
Long-Term Arsenic Sequestration in Biogenic Pyrite from Contaminated Groundwater: Insights from Field and Laboratory Studies



 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
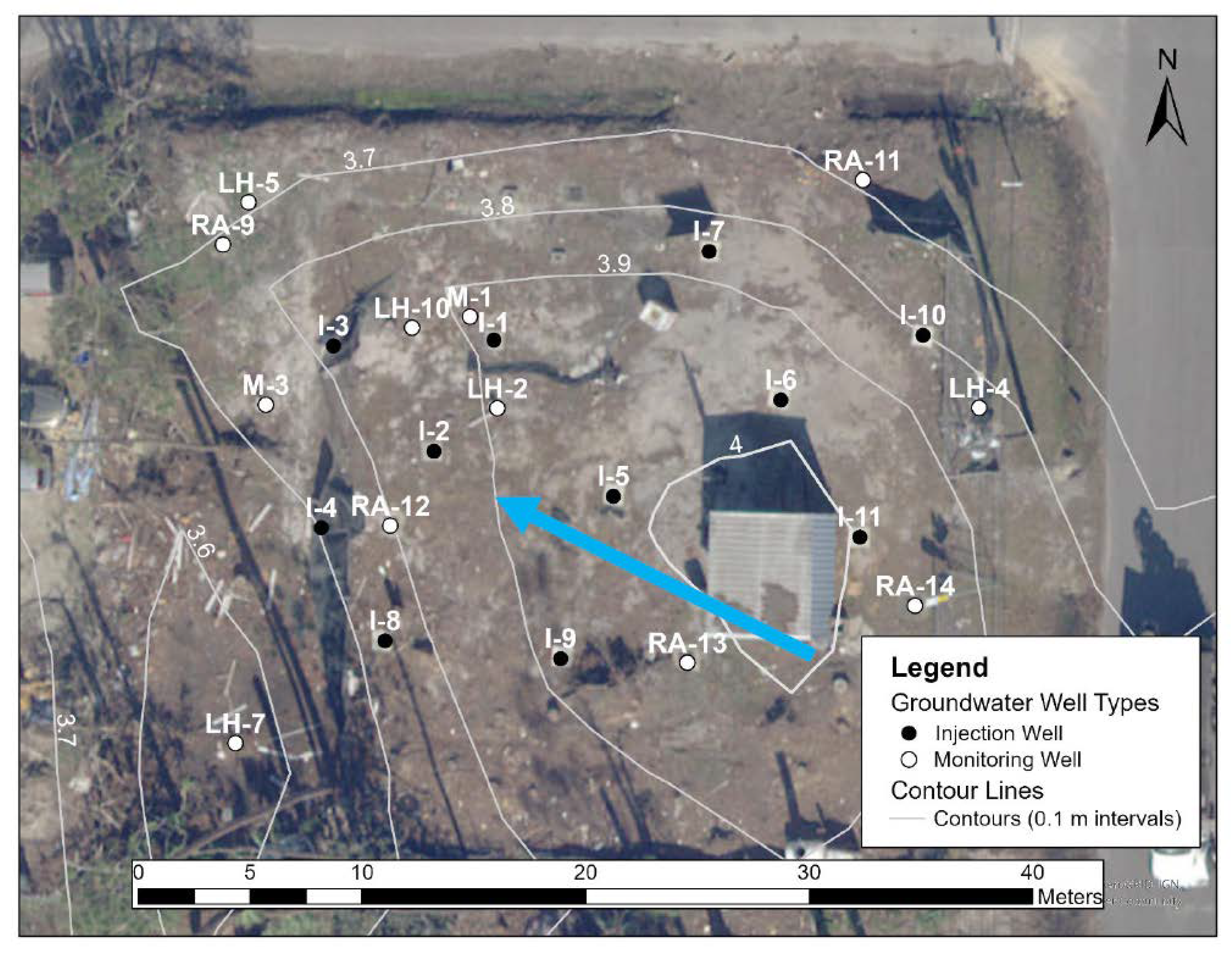
2.1. Study Site
