
The Uptake of Actinides by Hardened Cement Paste in High-Salinity Pore Water

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Stock Solutions
2.3. Batch Sorption Experiments
2.4. Analytical Methods
2.5. CE-ICP-MS Measurements
2.6. XAFS Measurements
2.7. Speciation Calculations
3. Results and Discussion
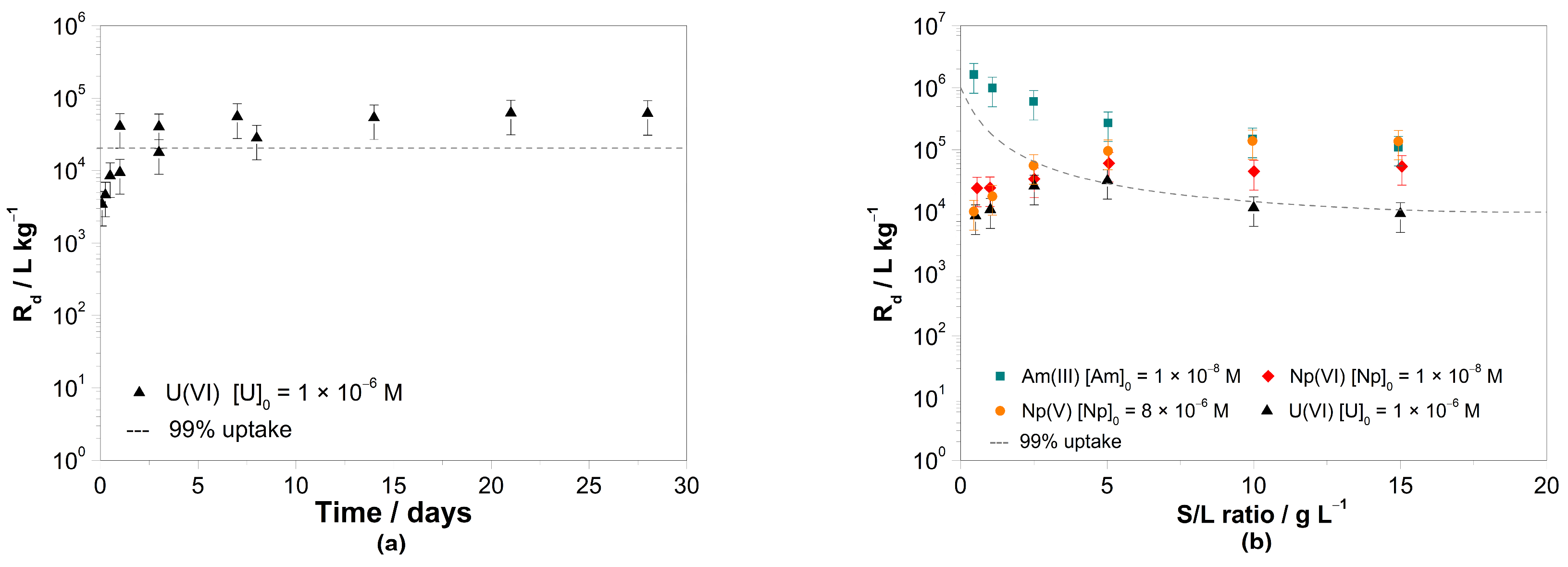
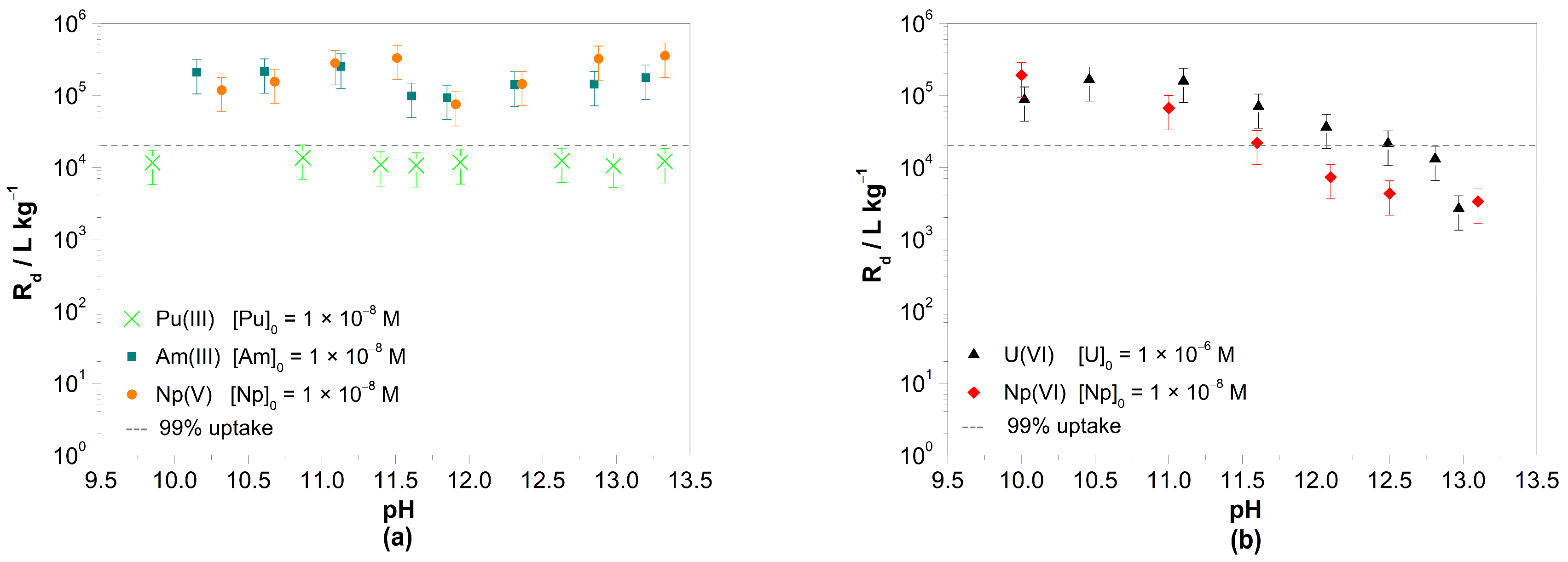
3.1. Batch Sorption Experiments
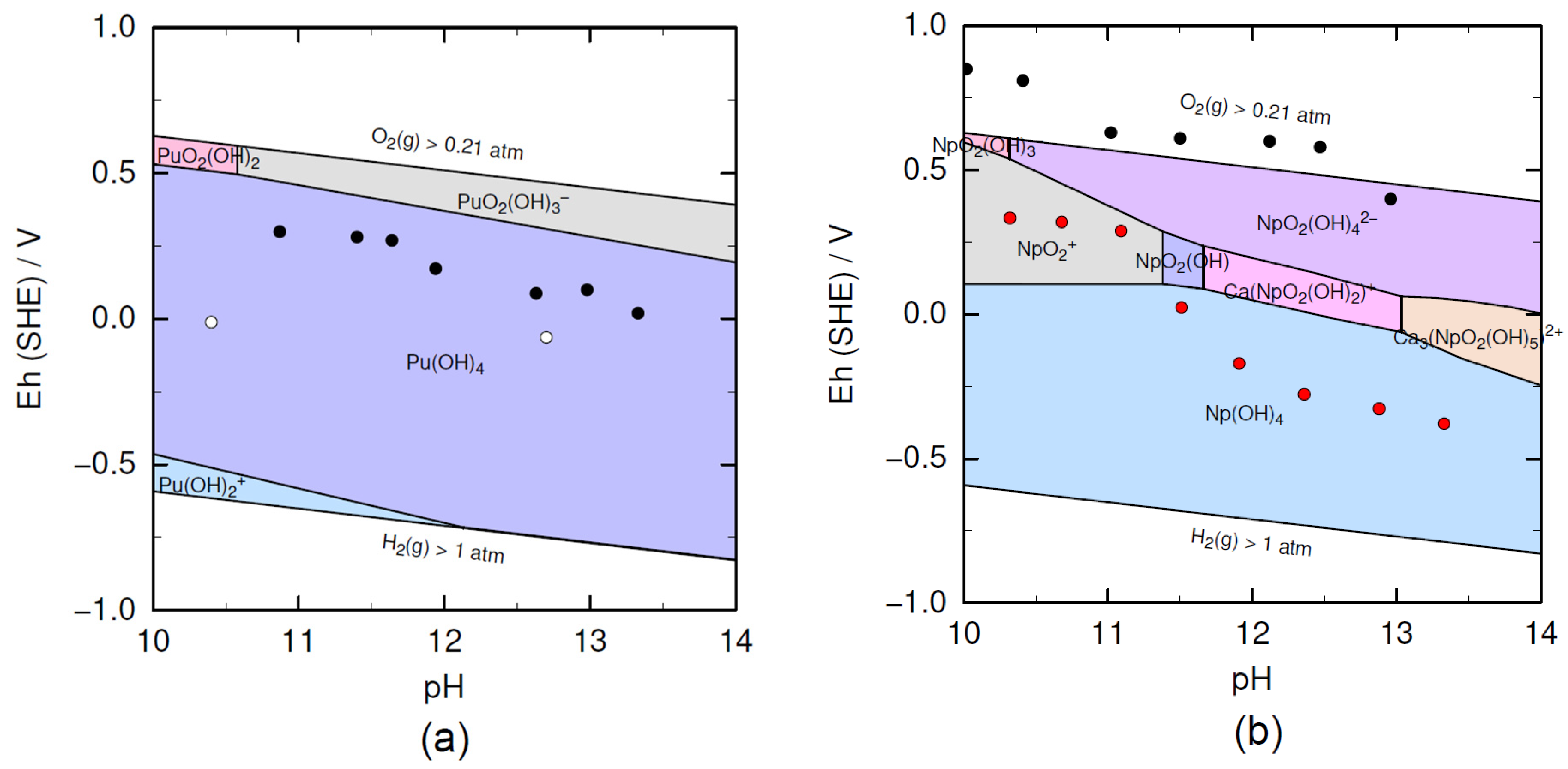
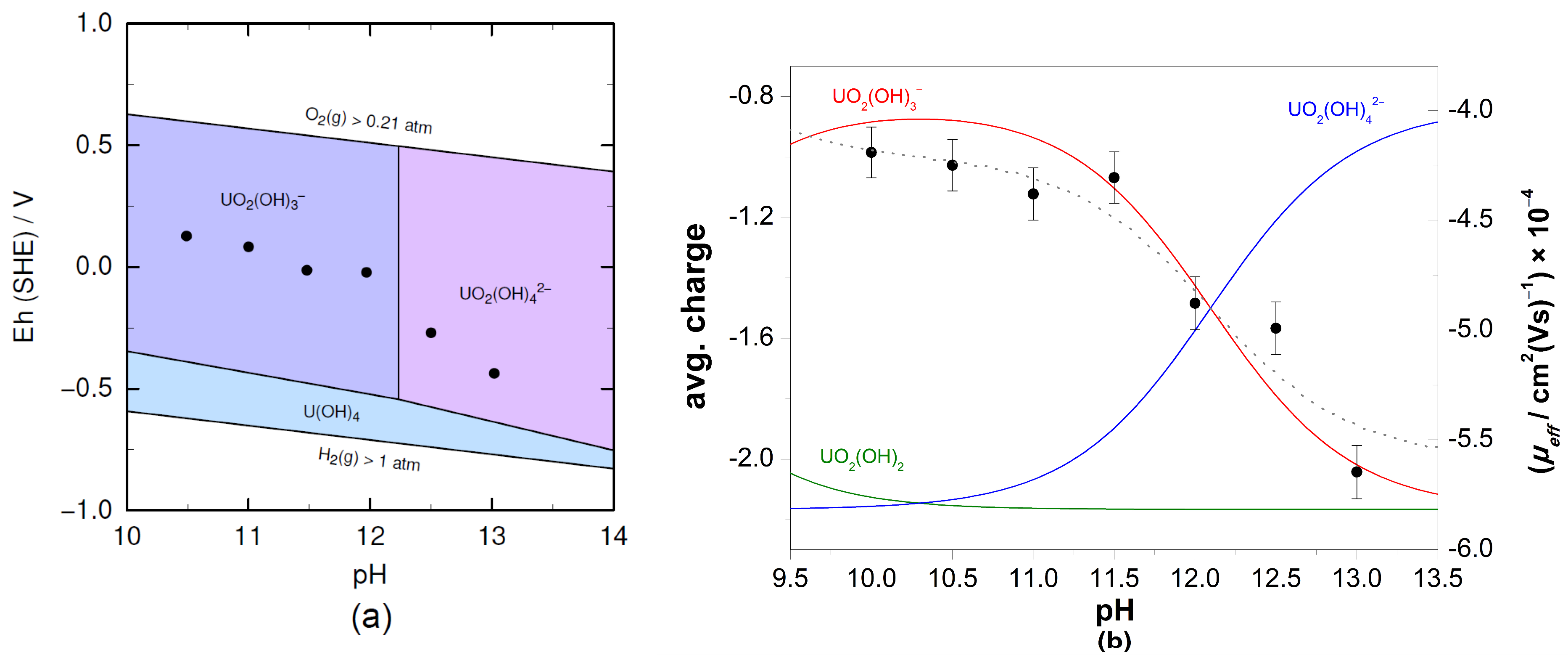
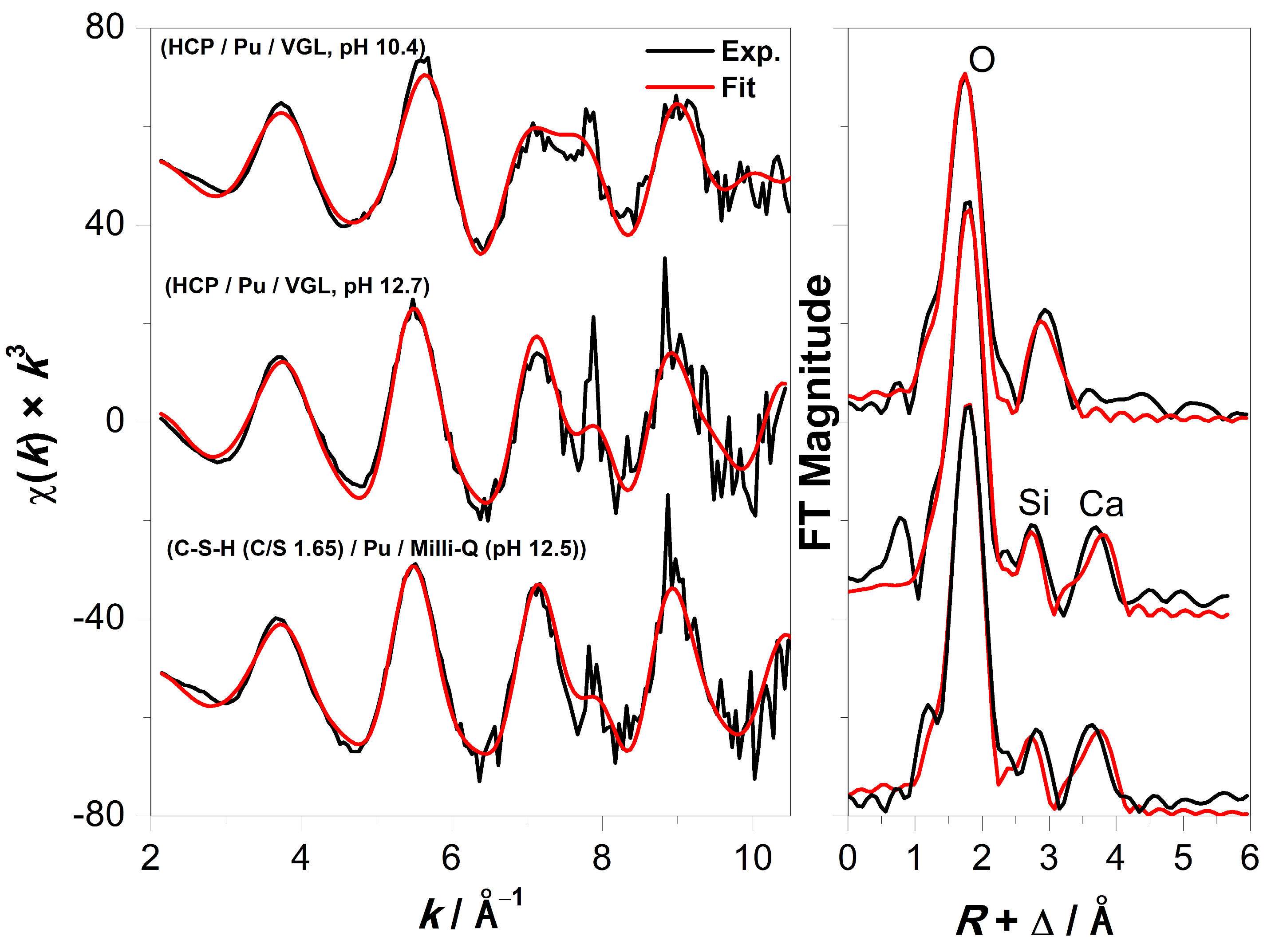
