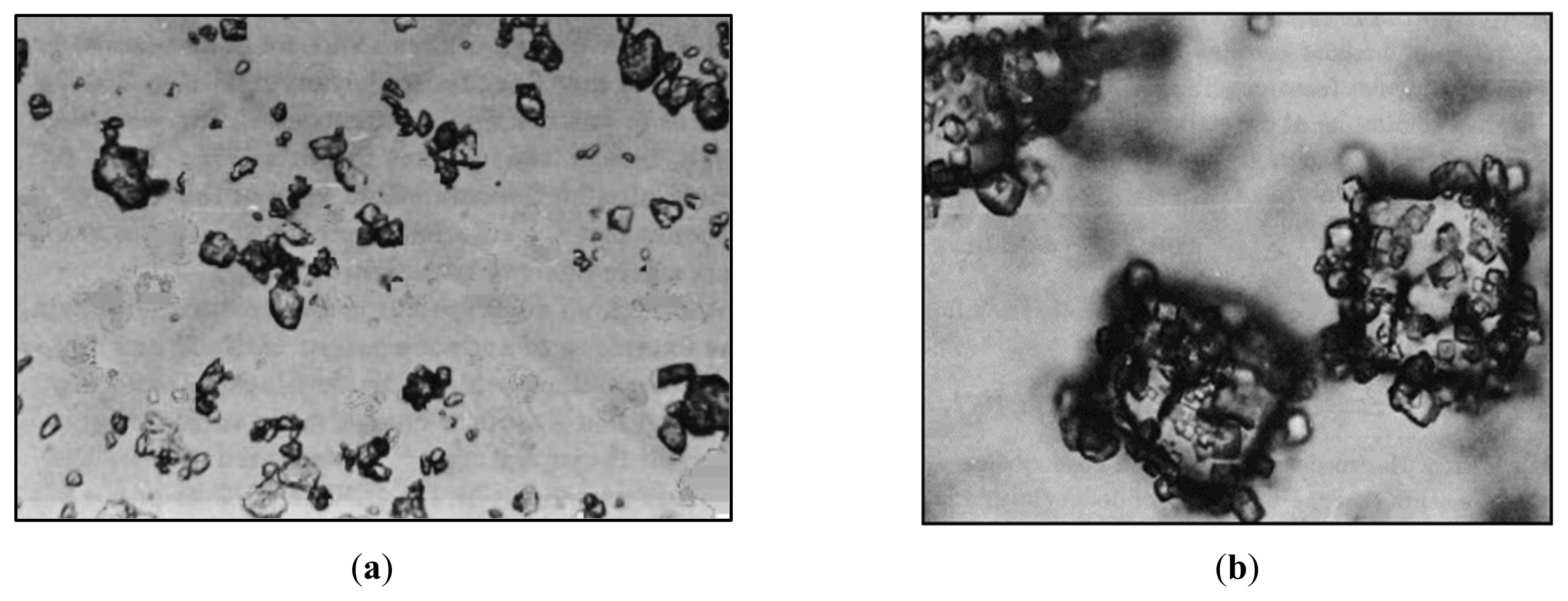
A micrograph is shown in
Figure 1 in which smaller KCl particles are attracted to the surfaces of larger NaCl particles because of their opposite charges. As a result, the smaller KCl particles cling to the larger NaCl particles.
The charge on the surface of salt has been determined by a variety of techniques, including membrane potentiometry and electrophoresis [
14,
15,
16,
17]. Industrially, the opposite charges on the surfaces of NaCl and KCl are exploited in the flotation of potash, where a positively charged collector attaches to a negatively charged surface and induces a flotation response [
14].
It is essential that the effect of the surface charge is coherently incorporated into the mechanism of dissolution because dissolution is the removal of charged particles from the surface and the presence of a net surface charge should affect the rate at which these particles are removed. It is argued in this paper that the surface charge is a ‘hidden variable’ in dissolution and crystallization kinetics. It is the key that unlocks the understanding of the mechanism of dissolution.
Two broad approaches to developing a fundamental mechanism of dissolution have been developed in the past. The first approach is to build a molecular model, for example, in which bonding atoms are removed according to their coordination with other members of the constituent solid. This approach has been pursued several times over the last few decades [
18,
19,
20]. However, this approach has only partially addressed the orders of reaction and has not addressed surface charge.
The second approach to developing a fundamental understanding of the kinetics of dissolution is the testing of kinetic mechanisms. In this approach, a mechanism is first proposed, then a rate law is derived from the proposed mechanism, and finally this rate law is compared with the experimental results. Before this approach is discussed, it is worth considering the experimental results that are used in the verification of the mechanism.
The experimental measurements are the effects of temperature and concentration on the rate of reaction, which can be summarized as an empirical expression of the form where represents the concentration of in solution, represents the order of reaction with respect to the species with concentration , and is the activation energy. The other symbols and represent the gas constant (8.314 J·K−1·mol−1) and the absolute temperature (K). The rate is usually expressed in mol·m−2·s−1, but an alternative unit that is used in fields such as corrosion is m·s−1. The former unit divided by the molar density of the solid yields the latter. The key experimental results are the orders of reaction, , with respect to each of the reactants, and the activation energy. These parameters quantify the effects of concentration and temperature.
A mechanism in chemical kinetics is a set of elementary reaction steps. By elementary steps, it is meant that the order of reaction reflects the molecularity of the reaction step. Thus, if the step
is an elementary step, the requirement is that the rate of reaction is proportional to the probability of a collision of two molecules of
and three molecules of
, which means the rate expression is
, where the order of reaction is 2 with respect to
and 3 with respect to
[
21]. Importantly, this means that the stoichiometric coefficients in an elementary step cannot be fractions. Thus, this approach requires the proposal of elementary reaction steps with integer-valued stoichiometry whose individual rate expressions combine so that the orders of reaction derived for the overall reaction agree with those obtained experimentally. Expressed differently, the orders of reaction provide researchers with a macroscopic window to the microscopic events occurring during reaction.
The rate of dissolution of solids varies over many orders of magnitude. For some solids, this rate is sufficiently fast that the overall rate is controlled by diffusion within the hydrodynamic boundary layer. In this study, we exclude data in which such film diffusion is rate-controlling because the focus is on the kinetics of the chemical reaction at the surface. In addition, diffusional processes to a planar non-porous surface are described by Fick’s law, which in an integrated form for diffusion control is . Clearly, this expression is first order with respect to the diffusing species. Diffusion alone cannot explain orders of reaction that are not equal to one.
Several proposals for mechanisms of reaction have been made for dissolution. The first proposal is referred to as the surface-complexation model, which proposes that a ‘complex’ forms on the surface between the reactant and the surface species, and that it is the departing of this complex that is this rate limiting step [
22,
23,
24]. The rate of reaction is given by the expression
, where
is the concentration of the surface complex, and
is the order of reaction. The concentration of
is not measured; rather, it is calculated by proposing multiple equilibria involving solution species and hypothetical surface species. Although Casey [
25] proposed an explanation for
, the underlying model introduces numerous adjustable parameters that have not been independently verified. In effect, the model creates a new order of reaction and ignores the fractional orders with respect to solution species.
An alternative proposal for the mechanism of dissolution is the precursor-species model, in which a precursor forms on the surface that then reacts to form an ‘activated complex’, which in turn decomposes to form the reaction products [
26,
27,
28,
29]. However, the proponents of this model make the mistake of using fractional stoichiometry to obtain fractional orders of reaction, which, as explained earlier, makes no sense in the context of kinetic mechanisms composed of elementary reactions.
Yet another proposed mechanism of dissolution uses crystallization theory [
30]. Rate expressions based on degree of saturation have been derived from crystal growth theories of the terrace-edge-kink type [
30]. These models are concerned mainly with the approach to equilibrium, and do not address any of the three challenges of dissolution reactions that are the subject of this paper, namely, fractional orders of reaction, accelerating or decelerating initial rates, and surface charge.
Thus, the current models of dissolution are deficient because they cannot explain (i) reaction orders, (ii) initial transients, and especially (iii) surface charge. The purpose of this paper is to provide a general theory of dissolution that accounts for these experimental observations in a holist manner. The objectives of the present work and the requirements for a general dissolution model are presented in the next section. Following this, the underlying physical foundations of the proposed model are described. From this physical model, kinetics expressions are derived. The correspondence between the model and the experimental data for the different observed parameters, such as the reaction orders, and phenomena, such as transient effects, is discussed. The implications of the model are discussed, and suggestions for independent testing are made.
2. Objectives of This Paper
The aim of this work is to outline a general model of the dissolution of salts and minerals. As discussed in the introduction, the most important requirements for a general model of dissolution are as follows:
It should be applicable to the (non-oxidative) dissolution of all minerals and salts.
It must provide a clear physical foundation from which the rate equations are derived.
It must coherently describe the effect of surface charge on the rate of removal of ions.
The rate expressions derived from the physical model must inherently and coherently describe the following dissolution phenomena:
- (a)
The effects of H+ and OH− on the rate of dissolution expressed in terms of the order of reaction with respect to these reactants;
- (b)
The approach to equilibrium, including the effects of constituent ions in solution on the rate of dissolution, for example, Na+ on the rate of dissolution of NaCl;
- (c)
The change in rate of dissolution, either acceleration or deceleration, during the initial stage of reaction;
- (d)
The non-stoichiometric dissolution of more complex solids, such as felspars.
The physical foundations of the model should be testable by independent means.
The model should make predictions that extend the field.
The surface-vacancy model is the only model that meets these requirements. (In previous papers, the author referred to this model as the surface-dissociation model, but the name surface-vacancy model is preferred because it better reflects the underlying physical features of the model.) The physical foundations of this model are laid down in the next section. In the sections that follow this foundational material, it is shown that the surface-vacancy model describes the orders of reaction with respect to H+ and OH−, that it describes the approach to equilibrium, that it describes the change in initial rate of dissolution, and that it describes the non-stoichiometric phases of dissolution. Importantly, the physical model provides predictions that allow the model to be independently and more thoroughly tested. Finally, and by no means least, are the predictions that the model makes that are not made by other models, and which allow the field to develop further.
As mentioned, the next section lays down the physical foundations of this model.