2.2. Spatial and Temporal Concentration Analyses
2.3. Mineralogical and Geochemical Analyses
2.4. Imaging and Modeling Analyses
3. Results
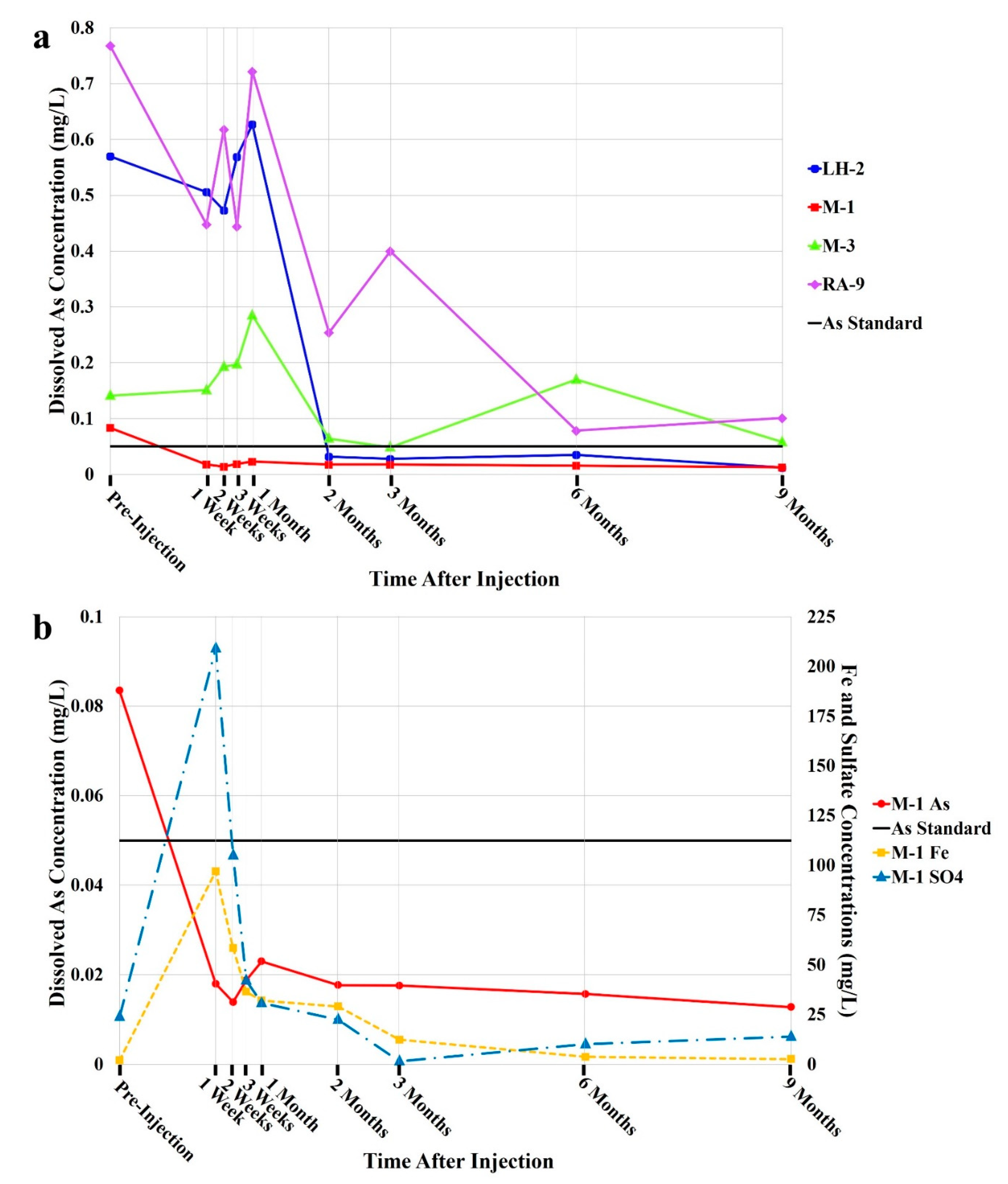
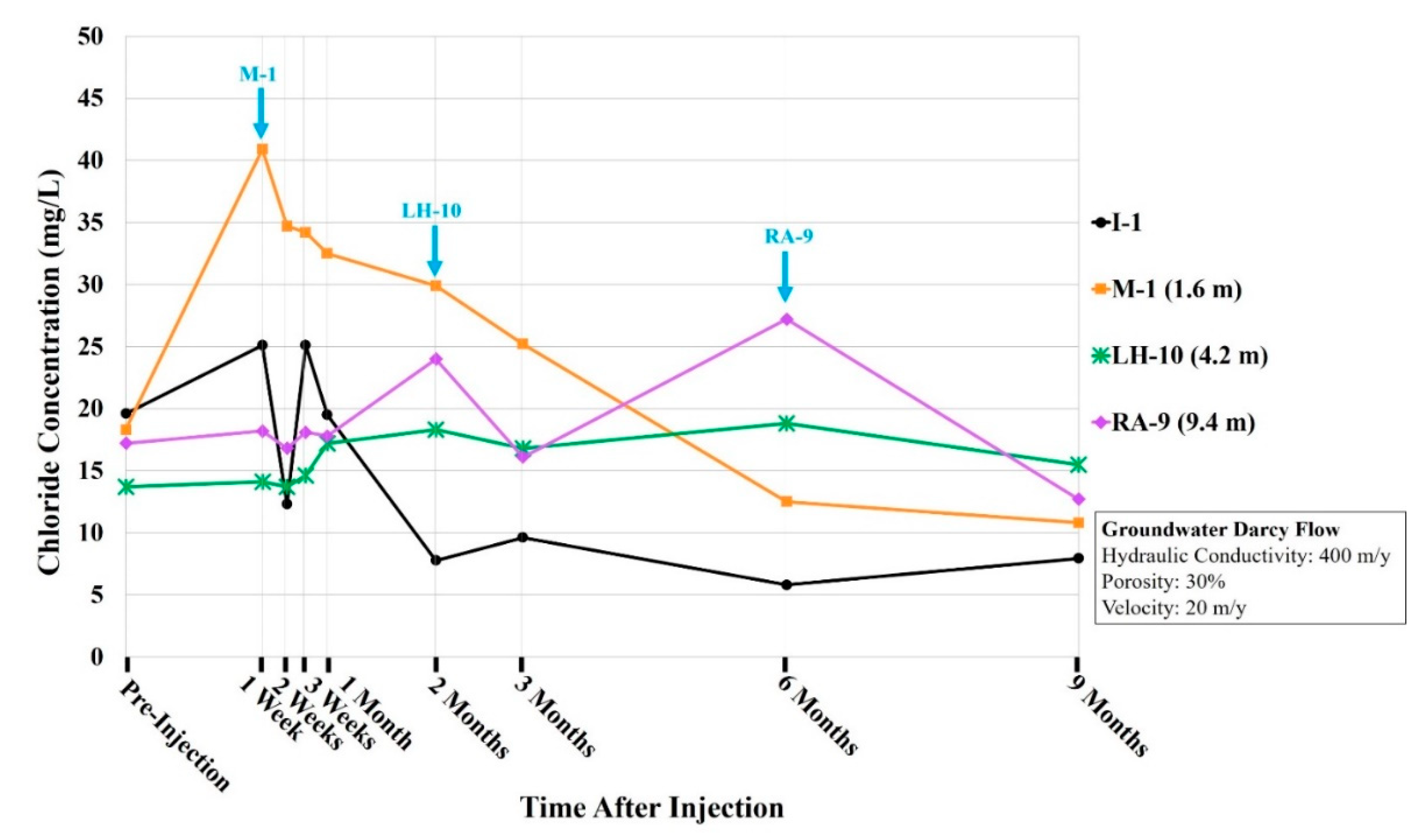
