
A Kinetic Monte Carlo Approach to Model Barite Dissolution: The Role of Reactive Site Geometry


Abstract
:1. Introduction

2. Materials and Methods
2.1. Simulated System
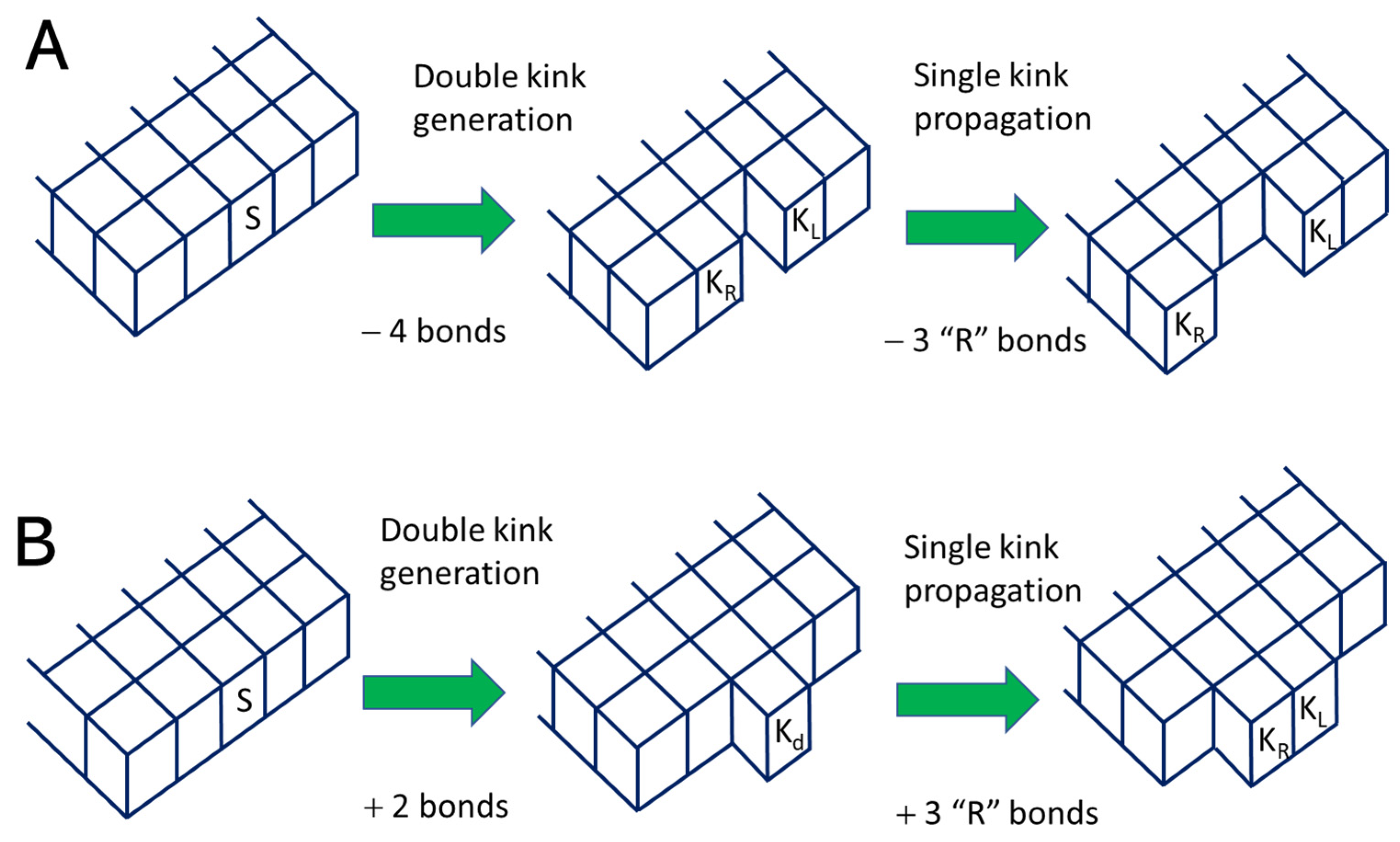
2.2. The Kinetic Monte Carlo Model
2.2.1. Reaction Rates and Probabilities
2.2.2. The Algorithm
2.2.3. Parameterization

2.2.4. Data Visualization
3. Results
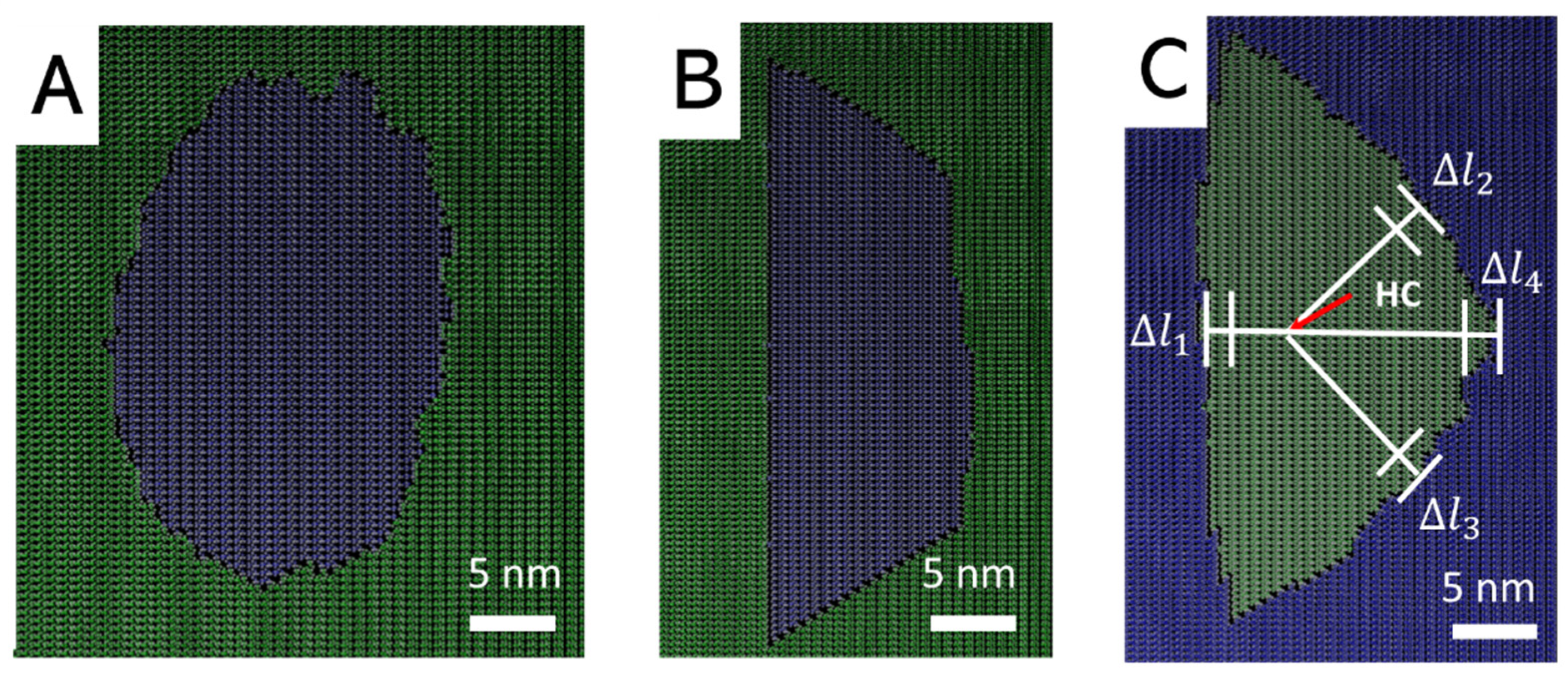
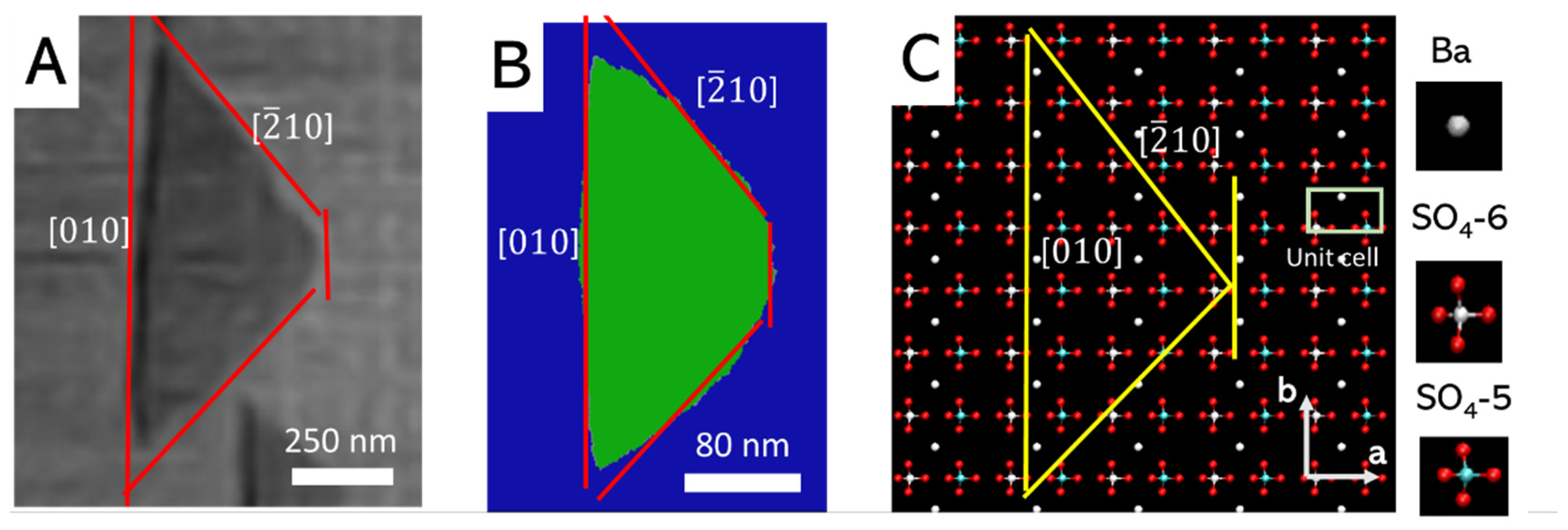
3.1. Morhoplogy of Monolayer Pits

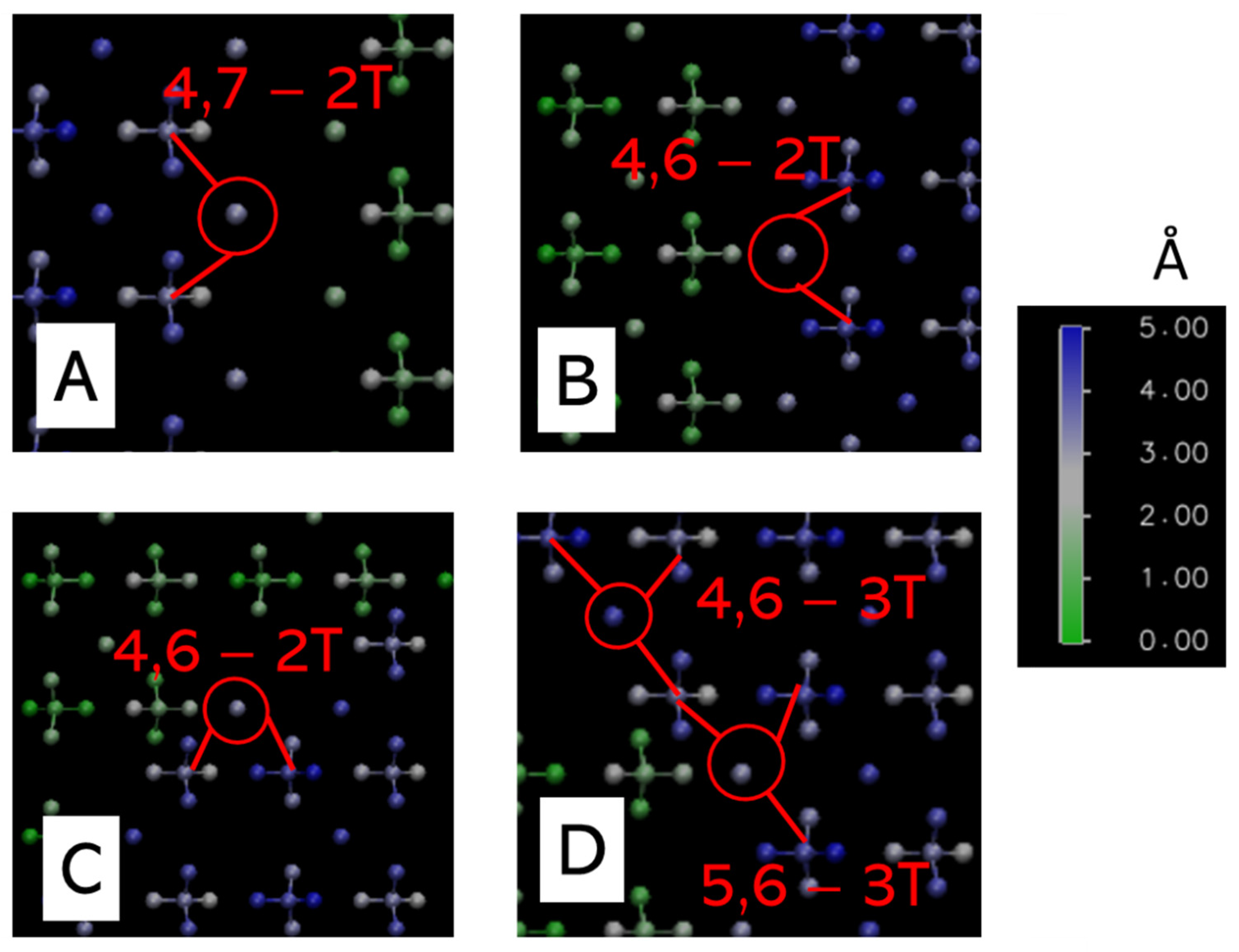
3.2. Reactive Sites Coordination
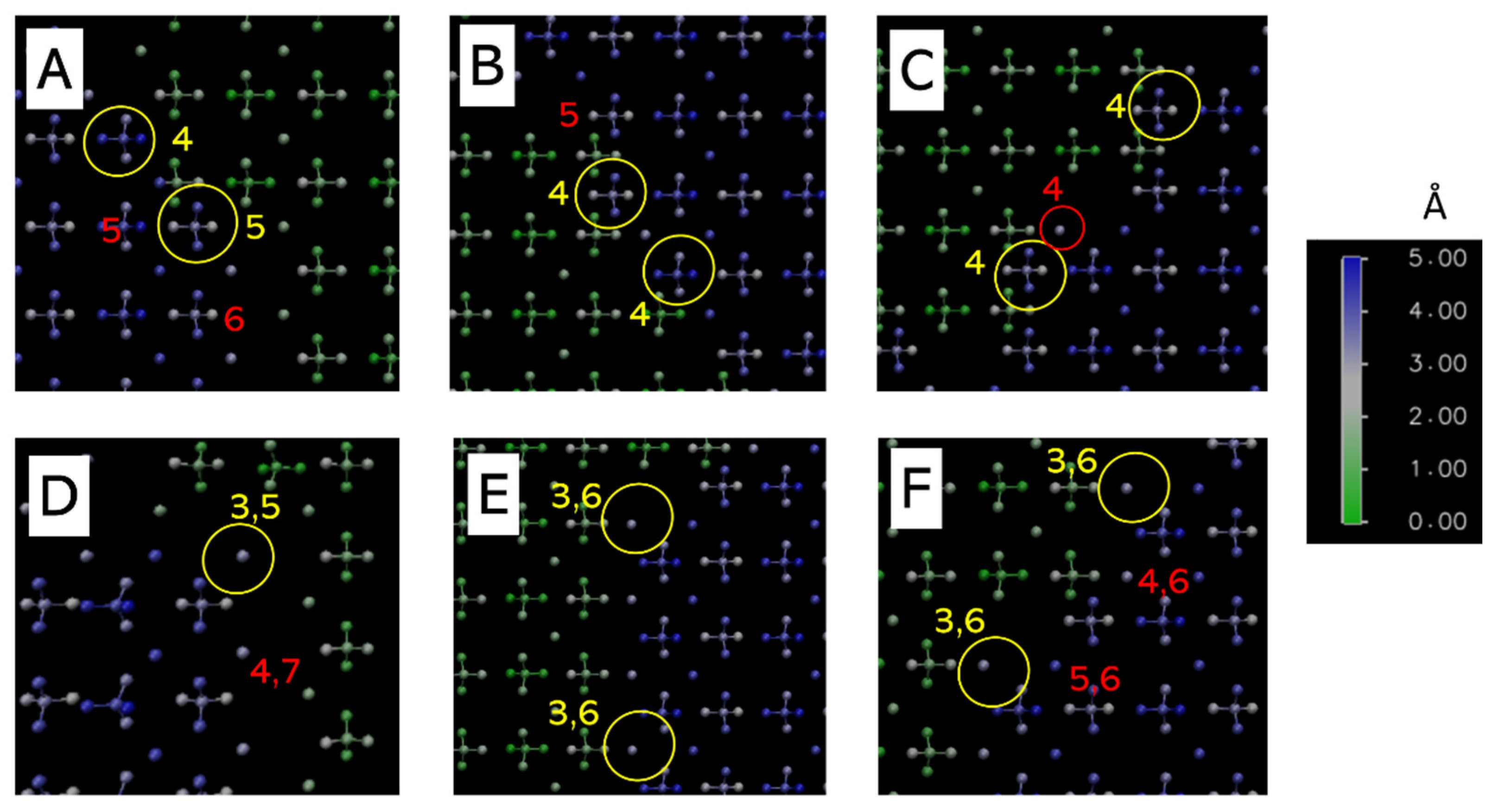
3.2.1. Step Sites
3.2.2. Kink Sites
3.3. Morphology of Mutlilayer Pits
4. Discussion
4.1. Rate Limiting Step
4.2. Recommendations for Molecular Dynamics Calculations
- (1)
- Dissolution of Ba step sites from the left [010] step where Ba has coordination (4,7) and from the right site where Ba has coordination (4,6);
- (2)
- Dissolution of Ba step site from the straight [120] step;
- (3)
- Dissolution of SO4 kink sites of coordination 5 at the left [010] step and coordination 4 at the right [010] step;
- (4)
- Extra calculations for checking preferential removal of SO4 and Ba at step sites. These calculations should ensure whether Ba or SO4 kink sites along each kinetically relevant atomic step influence onto step propagation rates.
5. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bleiwas, D.I.; Miller, M.M. Barite: A Case Study of Import Reliance on an Essential Material for Oil and Gas Exploration and Development Drilling; Scientific Investigations Report; U.S. Geological Survey: Reston, VA, USA, 2015; Volumes 2014–5230, p. 14.