3. Physical Foundations of the Surface-Vacancy Model of Dissolution
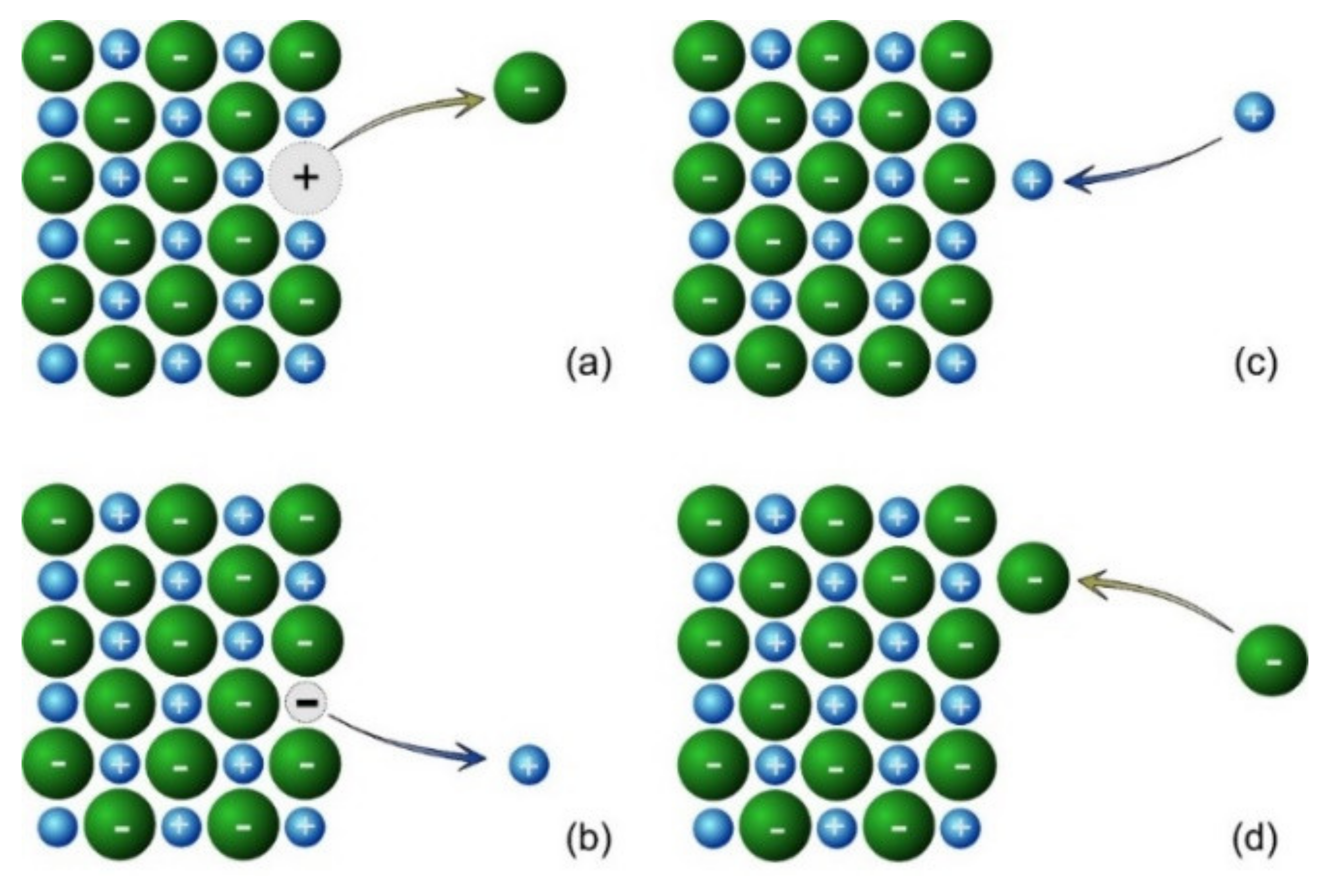
When ions depart from the surface to dissolve in the aqueous phase, they leave a vacancy of equal charge but of opposite sign to the ion. This results in a net excess charge on the surface, shown in
Figure 2a,b. Of course, the deposition of ions from the aqueous solution can also occur, also resulting in excess charge, as shown in
Figure 2c,d. Therefore, there are four events that must be considered in the kinetic model.
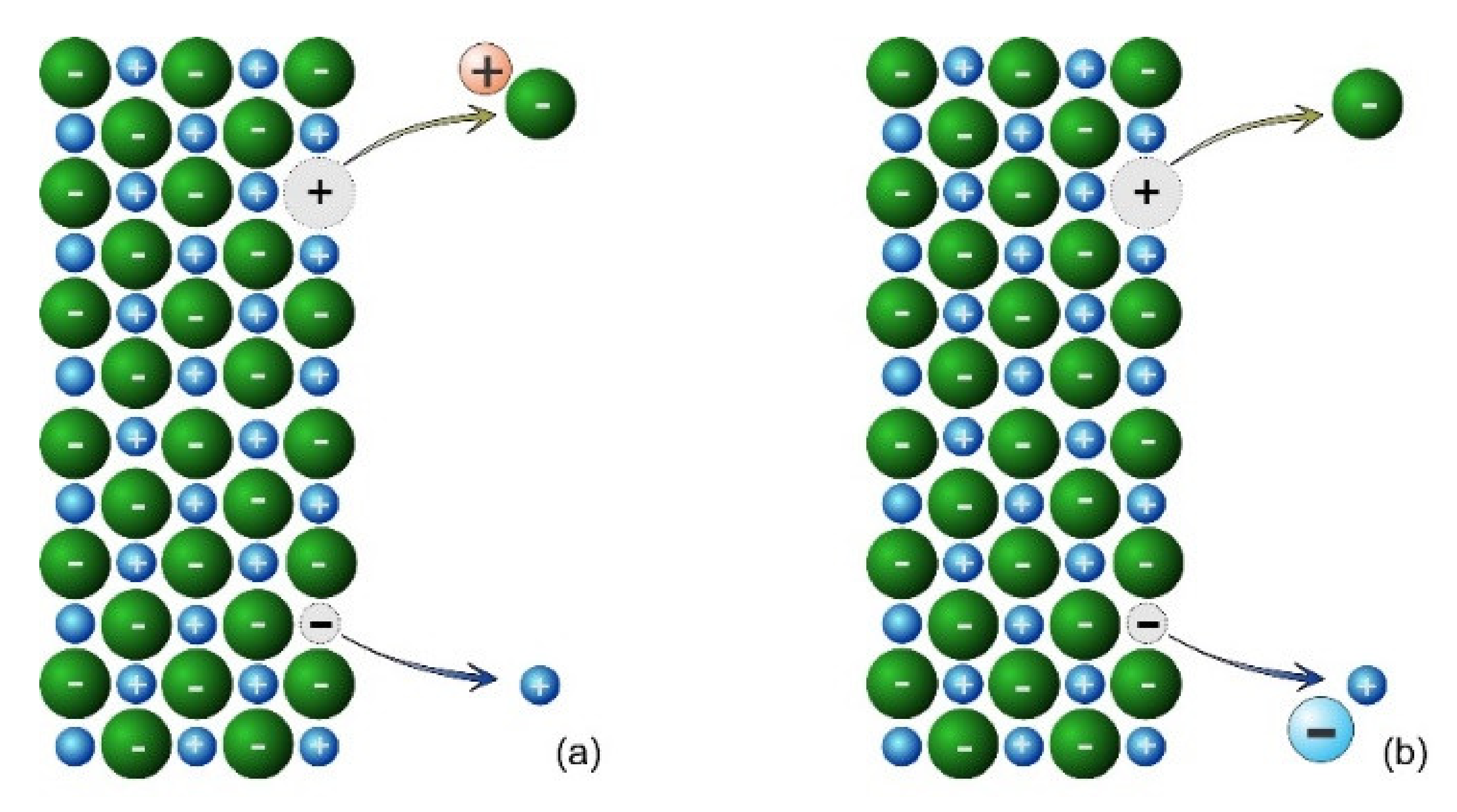
This situation describes the underlying physical model for salts, where the dissolution is an interaction between the solid and the solvent. For other minerals, the rate of dissolution is enhanced in either acid or alkaline solutions or both, so the physical model must be extended to include H
+ and OH
−. In this case, the underlying physical model is shown in
Figure 3. The removal of the anion from the surface occurs with the participation of a proton (or hydronium ion), shown in
Figure 3a. Likewise, the removal of the cation from the surface occurs with the participation of the hydroxide ion, shown in
Figure 3b. The impact of ligands occurs in a similar manner—anionic ligands assist in the removal of cations from the surface, leaving a negative vacancy, while cationic ligands assist in the removal of anions from the surface, leaving a positive vacancy.
Consider the dissolution of a solid
in acid according to the reaction (1). Furthermore, consider for the sake of simplicity only the forward reaction.
The symbol represents the stoichiometric coefficient for the number of protons (hydronium ions) that interact with the anion during its removal from the surface. The values of are restricted to 0, 1, 2, 3….
The individual steps corresponding to
Figure 2a,b can be expressed in chemical notation as Equations (2) and (3), where
and
represent the M-constituent and the
A-constituent of the solid surface, respectively. Likewise,
represents a surface vacancy with a positive charge, and
represents a vacancy with a negative charge.
The formulation is not restricted to minerals with 1:1 stoichiometric ratio—the reaction scheme can be extended to any stoichiometry.
If dissolution occurs in an alkaline medium by, for example,
, the dissolution steps are given by Equations (4) and (5).
Other extensions to these simple steps might be envisaged. For example, a similar scheme can be envisaged to account for ligand interactions. In electrochemical kinetics, it is often postulated that the first step is the adsorption of an ion, say a proton or a ligand, onto the surface before the rate-determining step. This might allow for a saturation effect of the ion by applying an isotherm, for example, the Langmuir isotherm. Such additional or prior steps can be incorporated.
Due to the removal of ions from the surface, there is a net charge on the surface. This net charge affects the affects the rate at which ions are removed. Thus, this forms a feedback mechanism: removal of ions causes a charge, but this charge affects the rate of removal. This means that the individual steps shown in Equations (2)–(5) are not chemical, but electrochemical.
To summarize, the physical foundations of the proposed model of dissolution are (i) the removal of ions creates a vacancy of charge opposite that of the departing ion on the surface, (ii) the removal of ions leaves a net excess charge on the surface, (iii) the net excess charge has a strong influence on the rate of removal of ions, and (iv) the rate of removal of ions might be enhanced by H+, OH− or some other ligand.
Questions that may come to mind from this formulation of dissolution might include, amongst others, the following: (a) If the rate of removal of anions and cations are independent, what principle determines that the rate of removal of anions is stoichiometrically equivalent to the rate of removal of cations? (b) What principle governs steady state dissolution? (c) Is this consistent with chemical thermodynamics? These questions are answered in the next section in which this physical foundation is transformed into mathematical expressions for the surface charge and for the rate of dissolution.
4. Mathematical Formulation of the Surface-Vacancy Model
The excess surface charge at any time is the difference in surface concentration of the vacancies and deposited ions on the surface, given by Equation (6). The surface charge is given by
, and the symbols
and
represent the surface concentrations of the anionic and cationic vacancies, respectively. The symbols
and
represent the absolute charge number of the anion and cation, respectively, so that for 1:1 salts such as NaCl,
.
represents the Faraday constant (C·mol
−1).
Equation (6) can be written as the difference in excess anions and cations using the symbols
and
[
31]. The symbol
is specifically used here to emphasize that the mechanism of surface charging is by surface vacancies and deposition.
Taking derivatives with respect to time, Equation (6) can be written as Equation (7).
The rate of change of the surface concentration of cationic vacancies is equal to the rate of removal of anionic species, that is,
is equal to
, and similarly,
is equal to
. Since the charge number of the departing ion is the absolute value of the vacancy left behind,
is the same as
and
is the same as
. The substitution of these relationships into Equation (7) yields Equation (8).
Equation (8) relates the change in the electrical conditions of the surface to the dissolution kinetics. If the rate of removal of the cations is not in stoichiometric proportion to the rate of removal of anions, the surface charge will change with time according to Equation (8). Therefore, Equation (8) encapsulates the cause of transient effects: the surface charge is yet to reach its stationary state, and changes in the rate of removal of anions and cations will be experienced until a stationary state is reached. This expression will be revisited when the transient effects are discussed later.
Assume for the time being that a stationary state has been reached, so that the left-hand side of Equation (8) is approximately zero. This then yields Equation (9), which is a statement of stoichiometry.
This result is crucial—Equation (9) states that the condition for stoichiometric dissolution is that the surface charge is stationary, and that dissolution occurs at a constant rate, while Equation (8) describes the path that is taken to reach this stationary state where stoichiometric dissolution takes place.
In addition, Equation (8) describes how the individual steps of Equations (2) and (3) are linked to the overall reaction. The connection is not through chemistry, nor with a surface with neutral charge (which is the tacit assumption of all previous models of dissolution and crystallization), but through the surface charge reaching a stationary state. There are, in fact, three obvious stationary states: (i) dissolution at a constant rate; (ii) equilibrium, in which the rates of dissolution are matched by the rates of crystallization; (iii) crystallization at a constant rate.