3.1. Spatial and Temporal Concentration Analyses
3.2. Mineralogical and Geochemical Analyses
3.3. Imaging Analysis
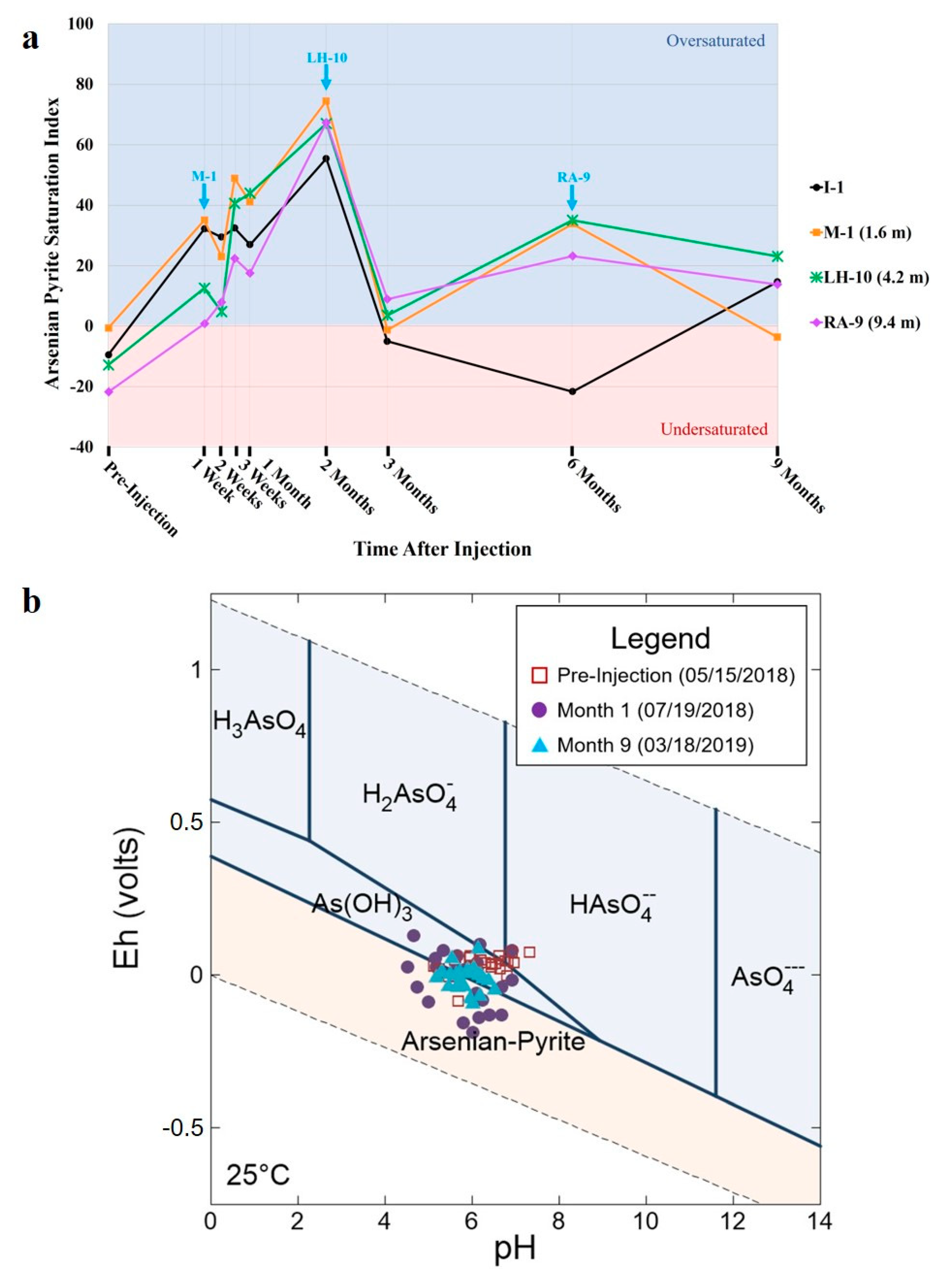
3.4. Geochemical Modeling Analysis
4. Discussion
4.1. Spatial and Temporal Concentration Analyses
4.2. Mineralogical and Geochemical Analyses
4.3. Imaging Analysis
4.4. Geochemical Modeling Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Exposure to Arsenic: A Major Public Health Concern (2019 Revision); WHO: Geneva, Switzerland, 2019.
- Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological Profile for Arsenic; ATSDR: Atlanta, GA, USA, 2007.
- Huerta-Diaz, M.A.; Morse, J.W. Pyritization of Trace Metals in Anoxic Marine Sediments. Geochim. Et Cosmochim. Acta 1992, 56, 2681–2702. [Google Scholar] [CrossRef]
- Gong, Z.; Lu, X.; Ma, M.; Watt, C.; Le, X.C. Arsenic Speciation Analysis. Talanta 2002, 58, 77–96. [Google Scholar] [CrossRef]
- Glodowska, M.; Stopelli, E.; Schneider, M.; Rathi, B.; Straub, D.; Lightfoot, A.; Kipfer, R.; Berg, M.; Jetten, M.; Kleindienst, S.; et al. Arsenic Mobilization by Anaerobic Iron-Dependent Methane Oxidation. Commun. Earth Environ. 2020, 1, 42. [Google Scholar] [CrossRef]
- Glodowska, M.; Stopelli, E.; Straub, D.; Thi, D.V.; Trang, P.T.K.; Viet, P.H.; AdvectAs Team Members; Berg, M.; Kappler, A.; Kleindienst, S. Arsenic Behavior in Groundwater in Hanoi (Vietnam) Influenced by a Complex Biogeochemical Network of Iron, Methane, and Sulfur Cycling. J. Hazard. Mater. 2021, 407, 124398. [Google Scholar] [CrossRef]
- Kontny, A.; Schneider, M.; Eiche, E.; Stopelli, E.; Glodowska, M.; Rathi, B.; Göttlicher, J.; Byrne, J.M.; Kappler, A.; Berg, M.; et al. Iron Mineral Transformations and Their Impact on As (Im)Mobilization at Redox Interfaces in As-Contaminated Aquifers. Geochim. Et Cosmochim. Acta 2021, 296, 189–209. [Google Scholar] [CrossRef]
- Woolson, E.A. Emissions, cycling and effects of arsenic in soil ecosystems. In Biological and Environmental Effects of Arsenic; Fowler, B.A., Ed.; Elsevier Ltd.: New York, NY, USA, 1983; pp. 51–139. [Google Scholar]
- Smedley, P.L.; Kinniburgh, D.G. A Review of the Source, Behaviour and Distribution of Arsenic in Natural Waters. Appl. Geochem. 2002, 17, 517–568. [Google Scholar] [CrossRef] [Green Version]
- Bencko, V.; Foong, F.Y.L. The History of Arsenical Pesticides and Health Risks Related to the Use of Agent Blue. Ann. Agric. Environ. Med. 2017, 24, 312–316. [Google Scholar] [CrossRef]
- Lee, M.-K.; Saunders, J.A.; Wilson, T.; Levitt, E.; Ghandehari, S.S.; Dhakal, P.; Redwine, J.; Marks, J.; Billor, Z.M.; Miller, B.; et al. Field-Scale Bioremediation of Arsenic-Contaminated Groundwater Using Sulfate-Reducing Bacteria and Biogenic Pyrite. Bioremed. J. 2018, 23, 1–21. [Google Scholar] [CrossRef]
- U.S. EPA. Organic Arsenicals; Notice of Receipt of Requests to Voluntarily Cancel or to Amend to Terminate Uses of Certain Pesticide Registrations; U.S. EPA: Washington, DC, USA, 2009; pp. 32596–32604.