- Abdou, M.I.; Al-Sabagh, A.M.; Ahmed, H.E.S.; Fadl, A.M. Impact of Barite and Ilmenite Mixture on Enhancing the Drilling Mud Weight. Egypt. J. Pet. 2018, 27, 955–967. [Google Scholar] [CrossRef]
- Todd, A.C.; Yuan, M. Barium and Strontium Sulfate Solid-Solution Formation in Relation to North Sea Scaling Problems. SPE Prod. Eng. 1990, 5, 279–285. [Google Scholar] [CrossRef]
- Evcin, O.; Evcin, A.; Bezir, N.; Akkurt, İ.; Günoğlu, K.; Ersoy, B. Production of Barite and Boroncarbide Doped Radiation Shielding Polymer Composite Panels. Acta Phys. Pol. A 2017, 132, 1145–1148. [Google Scholar] [CrossRef]
- Klinkenberg, M.; Brandt, F.; Breuer, U.; Bosbach, D. Uptake of Ra during the Recrystallization of Barite: A Microscopic and Time of Flight-Secondary Ion Mass Spectrometry Study. Environ. Sci. Technol. 2014, 48, 6620–6627. [Google Scholar] [CrossRef]
- Paytan, A.; Griffith, E.M. Marine Barite: Recorder of Variations in Ocean Export Productivity. Deep Sea Res. Part II Top. Stud. Oceanogr. 2007, 54, 687–705. [Google Scholar] [CrossRef]
- Griffith, E.M.; Paytan, A. Barite in the Ocean—Occurrence, Geochemistry and Palaeoceanographic Applications. Sedimentology 2012, 59, 1817–1835. [Google Scholar] [CrossRef]
- Christy, A.G.; Putnis, A. The Kinetics of Barite Dissolution and Precipitation in Water and Sodium Chloride Brines at 44–85 °C. Geochim. Cosmochim. Acta 1993, 57, 2161–2168. [Google Scholar] [CrossRef]
- Zhen-Wu, B.Y.; Dideriksen, K.; Olsson, J.; Raahauge, P.J.; Stipp, S.L.S.; Oelkers, E.H. Experimental Determination of Barite Dissolution and Precipitation Rates as a Function of Temperature and Aqueous Fluid Composition. Geochim. Cosmochim. Acta 2016, 194, 193–210. [Google Scholar] [CrossRef]