3.2. XAFS Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lommerzheim, A.; Jobmann, M. Projekt ANSICHT-Endlagerkonzept sowie Verfüll- und Verschlusskonzept für das Standortmodell NORD; TEC-14-2015-TB; DBE Technology GmbH: Peine, Germany, 2015. [Google Scholar]
- Jobmann, M.; Bebiolka, A.; Burlaka, V.; Herold, P.; Jahn, S.; Lommerzheim, A.; Maßmann, J.; Meleshyn, A.; Mrugalla, S.; Reinhold, K.; et al. Safety assessment methodology for a German high-level waste repository in clay formations. J. Rock Mech. Geotech. Eng. 2017, 9, 856–876. [Google Scholar] [CrossRef]
- Brewitz, W. Eignungsprüfung der Schachtanlage Konrad für die Endlagerung radioaktiver Abfälle: Abschlussbericht (GSF-T 136); GSF: Neuherberg, Germany, 1982. [Google Scholar]
- Pointeau, I.; Piriou, B.; Fedoroff, M.; Barthes, M.G.; Marmier, N.; Fromage, F. Sorption mechanisms of Eu3+ on CSH phases of hydrated cements. J. Colloid Interface Sci. 2001, 236, 252–259. [Google Scholar] [CrossRef]
- Almendros-Ginestà, O.; Missana, T.; García-Gutiérrez, M.; Alonso, U. Analysis of radionuclide retention by the cement hydrate phase portlandite: A novel modelling approach. Prog. Nucl. Energy 2023, 159, 104636. [Google Scholar] [CrossRef]
- Wieland, E. Sorption Data Base for the Cementious Near Field of L/ILW and ILW Repositories for Provisional Safety