- Pi, K.; Wang, Y.; Xie, X.; Ma, T.; Liu, Y.; Su, C.; Zhu, Y.; Wang, Z. Remediation of Arsenic-Contaminated Groundwater by in-Situ Stimulating Biogenic Precipitation of Iron Sulfides. Water Res. 2017, 109, 337–346. [Google Scholar] [CrossRef]
- Keimowitz, A.R.; Mailloux, B.J.; Cole, P.; Stute, M.; Simpson, H.J.; Chillrud, S.N. Laboratory Investigations of Enhanced Sulfate Reduction as a Groundwater Arsenic Remediation Strategy. Environ. Sci. Technol. 2007, 41, 6718–6724. [Google Scholar] [CrossRef] [Green Version]
- Saunders, J.; Cook, R.; Thomas, R.; Crowe, D. Coprecipitation of trace metals in biogenic pyrite: Implications for enhanced intrinsic bioremediation. In Proceedings of the Fourth International Symposium of the Geochemistry of the Earth’s Surface: Short Papers, Yorkshire, UK, 1996; Bottrell, S.H., Ed.; University of Leeds: Ilkley, UK, 1996; pp. 470–474. [Google Scholar]
- Barton, L.L.; Tomei, F.A. Characteristics and activities of sulfate-reducing bacteria. In Sulfate-Reducing Bacteria; Barton, L.L., Ed.; Springer: Boston, MA, USA, 1995; pp. 1–32. [Google Scholar] [CrossRef]
- Dyer, B.D. Field Guide to Bacteria; Cornell University Press: Ithaca, NY, USA, 2003. [Google Scholar]
- Donald, R.; Southam, G. Low Temperature Anaerobic Bacterial Diagenesis of Ferrous Monosulfide to Pyrite. Geochim. Et Cosmochim. Acta 1999, 63, 2019–2023. [Google Scholar] [CrossRef]
- Thiel, J.; Byrne, J.M.; Kappler, A.; Schink, B.; Pester, M. Pyrite Formation from FeS and H2S Is Mediated through Microbial Redox Activity. Proc. Natl. Acad. Sci. USA 2019, 116, 6897–6902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bostick, B.C.; Fendorf, S. Arsenite Sorption on Troilite (FeS) and Pyrite (FeS2). Geochim. Et Cosmochim. Acta 2003, 67, 909–921. [Google Scholar] [CrossRef]
- Saunders, J.; Lee, M.-K.; Dhakal, P. Bioremediation of Arsenic-Contaminated Groundwater by Sequestration of Arsenic in Biogenic Pyrite. Appl. Geochem. 2018, 98, 233–243. [Google Scholar] [CrossRef]
- DeFlaun, M.F.; Lanzon, J.; Lodato, M.; Henry, S.; Onstott, T.; Chan, E.W.; Otemuyima, B. Anaerobic Biostimulation for the In Situ Precipitation and Long-Term Sequestration of Metal Sulfides; Geosyntec Consultants: Tampa, FL, USA, 2009. [Google Scholar]