- Dunn, K.; Daniel, E.; Shuler, P.J.; Chen, H.J.; Tang, Y.; Yen, T.F. Mechanisms of Surface Precipitation and Dissolution of Barite: A Morphology Approach. J. Colloid Interface Sci. 1999, 214, 427–437. [Google Scholar] [CrossRef]
- Dove, P.M.; Czank, C.A. Crystal Chemical Controls on the Dissolution Kinetics of the Isostructural Sulfates: Celestite, Anglesite, and Barite. Geochim. Cosmochim. Acta 1995, 59, 1907–1915. [Google Scholar] [CrossRef]
- Higgins, S.R.; Jordan, G.; Eggleston, C.M.; Knauss, K.G. Dissolution Kinetics of the Barium Sulfate (001) Surface by Hydrothermal Atomic Force Microscopy. Langmuir 1998, 14, 4967–4971. [Google Scholar] [CrossRef]
- Kowacz, M.; Putnis, A. The Effect of Specific Background Electrolytes on Water Structure and Solute Hydration: Consequences for Crystal Dissolution and Growth. Geochim. Cosmochim. Acta 2008, 72, 4476–4487. [Google Scholar] [CrossRef]
- Kuwahara, Y. In Situ Hot-Stage AFM Study of the Dissolution of the Barite (001) Surface in Water at 30–55 °C. Am. Mineral. 2012, 97, 1564–1573. [Google Scholar] [CrossRef]
- Kuwahara, Y. In Situ Atomic Force Microscopy Study of Dissolution of the Barite (001) Surface in Water at 30 °C. Geochim. Cosmochim. Acta 2011, 75, 41–51. [Google Scholar] [CrossRef]
- Putnis, A.; Junta-Rosso, J.L.; Hochella, M.F. Dissolution of Barite by a Chelating Ligand: An Atomic Force Microscopy Study. Geochim. Cosmochim. Acta 1995, 59, 4623–4632. [Google Scholar] [CrossRef]
- Risthaus, P.; Bosbach, D.; Becker, U.; Putnis, A. Barite Scale Formation and Dissolution at High Ionic Strength Studied with Atomic Force Microscopy. Colloids Surf. A Physicochem. Eng. Asp. 2001, 3, 201–214. [Google Scholar] [CrossRef]