Consider for the sake of presentation, that the dissolution reaction is given by Equation (1). As mentioned previously, the rates of removal of the anions and cations must be affected by the surface charge. In electrochemistry, this dependence is expressed in the form of a Boltzmann-like function with the potential difference across the Helmholtz layer as the independent variable [
32]. If a double layer is assumed, then the surface charge and the potential difference are proportional to each other, with the constant of proportionality referred to as the surface capacity. This means that the rate of the removal of cations (Equation (2)) is given by Equation (10), and the rate of removal of anions, Equation (3), is given by Equation (11).
The symbols
and
represent the rate constants for the removal of anions and cations, respectively. The symbol
y represents the dimensionless factor
, which includes the surface potential difference,
. The terms
and
are the Boltzmann-like factors that account for the effect of surface potential on the rate. This effect arises from the effect of potential on the activation barrier, identical to those found in electrochemical reactions. The factor 0.5 arises because the activated state (transition state) occurs approximately halfway between the surface and the outer Helmholtz plane [
32].
Equations (9)–(11) represent three equations in the three unknowns,
,
and
, which can be easily solved by substituting both Equations (10) and (11) in Equation (9), and making
the subject of the formula. This operation yields Equation (12).
Equation (12) describes the effect of pH on the potential difference across the Helmholtz double layer, which can be determined by zeta potential and other electrochemical techniques.
The rate of dissolution is obtained by substituting Equation (12) into either Equation (10) or (11), which yields Equation (13).
Thus, Equation (13) represents the simplest rate expression derived from the physical foundations presented in the previous section. Only dissolution due to acid, at steady conditions of dissolution, and at conditions far from equilibrium have been considered. Of course, these other cases can be accounted for, some of which will be addressed later in this paper, but Equation (13) combines the main features of the physical foundations into its simplest form so that it can be tested.
The significance of Equation (13) is that it predicts that if one proton is involved in the elementary step given by Equation (3), then , and the order of reaction is equal to one-half. This demonstrates that the surface vacancy model correctly predicts one of the most difficult aspects of dissolution reactions.
Furthermore, the values of in Equation (13) take on values of 0, 1, 2, 3,…, which means that the order of reaction with respect to H+ can take on values of 0, 0.5, 1.0, 1.5,…. If a salt dissolves without the assistance of acid or alkali, the value of is zero, and the order of reaction is zero.
From kinetic theory, the formation of an activated complex becomes less likely as the number of species forming the activated complex increases. This means that the likelihood of finding minerals with an order of reaction of 0.5 is greater (because only one proton is involved) than finding those with an order of reaction of 1.0 (where two protons are involved), etc., and so forth.
This means that the expectation from the surface-vacancy model is that values of about one-half will be encountered most frequently. Values of one are expected less frequently than values of one-half, and values of one-and-a-half are expected at lower than this. This prediction of the orders of reaction is examined in the next section.
5. Testing the Surface-Vacancy Model—Orders of Reaction
Generally, the approach in dissolution studies is to focus on a single mineral reaction and examine its behavior in detail. Specific applications of the surface-vacancy model have been made for silica and quartz [
2,
7], feldspars [
10,
11], forsterite [
9], oxides [
3] and halite [
8].
However, a broad approach is required for testing a general model such as the surface-vacancy model, and this broad approach is adopted in this paper. In this section, the predictions of the model are tested against data for the orders of reaction for metal oxides, orthosilicates and metal sulphides. Following this, the scenario of equilibrium is considered, after which the phenomenon of decelerating and accelerating dissolutions rates is analyzed.
5.1. Overview of Orders of Reaction
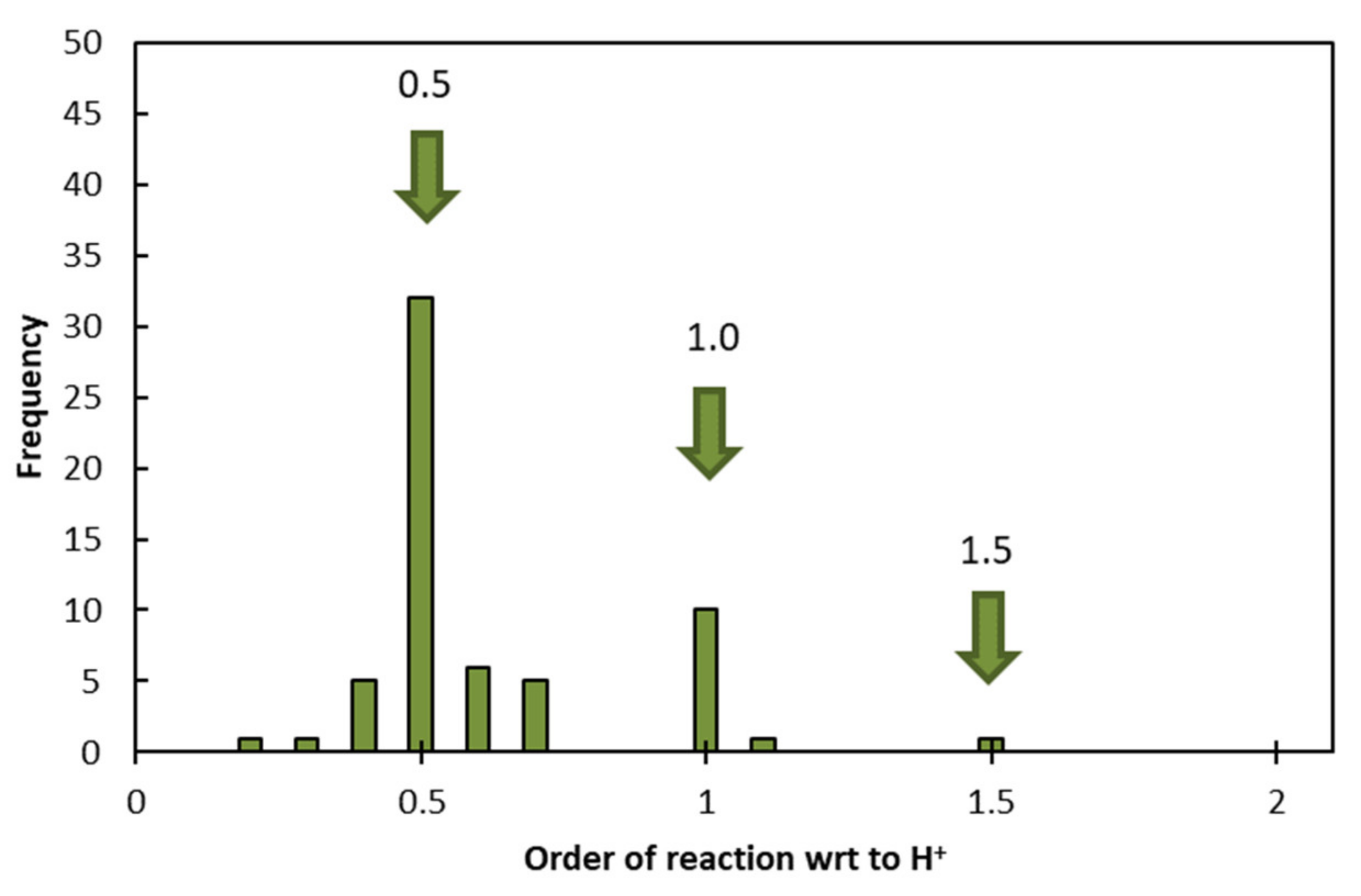
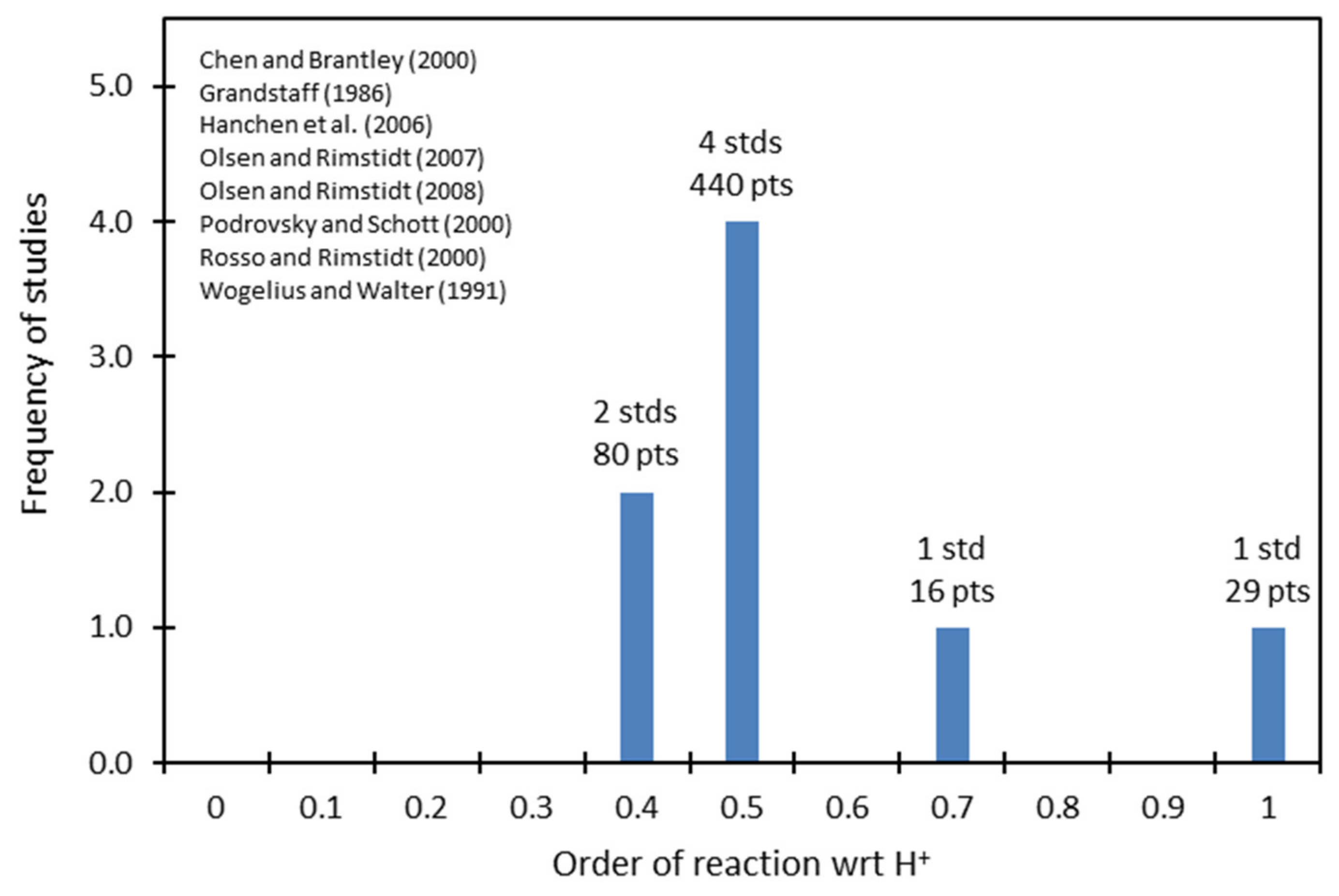
The orders of reaction with respect to H
+ reported by 62 independent studies of different minerals are shown in
Figure 4. These results suggest the clustering of the data about 0.5, at 1.0 with a lower frequency, and at 1.5 with an even lower frequency. These results agree with the surface-vacancy model presented in the previous section: as
in Equation (13) takes on values of 0, 1, 2, 3,…
, the order of reaction with respect to H
+ takes on values of 0, 0.5, 1.0, 1.5,…
, with diminishing frequency. No previously proposed model has been able to derive a relationship of this nature, and these results provide a powerful indication of the validity of the surface-vacancy model.