- Onstott, T.C.; Chan, E.; Polizzotto, M.L.; Lanzon, J.; DeFlaun, M.F. Precipitation of Arsenic under Sulfate Reducing Conditions and Subsequent Leaching under Aerobic Conditions. Appl. Geochem. 2011, 26, 269–285. [Google Scholar] [CrossRef]
- Saunders, J.A.; Lee, M.-K.; Wolf, L.W.; Morton, C.M.; Feng, Y.; Thomson, I.; Park, S. Geochemical, Microbiological, and Geophysical Assessments of Anaerobic Immobilization of Heavy Metals. Bioremed. J. 2005, 9, 33–48. [Google Scholar] [CrossRef]
- Saunders, J.A.; Lee, M.K.; Shamsudduha, M.; Dhakal, P.; Uddin, A.; Chowdury, M.T.; Ahmed, K.M. Geochemistry and Mineralogy of Arsenic in (Natural) Anaerobic Groundwaters. Appl. Geochem. 2008, 23, 3205–3214. [Google Scholar] [CrossRef]
- Fischer, A.; Lee, M.K.; Ojeda, A.S.; Rogers, S.R. GIS Interpolation Is Key in Assessing Spatial and Temporal Bioremediation of Groundwater Arsenic Contamination. J. Environ. Manag. 2021, 280, 111683. [Google Scholar] [CrossRef]
- Mintz, J.; Miller, J. Lynn Haven Retired Substation Remedial Action Plan; 1983. [Google Scholar]
- Schmidt, W.; Clark, M.W. Geology of Bay County, Florida (No. 57); Bureau of Geology, Florida Department of Natural Resources: Tallahassee, FL, USA, 1980.
- Wilson, T.J. Pyrite Biomineralization and Arsenic Sequestration at a Florida Industrial Site: Imaging and Geochemical Analysis. Master’s Thesis, Auburn University, Auburn, AL, USA, 2018. [Google Scholar]
- Fischer, A.B. Field and Laboratory Investigations of Groundwater Arsenic Sequestration in Biogenic Pyrite at an Industrial Site in Florida. Master’s Thesis, Auburn University, Auburn, AL, USA, 2020. [Google Scholar]
- Ben-Dov, E.; Brenner, A.; Kushmaro, A. Quantification of sulfate-reducing bacteria in industrial wastewater, by real-time polymerase chain reaction (PCR) using dsrA and apsA genes. Microb. Ecol. 2007, 54, 439–451. [Google Scholar] [CrossRef]
- Rajaković, L.V.; Todorović, Ž.N.; Rajaković-Ognjanović, V.N.; Onjia, A.E. Analytical Methods for Arsenic Speciation Analysis. J. Serb. Chem. Soc. 2013, 78, 1461–1479. [Google Scholar] [CrossRef] [Green Version]
- Rasul, S.B.; Munir, A.K.M.; Hossain, Z.A.; Khan, A.H.; Alauddin, M.; Hussam, A. Electrochemical Measurement and Speciation of Inorganic Arsenic in Groundwater of Bangladesh. Talanta 2002, 58, 33–43. [Google Scholar] [CrossRef]
- Bruker Tracer Series User Guide: Hand-Held XRF Analyzers; Bruker: Billerica, MA, USA, 2010; pp. 169–232.