- Bracco, J.N.; Lee, S.S.; Stubbs, J.E.; Eng, P.J.; Heberling, F.; Fenter, P.; Stack, A.G. Hydration Structure of the Barite (001)–Water Interface: Comparison of X-Ray Reflectivity with Molecular Dynamics Simulations. J. Phys. Chem. C 2017, 121, 12236–12248. [Google Scholar] [CrossRef]
- Stack, A.G.; Raiteri, P.; Gale, J.D. Accurate Rates of the Complex Mechanisms for Growth and Dissolution of Minerals Using a Combination of Rare-Event Theories. J. Am. Chem. Soc. 2012, 134, 11–14. [Google Scholar] [CrossRef]
- Stack, A.G. Molecular Dynamics Simulations of Solvation and Kink Site Formation at the {001} Barite−Water Interface†. J. Phys. Chem. C 2009, 113, 2104–2110. [Google Scholar] [CrossRef]
- Stack, A.G.; Rustad, J.R. Structure and Dynamics of Water on Aqueous Barium Ion and the {001} Barite Surface. J. Phys. Chem. C 2007, 111, 16387–16391. [Google Scholar] [CrossRef]
- Wang, K.-S.; Resch, R.; Dunn, K.; Shuler, P.; Tang, Y.; Koel, B.E.; Fu Yen, T. Dissolution of the Barite (001) Surface by the Chelating Agent DTPA as Studied with Non-Contact Atomic Force Microscopy. Colloids Surf. A Physicochem. Eng. Asp. 1999, 160, 217–227. [Google Scholar] [CrossRef]
- Fenter, P.; McBride, M.T.; Srajer, G.; Sturchio, N.C.; Bosbach, D. Structure of Barite (001)− and (210)−Water Interfaces. J. Phys. Chem. B 2001, 105, 8112–8119. [Google Scholar] [CrossRef] [Green Version]
- Gilmer, G.H. Computer Models of Crystal Growth. Science 1980, 208, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Lasaga, A.C.; Luttge, A. Mineralogical Approaches to Fundamental Crystal Dissolution Kinetics. Am. Mineral. 2004, 89, 527–540. [Google Scholar] [CrossRef]