There is scatter of the data about these values, particularly around the value of 0.5. Such scatter is expected due to random experimental errors. For example, shown in
Figure 5 are the frequencies of reported values for the order of reaction with respect to H+ for forsterite, Mg
2SiO
4, in the acidic region. This figure indicates that although six studies reported values close to 0.5, two were significantly distant from this value. As the number of data points (tests of the rate of dissolution) accumulate, so the confidence increases in the reported value. Thus, the value for forsterite 520 tests attested to a value close to 0.5, while 45 tests suggest a significant deviation from this value.
The results shown in
Figure 4 correspond with the predictions of Equation (13), the simplest form of the surface-vacancy model. As mentioned in the previous section, one would expect from kinetic theory that the frequency of a value of one for
would be higher than a value of 2, and that of two would be higher than that of 3, etc. This means that it is expected that the frequency of the order of reaction with respect to H+ of 0.5 would higher than 1.0, which in turn would be higher than that of 1.5. (This is subject to the obvious proviso that the measurements are obtained under conditions that are controlled by reaction kinetics and are not under diffusional control or mixed control.)
5.2. Orders of Reaction for Mineral Oxides
Consider the dissolution of a mineral oxide in an acidic aqueous solution for the conditions of steady-state dissolution at conditions far from equilibrium. The orders of reaction for mineral oxides under these conditions are given in
Table 1. Many of the orders of reaction with respect to H
+ are close to 0.5, while fewer are close to 1.0, and only one has a value close to 1.5.
5.3. Orders of Reaction for Orthosilicates
The orders of reaction for orthosilicates are given in
Table 2. All the reported values are close to 0.5, in agreement with the surface-vacancy model.
5.4. Orders of Reaction for Feldspars
The orders of reaction with respect to H
+ for feldspars are given in
Table 3. All these values are close to 0.5, except for bytownite and anorthite, which have values of 1.0 and 1.5. These values are in agreement with the surface-vacancy model.
5.5. Orders of Reaction for Metal Sulphides
The values reported for the order of reaction with respect to H
+ for some metal sulphides are given in
Table 4. These values are close to 0.5 for some reactions and close to 1.0 for others. Importantly, in several cases in which the order of reaction with respect to H
+ are close to 1.0, the orders of reaction with respect to constituent ions for the reverse reaction (precipitation) are close to 0.5. This leads the discussion to the topic of equilibrium, crystallization and precipitation, which is discussed next in the section.
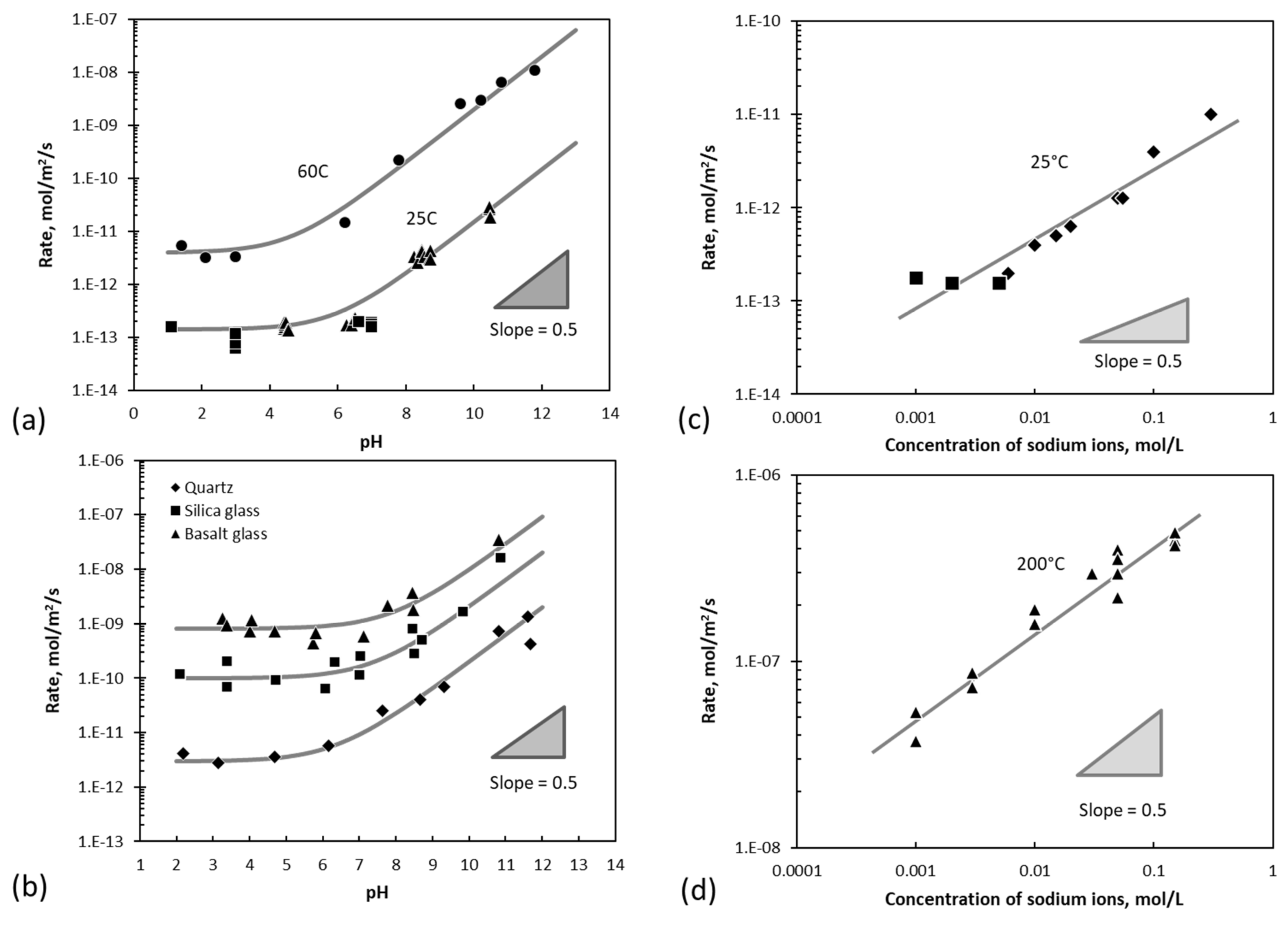
6. Testing the Surface-Vacancy Model—Quartz
The dissolution of quartz provides an example of more complex kinetic behavior that can be used to test the surface-vacancy model [
7]. Data for the dissolution behavior of quartz has been compiled by Bickmore [
69], and Rimstidt [
70] analyzed this data statistically and showed that the order of reaction with respect to H
+ is zero in the acid region, while the order of reaction with respect to OH
- is 0.5 in the alkaline region. In addition, it was found that, in the alkaline region, the order of reaction with respect to Na
+ is 0.5. Some of the data from the Bickmore compilation are shown in
Figure 6.
Crundwell [
7] proposed that the dissolution of quartz (SiO
2) occurs due to the removal of a ‘cation’ species by Reaction (14) for which the rate expression is Equation (15).
The removal of the ‘anion’ species was proposed to be by the parallel reaction with H
+ and a Na
+ catalyzed step, as shown in Reactions (16) and (17).
The rate expression for these two parallel reactions is given by Equation (18).
Equations (15) and (18) are substituted in Equation (9) and solved for the unknown variable
, which is a dimensionless form of the potential difference across the surface.
If Equation (19) is substituted back into Equation (15), the rate of dissolution is given by Equation (20).
The term
is constant (because it represents the dissociation of water). In the acidic region, the rate is not dependent on pH while in the alkaline region the rate of dissolution is dependent on the pH and the concentration of sodium ions. The lines shown in the graphs in
Figure 6 represent the fit of this equation to the data, and a full fit of 285 data points from fourteen different studies shows that this is an excellent description of the data [
7].
7. Testing the Surface-Vacancy Model—Thermodynamics
Equilibrium is accounted for by including the kinetics of the reverse reactions for the steps given in Equations (2) and (3). This means that Equations (10) and (11) are expanded as Equations (21) and (22) by including the reverse terms.
Combining these expressions with Equation (9) yields, after some algebraic rearrangement, the rate expression given in Equation (23).
At equilibrium, the rate is zero. Therefore, the equilibrium condition for this surface-vacancy model can be obtained by setting the left-hand side of Equation (23) to zero. This yields Equation (23).
is clearly the equilibrium constant.
This clearly demonstrates that the proposed mechanism is consistent with chemical thermodynamics.
8. Testing the Surface-Vacancy Model—Reverse Reaction
8.1. Orders of Reaction for the Reverse Reaction
The reverse reaction, that is, deposition, crystallization and precipitation, can be considered either separately, or in conjunction with Equation (24). Specific orders of reaction have been reported for the reverse reaction for sulfides and for calcite, and the application of the surface-vacancy model to these cases is examined in this section.
Consider the reverse reaction for the dissolution of ZnS given by Equation (25).
Both Locker and de Bruyn [
62] and Crundwell and Verbaan [
68] reported that the orders of the reverse reaction had values of 0.5 with respect to both Zn
2+ and H
2S (see
Table 4). To describe these results, select a value of 2 for
and consider only terms for the reverse reactions in Equation (16). This yields that the rate of precipitation of ZnS is given by Equation (26).
This mechanism clearly accounts for the results presented in
Table 4 for ZnS and CdS [
62].
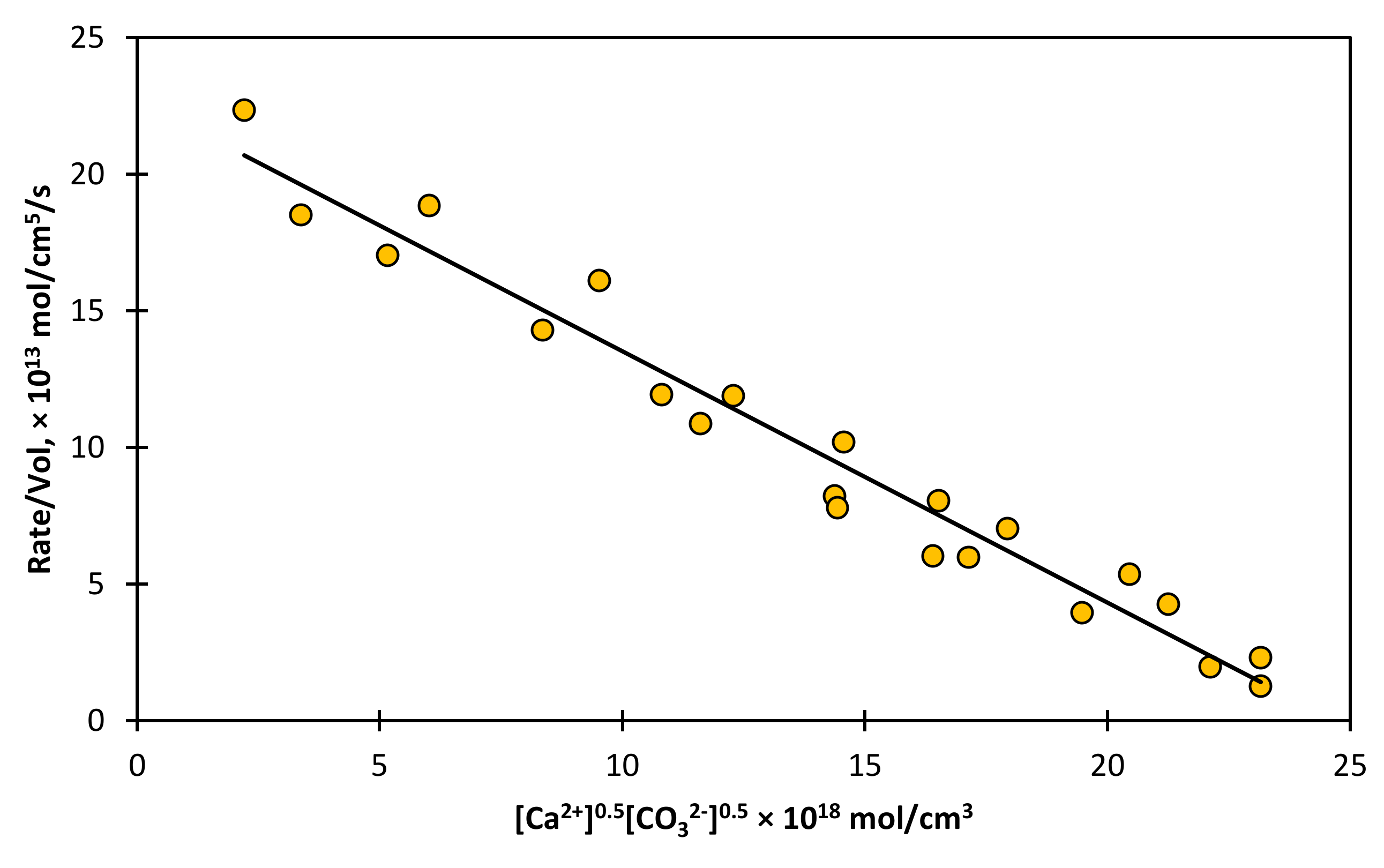
The dissolution of carbonates presents a similar set of kinetics. Consider the dissolution of calcium carbonate, given by Equation (27).