- Reed, S.J.B. Electron Microprobe Analysis and Scanning Electron Microscopy in Geology; Cambridge University Press: Cambridge, UK, 2005; ISBN 9780521848756. [Google Scholar]
- Bethke, C.M. Geochemical and Biogeochemical Reaction Modeling; Cambridge University Press: Cambridge, UK, 2008. [Google Scholar]
- Baker, B.J.; Banfield, J.F. Microbial communities in acid mine drainage. FEMS Microbiol. Ecol. 2003, 44, 139–152. [Google Scholar] [CrossRef] [Green Version]
- Berg, J.S.; Duverger, A.; Cordier, L.; Laberty-Robert, C.; Guyot, F.; Miot, J. Rapid pyritization in the presence of a sulfur/sulfate-reducing bacterial consortium. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Deditius, A.P.; Utsunomiya, S.; Renock, D.; Ewing, R.C.; Ramana, C.V.; Becker, U.; Kesler, S.E. A Proposed New Type of Arsenian Pyrite: Composition, Nanostructure and Geological Significance. Geochim. Et Cosmochim. Acta 2008, 72, 2919–2933. [Google Scholar] [CrossRef]
- Neumann, R.B.; Ashfaque, K.N.; Badruzzaman, A.B.M.; Ashraf Ali, M.; Shoemaker, J.K.; Harvey, C.F. Anthropogenic Influences on Groundwater Arsenic Concentrations in Bangladesh. Nat. Geosci. 2010, 3, 46–52. [Google Scholar] [CrossRef]
- Gao, X.; Wang, Y.; Hu, Q.; Su, C. Effects of Anion Competitive Adsorption on Arsenic Enrichment in Groundwater. J. Environ. Sci. Health Part A 2011, 46, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Su, C.; Wang, Y.; Hu, Q. Mobility of Arsenic in Aquifer Sediments at Datong Basin, Northern China: Effect of Bicarbonate and Phosphate. J. Geochem. Explor. 2013, 135, 93–103. [Google Scholar] [CrossRef]
- Aziz, Z.; Bostick, B.C.; Zheng, Y.; Huq, M.R.; Rahman, M.M.; Ahmed, K.M.; Geen, A. van Evidence of Decoupling between Arsenic and Phosphate in Shallow Groundwater of Bangladesh and Potential Implications. Appl. Geochem. 2017, 77, 167–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieder, M.; Crelling, J.C.; Šustai, O.; Drábek, M.; Weiss, Z.; Klementová, M. Arsenic in iron disulfides in a brown coal from the North Bohemian Basin, Czech Republic. Int. J. Coal Geol. 2007, 71, 115–121. [Google Scholar] [CrossRef]
- Gartman, A.; Luther, G. Comparison of Pyrite (FeS2) Synthesis Mechanisms to Reproduce Natural FeS2 Nanoparticles Found at Hydrothermal Vents. Geochim. Et Cosmochim. Acta 2013, 120, 447–458. [Google Scholar] [CrossRef]
- Couture, R.M.; Gobeil, C.; Tessier, A. Arsenic, iron and sulfur co-diagenesis in lake sediments. Geochim. Cosmochim. Acta 2010, 74, 1238–1255. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fischer, A.; Saunders, J.; Speetjens, S.; Marks, J.; Redwine, J.; Rogers, S.R.; Ojeda, A.S.; Rahman, M.M.; Billor, Z.M.; Lee, M.-K. Long-Term Arsenic Sequestration in Biogenic Pyrite from Contaminated Groundwater: Insights from Field and Laboratory Studies. Minerals 2021, 11, 537. https://doi.org/10.3390/min11050537
Fischer A, Saunders J, Speetjens S, Marks J, Redwine J, Rogers SR, Ojeda AS, Rahman MM, Billor ZM, Lee M-K. Long-Term Arsenic Sequestration in Biogenic Pyrite from Contaminated Groundwater: Insights from Field and Laboratory Studies. Minerals. 2021; 11(5):537. https://doi.org/10.3390/min11050537
Chicago/Turabian StyleFischer, Alicia, James Saunders, Sara Speetjens, Justin Marks, Jim Redwine, Stephanie R. Rogers, Ann S. Ojeda, Md Mahfujur Rahman, Zeki M. Billor, and Ming-Kuo Lee. 2021. "Long-Term Arsenic Sequestration in Biogenic Pyrite from Contaminated Groundwater: Insights from Field and Laboratory Studies" Minerals 11, no. 5: 537. https://doi.org/10.3390/min11050537
APA StyleFischer, A., Saunders, J., Speetjens, S., Marks, J., Redwine, J., Rogers, S. R., Ojeda, A. S., Rahman, M. M., Billor, Z. M., & Lee, M. -K. (2021). Long-Term Arsenic Sequestration in Biogenic Pyrite from Contaminated Groundwater: Insights from Field and Laboratory Studies. Minerals, 11(5), 537. https://doi.org/10.3390/min11050537