- Blum, A.E.; Lasaga, A.C. Monte Carlo Simulations of Surface Reaction Rate Laws. In Aquatic Surface Chemistry; Chemical Processes at the Particle-Water Interface; Stumm, W., Ed.; Wiley: New York, NY, USA, 1987; pp. 255–292. [Google Scholar]
- Gilmer, G.H.; Bennema, P. Simulation of Crystal Growth with Surface Diffusion. J. Appl. Phys. 1972, 43, 1347–1360. [Google Scholar] [CrossRef]
- Kurganskaya, I.; Rohlfs, R.D. Atomistic to Meso Scale Modelling of Mineral Dissolution: Methods, Challenges and Prospects. Am. J. Sci. 2020, 320, 1–26. [Google Scholar] [CrossRef]
- Kossel, W. Extending the Law of Bravais. Nachr. Ges. Wiss. Göttingen Math. Phys. Kl. 1927, 135–143. [Google Scholar]
- Stranski, I.N. Zur Theorie Des Kristallwachstums. Zeit. Phys. Chem. 1928, 136, 259–278. [Google Scholar] [CrossRef]
- Hess, F.; Over, H. Rate-Determining Step or Rate-Determining Configuration? The Deacon Reaction over RuO2(110) Studied by DFT-Based KMC Simulations. ACS Catal. 2017, 7, 128–138. [Google Scholar] [CrossRef]
- Exner, K.S.; Hess, F.; Over, H.; Seitsonen, A.P. Combined Experiment and Theory Approach in Surface Chemistry: Stairway to Heaven? Surf. Sci. 2015, 640, 165–180. [Google Scholar] [CrossRef]
- Kurganskaya, I.; Luttge, A. Kinetic Monte Carlo Simulations of Silicate Dissolution: Model Complexity and Parametrization. J. Phys. Chem. C 2013, 117, 24894–24906. [Google Scholar] [CrossRef]
- Pineda, M.; Stamatakis, M. Kinetic Monte Carlo Simulations for Heterogeneous Catalysis: Fundamentals, Current Status, and Challenges. J. Chem. Phys. 2022, 156, 120902. [Google Scholar] [CrossRef] [PubMed]