The results of the experiments are shown in
Figure 7, which show that the orders of reaction are 0.5 with respect to
and 0.5 with respect to
. Bearing in mind the complexities of this reaction, the similarities with ZnS under the conditions used by Sjöberg [
71] are quite remarkable.
The rate of the reverse reaction can be obtained directly from Equation (16) by choosing of value of zero for
t and considering the cation and anion to be Ca
2+ and CO
32−. This choice yields Equation (28) for the reverse reaction only. This expression is clearly in agreement with the experimental results shown in
Figure 7.
The full reaction path between reaction control and equilibrium is discussed next [
72].
8.2. Dissolution of Salts between Reaction Control and Equilibrium
The rate expression for the forward reaction for the dissolution of a salt is trivial since the salt only interacts with water, whose concentration is high and nearly always approximately constant. This means that the forward rate is constant with concentration. However, as the concentration of salt in solution increases, the reverse reaction plays a role. As mentioned in the introduction, the reverse reaction is most often accounted for in rate expressions of the form , where is equal to activity product divided by the equilibrium constant. The term reaction order has been used for the term in the rate expression where is the degree of saturation. This is an empirical rate expression, and the term ‘reaction order’ for is a misnomer.
For a salt such as NaCl,
is given by
. If the rate is expressed in terms of the concentration of salt,
, then this form of the rate expression is given by
, where
, and the value of
is taken as 1 for the sake of simplicity. Thus, this expression means that the rate decreases as a parabolic function of increasing salt concentration,
, representing the path from rate control to equilibrium [
8,
72].
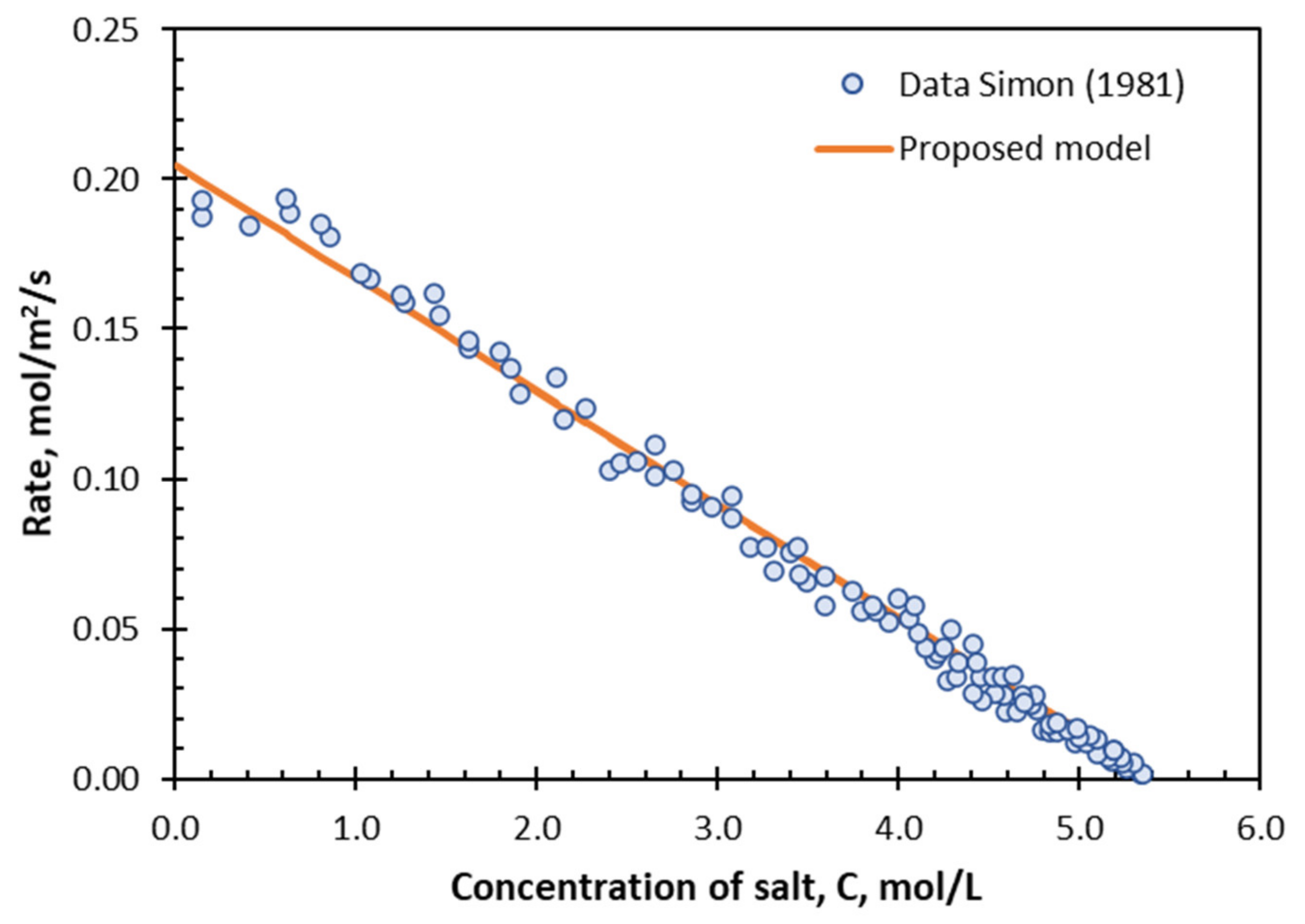
The rate of dissolution of NaCl is shown in
Figure 8 as a function of the concentration of NaCl in solution,
. This is not a parabolic function—in contrast, it is linear.
The linear relationship between the rate of dissolution and the concentration of NaCl means that the rate expression is . The similarities with the results for ZnS, CdS, and CaCO3 in the previous sections are difficult to miss.
The full rate expression is derived from Equation (23) by choosing the value of zero for
t and using the Equation (24) for the equilibrium concentration. This yields Equation (29)
The rate constant
is given by
and
is given by
. The line in
Figure 8 represents the best fit of Equation (29) to the data.
This linear dependence on
C, and hence half-order kinetics, is echoed in crystallization studies.
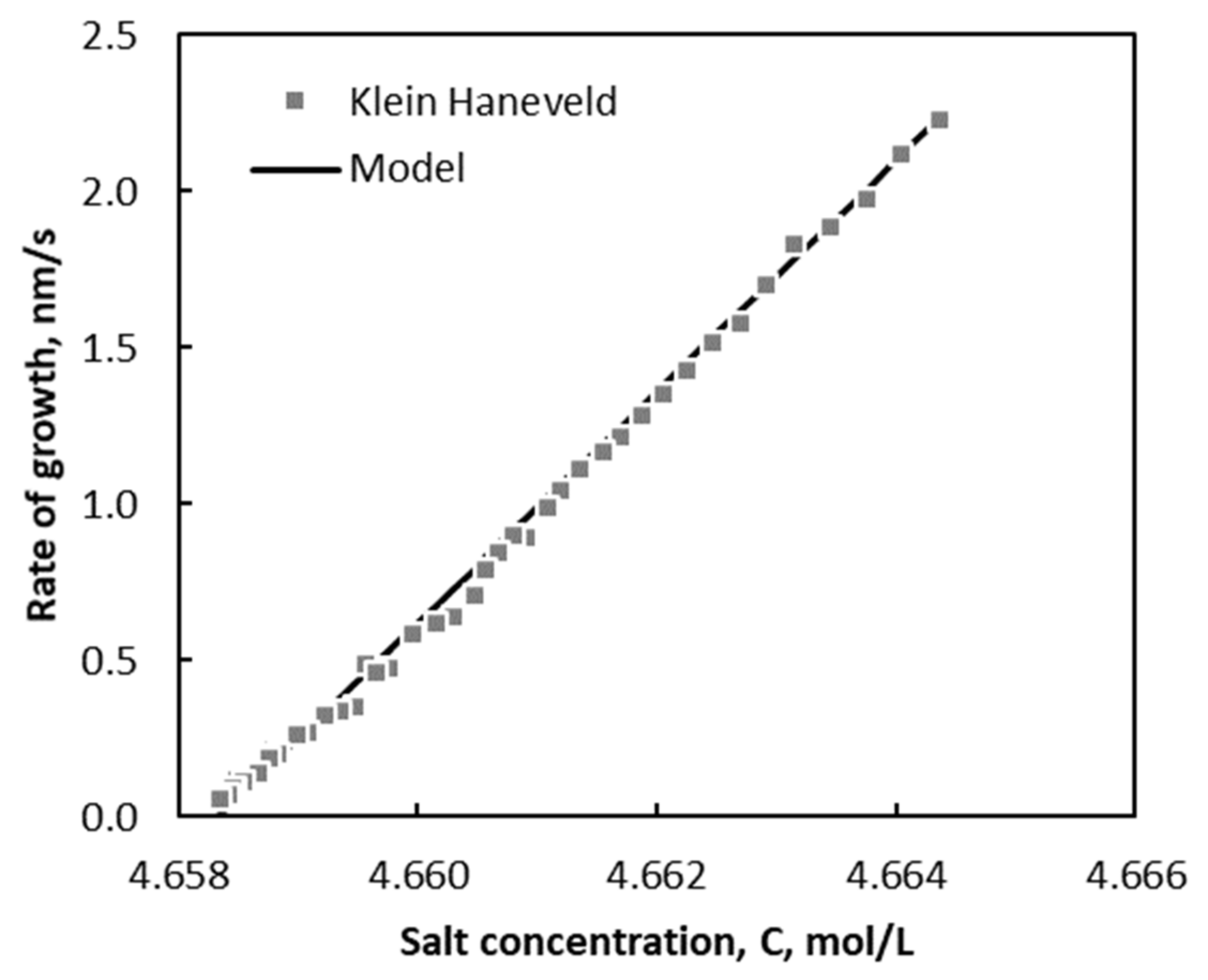
Figure 9 shows the growth of KCl as a function of the concentration of salt,
C, divided by the saturated concentration of salt,
. The line in
Figure 9 represents the fit of Equation (29) to this data set.
8.3. Rate Formulation Based on Chemical Affinity
The approach to equilibrium is sometimes modelled in dissolution studies using the chemical affinity [
75]. For a solid MA, the chemical affinity,
, is defined by Equation (30).
In some cases, the plot of
against
gives an S-shape, which is difficult to explain with a simple reverse reaction. An example of such an S-shape is shown in
Figure 10 for the feldspar albite.
The surface-vacancy model describes this observation. The substitution of Equation (30) into Equation (23) yields Equation (31) for the generic solid MA.
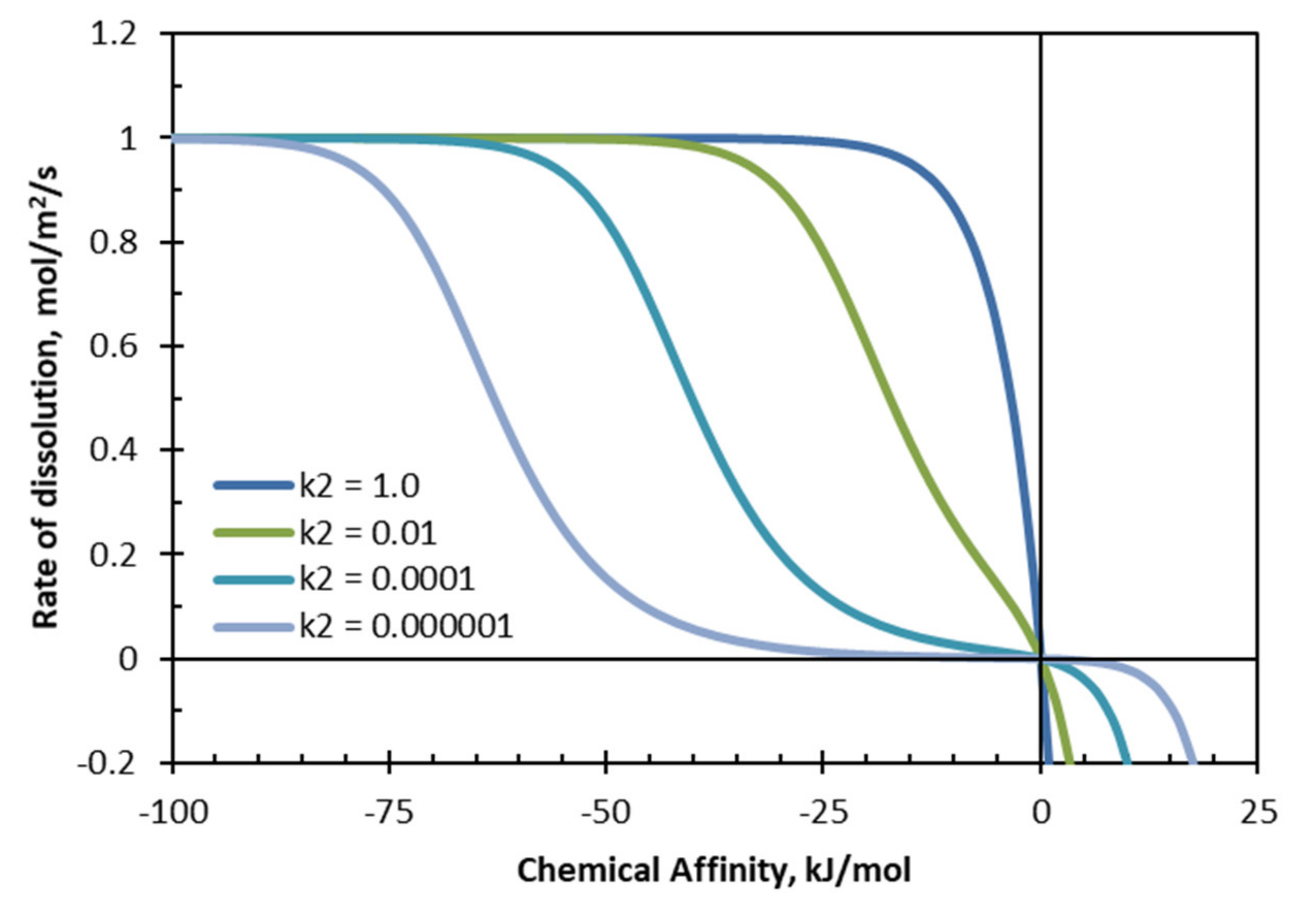
The rate for the surface-vacancy model as a function of chemical affinity is plotted in
Figure 11 for different values of
. The chemical affinity in
Figure 11 was varied by simultaneously changing the value of the concentrations of
and
so that
is equal to
at each position on the curve. Clearly, the surface-vacancy model can account for the change from monotonic form to the S-shape found for some minerals such as albite.
The line in
Figure 10 represents a fit of the surface-vacancy model to the data for albite, showing that the surface-vacancy model is a good description of this reaction under these conditions.
8.4. Rate Formulation Based on Degree of Saturation
The rate of dissolution over the range between reaction control and equilibrium has also been analyzed in terms of the degree of saturation,
, which is equal to the activity product divided by the equilibrium constant. Equation (31) can be written in this form as Equation (32).
The rate is plotted as a function of
in
Figure 12 assuming that
is varied by simultaneously changing the value of the concentrations of
and
so that
is equal to
at each point (yielding the special case that
).
Note the deviation from linearity in
Figure 12a as
tends towards 0 that is, the right-hand side of
Figure 12a,b as
tends to 1. This non-linearity is due to the contribution of the denominator in Equation (32), which is only predicted by the surface vacancy model.
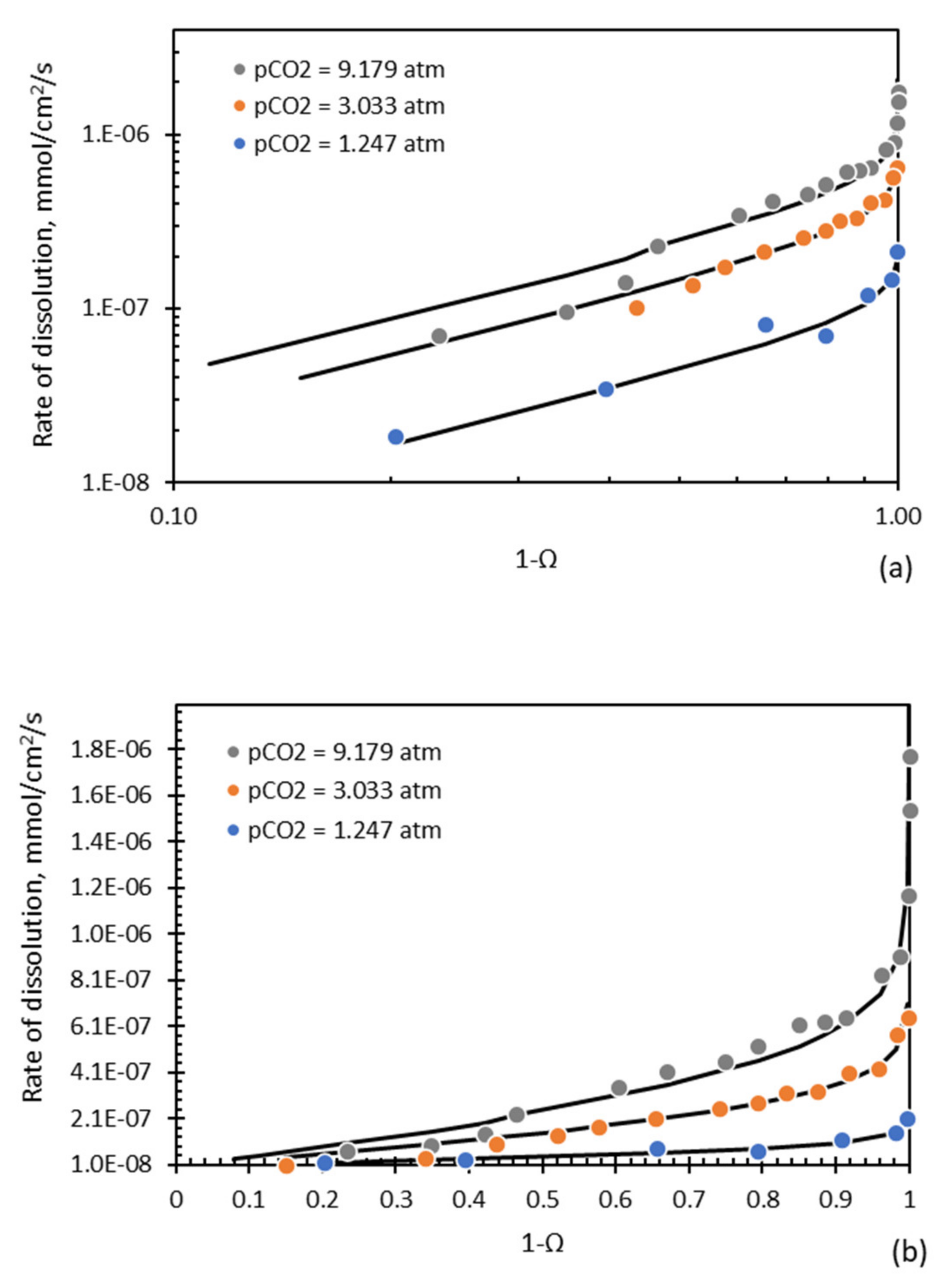
There is evidence to support this non-linearity. Consider the results reported by Busenberg and Plummer [
77] for the dissolution of calcite shown in
Figure 13 as a function of
. These results clearly display the non-linearity of the curve as
approaches 1. As the partial pressure of CO
2 was maintained in these tests, the results are modelled by Equation (33), where
accounts for the anion terms for either carbonate or bicarbonate or both. Busenberg and Plummer [
77] reported values for both
and
.
This simplified model clearly accounts for the results of Busenberg and Plummer [
76], especially the non-linearity as
approaches 1.
For the results of Sjoberg [
71] and these results to both hold true, it would mean that Sjoberg’s investigations were carried out in this non-linear region. Obviously, there are more details of this reaction than have been presented here and further investigation is required; however, these examples demonstrate the promise of the surface-vacancy model that might allow different aspects of the puzzle of calcite dissolution to be reconciled.
9. Testing the Surface-Vacancy Model—Initial Rates of Dissolution
When a solid is added to an aqueous solution in which it can dissolve, the rate of dissolution is initially fast and then slows. This is frequently interpreted as ‘passivation’, or a limitation posed by a surface layer—but this is not always the case.