- Reuter, K.; Scheffler, M. First-Principles Kinetic Monte Carlo Simulations for Heterogeneous Catalysis: Application to the CO Oxidation at RuO2 (110). Phys. Rev. B 2006, 73, 045433. [Google Scholar] [CrossRef] [Green Version]
- Colville, A.A.; Staudhammer, K. A Refinement of the Structure of Barite. Locality: Cow Green Mine, Teesdale, Durham, England. Am. Mineral. 1967, 52, 1877–1880. [Google Scholar]
- Downs, B.; Bartelmehs, K.; Sinnaswamy, K. XtalDraw; Mineralogy and Crystallogrpahy; University of Arizona: Tucson, AZ, USA, 2003; Available online: https://www.geo.arizona.edu/xtal/group/software.htm (accessed on 12 May 2022).
- Pelmenschikov, A.; Leszczynski, J.; Pettersson, L.G.M. Mechanism of Dissolution of Neutral Silica Surfaces: Including Effect of Self-Healing. J. Phys. Chem. A 2001, 105, 9528–9532. [Google Scholar] [CrossRef]
- Bortz, A.B.; Kalos, M.H.; Lebowitz, J.L. A New Algorithm for Monte Carlo Simulation of Ising Spin Systems. J. Comput. Phys. 1975, 17, 10–18. [Google Scholar] [CrossRef]
- Meakin, P.; Rosso, K.M. Simple Kinetic Monte Carlo Models for Dissolution Pitting Induced by Crystal Defects. J. Chem. Phys. 2008, 129, 204106. [Google Scholar] [CrossRef]
- Voter, A.F. Introduction to the Kinetic Monte Carlo Method. In Radiation Effects in Solids; Sickafus, K.E., Kotomin, E.A., Uberuaga, B.P., Eds.; Springer: Dordrecht, The Netherlands, 2007; pp. 1–23. [Google Scholar]
- Wehrli, B. Monte Carlo Simulations of Surface Morphologies during Mineral Dissolution. J. Colloid Interface Sci. 1989, 132, 230–242. [Google Scholar] [CrossRef]
- Kohli, C.S.; Ives, M.B. Computer Simulation of Crystal Dissolution Morphology. J. Cryst. Growth 1972, 16, 123–130. [Google Scholar] [CrossRef]
- Nangia, S.; Garrison, B.J. Advanced Monte Carlo Approach to Study Evolution of Quartz Surface during the Dissolution Process. J. Am. Chem. Soc. 2009, 131, 9538–9546. [Google Scholar] [CrossRef]
- Kurganskaya, I.; Luttge, A. A Comprehensive Stochastic Model of Phyllosilicate Dissolution: Structure and Kinematics of Etch Pits Formed on Muscovite Basal Face. Geochim. Cosmochim. Acta 2013, 120, 545–560. [Google Scholar] [CrossRef]
- Liang, Y.; Baer, D.R.; McCoy, J.M.; Amonette, J.E.; Lafemina, J.P. Dissolution Kinetics at the Calcite-Water Interface. Geochim. Cosmochim. Acta 1996, 60, 4883–4887. [Google Scholar] [CrossRef]
- McCoy, J.M.; LaFemina, J.P. Kinetic Monte Carlo Investigation of Pit Formation at the CaCO3 (10-14) Surface-Water Interface. Surf. Sci. 1997, 373, 288–299. [Google Scholar] [CrossRef]
- Kurganskaya, I.; Luttge, A. Kinetic Monte Carlo Approach to Study Carbonate Dissolution. J. Phys. Chem. C 2016, 120, 6482–6492. [Google Scholar] [CrossRef]
- Liang, Y.; Baer, D.R.; McCoy, J.M.; Lafemina, J.P. Interplay between Step Velocity and Morphology during the Dissolution of CaCO3 Surface. J. Vac. Sci. Technol. A Vac. Surf. Film. 1996, 14, 1368–1375. [Google Scholar] [CrossRef]
- Angus, J.C.; Ponton, J.W. Modelling of Kink Nucleation and Propagation along Steps of Finite Length. Surf. Sci. 1976, 61, 451–467. [Google Scholar] [CrossRef]