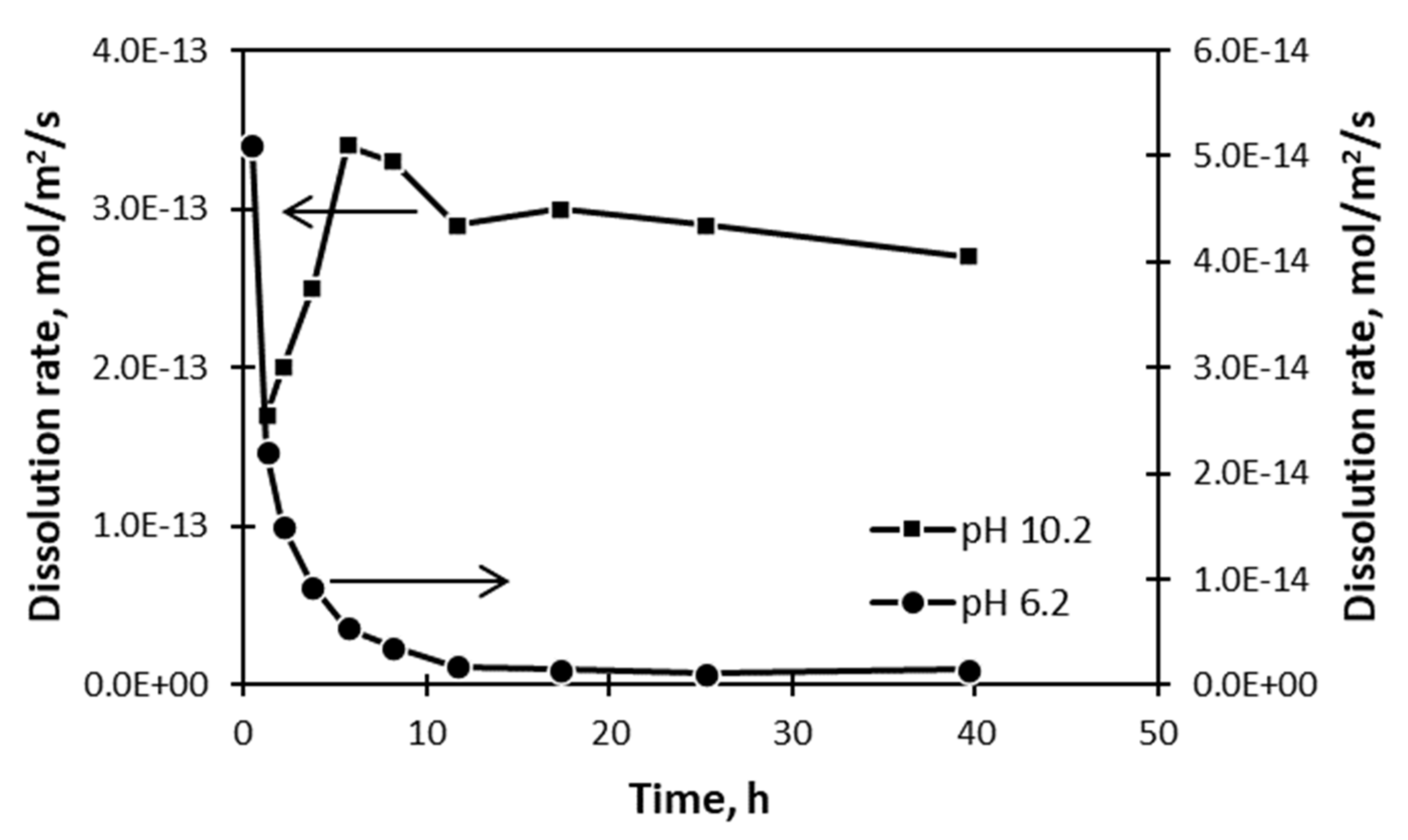
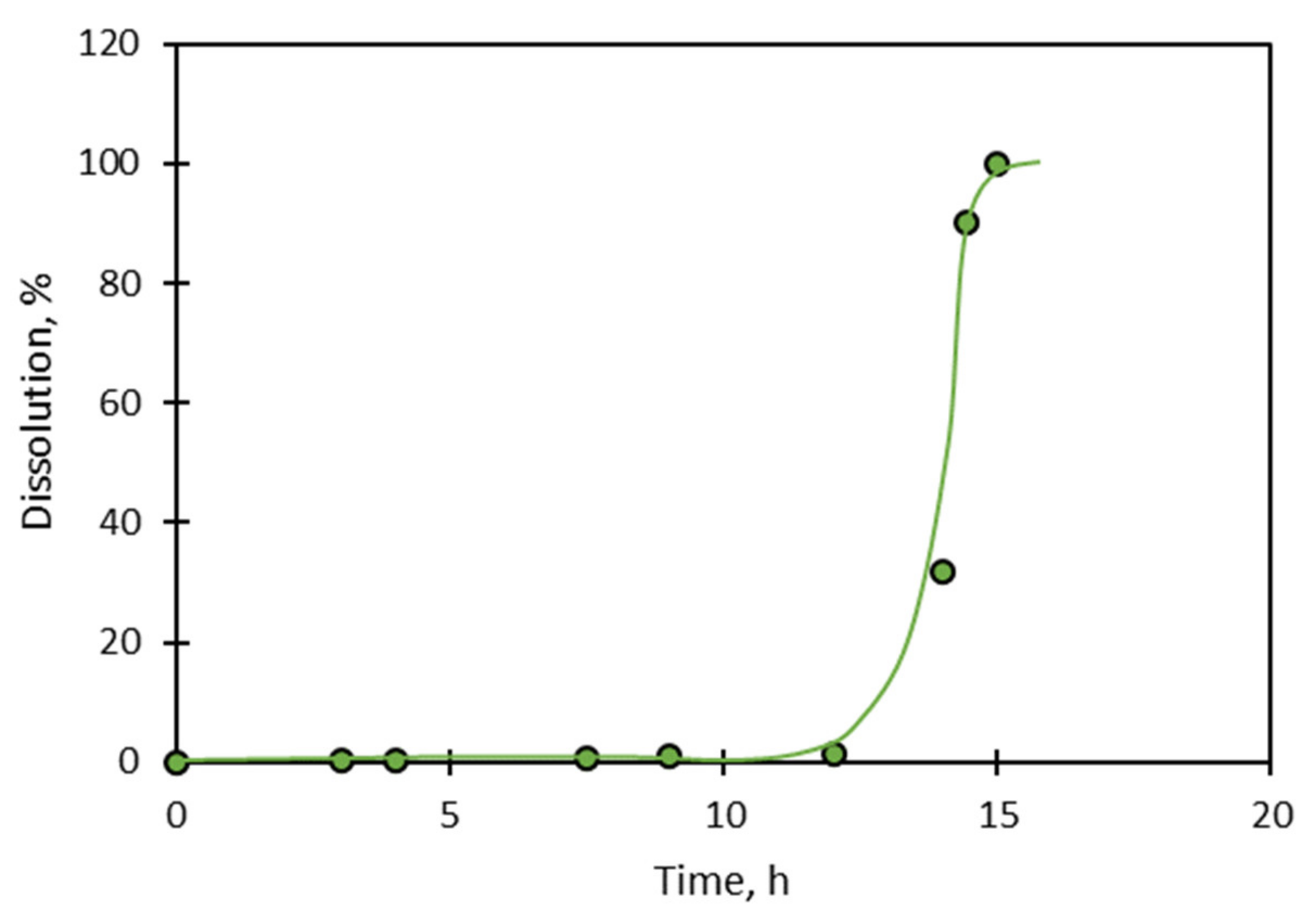
Consider the dissolution of quartz at pH 6.2, as shown in
Figure 14. Even though the rate of dissolution is initially fast and then slows, it reaches a steady state rate at which it continues to dissolve. That the solid continues to dissolve creates difficulties for the surface layer proposal, which means that it must be modified that limit growth of the layer once a certain thickness has been attained. No conditions have been set or derived by these layer models that determine such thickness, or how steady-state dissolution continues in its presence.
These difficulties with models based on surface layers are compounded when it is considered that the rate of dissolution of these same solids might initially accelerate in different circumstances.
Figure 14 also shows that the rate of dissolution of quartz at pH 10.2 is initially slow, and then accelerates to a steady or stationary value. Layer formation cannot explain such an accelerating rate of dissolution.
Similar results have been reported for NiO and AgBr. The acceleration of the dissolution of NiO is shown in
Figure 15.
The surface vacancy model accounts for this phenomenon without any additional physical modifications. In order to progress from Equation (8) to Equation (9), stationary conditions were assumed. To account for non-stationary effects, Equation (8) should be used instead of Equation (9).
The surface charge,
, is a proportional to the potential difference across the Helmholtz layer, with the proportionality constant referred to as the electrical capacitance of the layer. This means that Equation (8) can be written as Equation (34).
If the reaction is far from equilibrium,
and
are given by Equations (10) and (11). Substituting these values into Equation (34) and taking note that
is a dimensionless form of the potential difference, the expression is written in terms of
.
This equation needs to be combined with the batch mass balances for the concentrations of ions in solution. These mass balances are given by Equations (36) and (37).
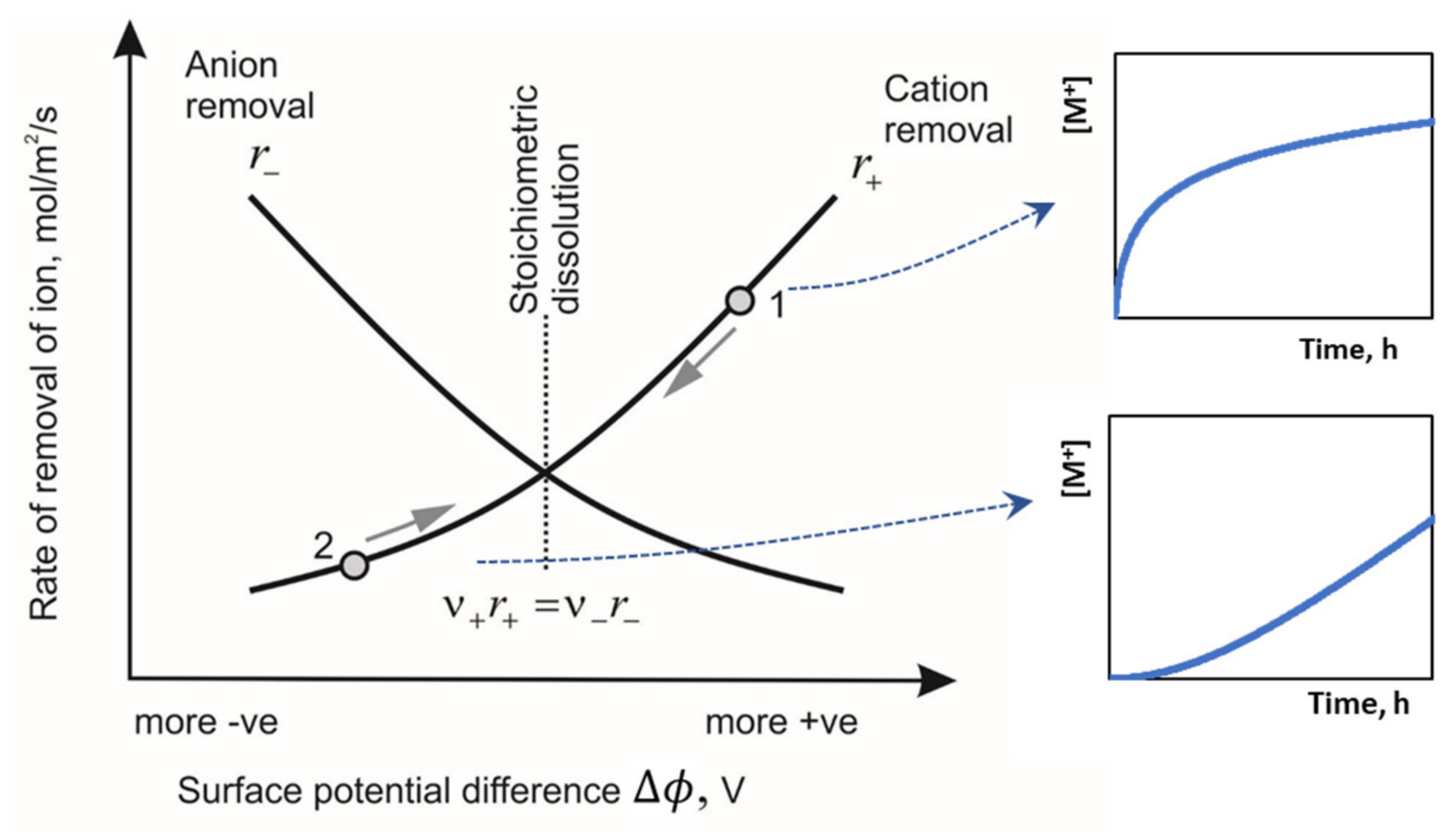
The curves for the rate of dissolution of the cation and the anion, given by Equations (10) and (11), are shown in
Figure 16 as a function of potential difference across the Helmholtz layer, noting that
is a dimensionless form of potential difference. The point at which stationary dissolution occurs is where Equation (9) is applicable and is at the intersection of the two curves. Most of the analysis on the orders of reaction and equilibrium has assumed that the experiments in question have been conducted at this stationary point.
The rate of dissolution is typically tracked by measuring only one of the constituent ions. Consider the case in which the cation is measured as it accumulates in solution. If the solid has an initial surface charge on immersion that is more positive than point of stationary dissolution, illustrated as point 1 in
Figure 16, then the positive charge will favor the removal of cations. The rate at potential 1 for cations is higher than that for anions, and as more cations are removed the surface becomes more negative, in turn lowering the potential and surface charge. This means that there is a driving force towards the stationary point. Turning attention to the rate of dissolution of cations, we see that it is initially highest, and then decreases until it reaches the stationary point where stoichiometric dissolution takes place.
If, on the other hand, the potential is initially more negative than the stationary point, then the removal of anions is more favorable, which makes the surface more positive, and this creates a driving force towards the stationary point. The dissolution of cations, though, is initially slow, and accelerates as the potential moves to the stationary point.
Thus, the surface-vacancy model describes both the acceleration and deceleration of the initial rates of dissolution.
10. Testing the Surface-Vacancy Model—Non-Stoichiometric Dissolution
Non-stoichiometric dissolution occurs by the same mechanism as discussed in the previous section, except that both anion and cation are measured, and the fact that the rates of dissolution are different is exposed. There is always a non-stoichiometric period initially before the surface charge has reached a stationary state. Stoichiometric is guaranteed only at the stationary point described by Equation (9), which incidentally is a stable operating point. Of course, for solids that dissolve quickly, the non-stoichiometric period might be very short.
11. Independent Testing of the Surface—Vacancy Model
The surface-vacancy model proposed here is based on a clearly described physical model of the surface. It posits that the surface is charged because the removal of ions leaves vacancies of opposite charge to the ions themselves. Thus, the predictions of the surface-vacancy model can be tested by measurements of the surface charge.
The surface-vacancy model has been applied to the data for the zeta potential of several minerals that are of importance to flotation has been examined [
79]. In that work, it was shown that the surface-vacancy model successfully described the measurements of the zeta potential. In other work, the surface-vacancy model has been shown to describe the measurements of zeta potential in a manner that is consistent with the mechanism of dissolution of quartz and silica [
7] and forsterite [
9].
The most common explanation for the surface charge is the multisite complexation model of Hiemstra et al. [
80,
81]. This model is a combination of the ‘amphoteric model’ and the ‘one-pK model’, both of which suffer from unclear and possibly indefensible foundations. For example, the multi-site complexation model assigns arbitrary values to the charge of a surface ion. Thus, it is proposed that the surface vacancy model is not only a model of dissolution but could be a foundational model for surface charge.
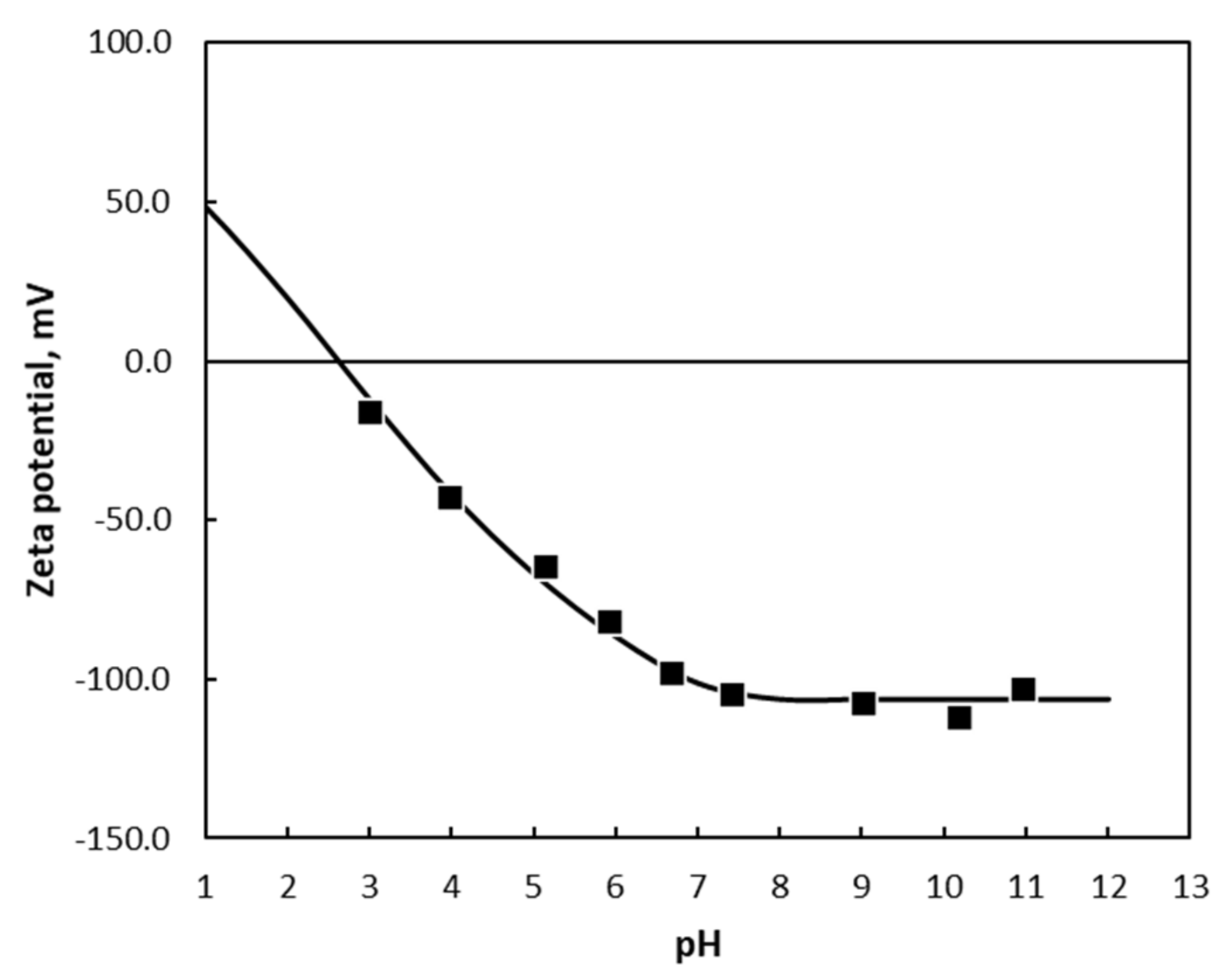
Surface charge, through independent measurements such as zeta potential, can be used as a clear and independent verification of the proposed model. As an example of this, consider the zeta potential of quartz, shown in
Figure 17.
The potential difference across the interface for quartz during dissolution is given by Equation (19). Examination of this equation reveals that the potential difference is proportional to the pH in the acid region. As the concentration of Na
+ is close to the concentration of OH
− in the alkaline region, it is independent of the pH in the alkaline region. This is precisely what is given by the data of James and Healy [
82] shown in
Figure 17. This demonstrates that the mechanism proposed for dissolution is effective in describing the surface charge.
12. Predictions from the Surface—Vacancy Model
The surface-vacancy model envisages the chemically independent removal of cations and anions, with the relationship between them governed only by surface charge (or the potential difference across the Helmholtz layer). Equilibrium, therefore, can be achieved in a partial manner [
4,
72]. A set of conditions can be envisaged in which one of the ‘partial reactions’ is in equilibrium, while the other is not. This situation is referred to as partial equilibrium, and it is possible that laboratory experiments in which it was assumed that the conditions were at equilibrium are in fact only at ‘partial equilibrium’. This is a unique view of equilibrium conditions and may have far reaching implications for the interpretation of experimental work in this field.
The surface vacancy model can account for the effect of other ligands. This has been treated in mechanisms for forsterite dissolution [
9].
The model can also account for the effect of impurities in solution on the rate of dissolution by including their deposition onto and their removal from the surface. This is an area for future work.
13. Oxidative and Reductive Dissolution
The discussion in this paper has focused on the dissolution of minerals in which no oxidation or reduction in the mineral takes place. Specifically, this is referred to as non-oxidative dissolution. Oxidation and reductive dissolution reactions are those in which oxidation or reduction in the mineral occurs during dissolution. Oxidative and reductive dissolution reactions are industrially important because the rates of these reactions are generally faster than the non-oxidation dissolution for the corresponding mineral.
Consequently, there has been extensive examination of the mechanism of oxidative dissolution in the metallurgical literature [
83,
84,
85,
86]. The influence of oxidants and reductants in solution is well documented. Research on the influence of the electronic structure of the mineral is beginning to make progress [
84,
85,
86], while advanced models for the effect of the content of iron in sphalerite, (Zn,Fe)S, on both the rate and activation energy of dissolution have been developed [
84,
86].
There are aspects of oxidative and reductive dissolution reaction that share features with the non-oxidative reactions analyzed in this paper. Most obvious is that oxidative and reductive dissolution frequently exhibit half-order kinetics with respect to oxidant or reductant. This half-order is described by the mixed-potential model [
87]. Many of the features of non-oxidative dissolution highlighted here might be applicable to oxidative dissolution reactions. Despite possible similarities between the mixed-potential model and the surface-vacancy model, there are major differences in their physical foundations. In oxidative and reductive dissolution, the surface charge is envisaged to be caused by excess electrons, whereas in the surface-vacancy model, the surface charge is envisaged to be caused by ionic vacancies on the surface.
This difference in nature of charge formation might seem immaterial. On the contrary, it is profound—it is not possible to understand the formation of surface charge during non-oxidative dissolution without the surface-vacancy model. Indeed, the surface-vacancy model is primarily a model of surface charge, with dissolution both the cause and the consequence.
14. Conclusions
This paper has demonstrated that there is a need for a new model of dissolution and has reviewed the novel proposal of the surface-vacancy model. The attention of this paper has focused on the physical foundations of this model, which provide clear independent tests of the model. Specifically, it has been shown (i) that the surface-vacancy model is applicable to the non-oxidative dissolution of all minerals and salts, (ii) that it provides a clear physical foundation from which the rate equations are derived, (iii) the rate expressions derived from this physical model describe the effects of H+ and OH- on the rate of dissolution expressed in terms of the order of reaction with respect to these reactants, (iv) that it describes the approach to equilibrium, (iv) that it describes the change in rate of dissolution during the initial stage of reaction and (v) that it describes the non-stoichiometric dissolution of more complex solids.
Most importantly, this model inherently and coherently describes the formation surface charge and the effect of this charge on the rate of removal and deposition of ions and demonstrates how this leads to orders of reaction that are multiples of one-half.
It has been demonstrated that the physical foundations of the model are testable by independent means, principally by measurements of the surface charge or surface potential difference. Finally, the model predicts a state of ‘partial equilibrium’ that is not contemplated other models.
Further work should be focused on assessing the foundations of the model, that is, the mechanism of surface charging due to surface vacancies, and the application of this novel physical foundation to other fields, such as crystallization, flotation, and colloid science.





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}