- Vekilov, P.G. What Determines the Rate of Growth of Crystals from Solution? Cryst. Growth Des. 2007, 7, 2796–2810. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Jang, Y.H.; Chang, X.Y.; Blanco, M.; Hwang, S.; Tang, Y.; Shuler, P.; Goddard, W.A. The MSXX Force Field for the Barium Sulfate−Water Interface. J. Phys. Chem. B 2002, 106, 9951–9966. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Set I | Set II | Set III | Set IV 1 |
|---|---|---|---|---|
| 1 | 1012 | 1012 | 1012 | 1012 |
| 30 | 30 | 30 | 30 | |
| 7 | 7 | 7 | 7.7 | |
| 7 | 7 | 7 | 7.7 | |
| 0 | 1 | 1 | 1.2 | |
| 0 | 0 | −5 | −5 | |
| 0 | 0 | −5 | −7 | |
| 0 | 0 | −3 | −6 | |
| 0 | 0 | −5 | −7 |
| [010] Slow | [010] Fast | [120] | |
|---|---|---|---|
| Pure water, experiments [14] | 0.09 ± 0.01 | - | 0.18 0.01 |
| Pure water, experiments [17] | 0.3 ± 0.1 | 0.8 ± 0.2 | - |
| Pure water, simulations 1 | 0.16 ± 0.04 2 | 0.29 ± 0.06 | 0.30 ± 0.05 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurganskaya, I.; Trofimov, N.; Luttge, A. A Kinetic Monte Carlo Approach to Model Barite Dissolution: The Role of Reactive Site Geometry. Minerals 2022, 12, 639. https://doi.org/10.3390/min12050639
Kurganskaya I, Trofimov N, Luttge A. A Kinetic Monte Carlo Approach to Model Barite Dissolution: The Role of Reactive Site Geometry. Minerals. 2022; 12(5):639. https://doi.org/10.3390/min12050639
Chicago/Turabian StyleKurganskaya, Inna, Nikolay Trofimov, and Andreas Luttge. 2022. "A Kinetic Monte Carlo Approach to Model Barite Dissolution: The Role of Reactive Site Geometry" Minerals 12, no. 5: 639. https://doi.org/10.3390/min12050639
APA StyleKurganskaya, I., Trofimov, N., & Luttge, A. (2022). A Kinetic Monte Carlo Approach to Model Barite Dissolution: The Role of Reactive Site Geometry. Minerals, 12(5), 639. https://doi.org/10.3390/min12050639