Analyses for SGT-E2; Technical Report 14-08; NAGRA: Wettingen, Switzerland, 2014. [Google Scholar]
- Taylor, H.F.W. Cement Chemistry; Springer Inernational Publishing: London, UK, 1997. [Google Scholar]
- Schlegel, M.L.; Pointeau, I.; Coreau, N.; Reiller, P. Mechanism of europium retention by calcium silicate hydrates: An EXAFS study. Environ. Sci. Technol. 2004, 38, 4423–4431. [Google Scholar] [CrossRef]
- Stumpf, T.; Tits, J.; Walther, C.; Wieland, E.; Fanghänel, T. Uptake of trivalent actinides (curium(III)) by hardened cement paste: A time-resolved laser fluorescence spectroscopy study. J. Colloid Interface Sci. 2004, 276, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Tits, J.; Stumpf, T.; Rabung, T.; Wieland, E.; Fanghänel, T. Uptake of Cm(III) and Eu(III) by calcium silicate hydrates: A solution chemistry and time-resolved laser fluorescence spectroscopy study. Environ. Sci. Technol. 2003, 37, 3568–3573. [Google Scholar] [CrossRef]
- Gaona, X.; Dähn, R.; Tits, J.; Scheinost, A.C.; Wieland, E. Uptake of Np(IV) by C-S-H phases and cement paste: An EXAFS study. Environ. Sci. Technol. 2011, 45, 8765–8771. [Google Scholar] [CrossRef]
- Macé, N.; Wieland, E.; Dähn, R.; Tits, J.; Scheinost, A.C. EXAFS investigation on U(VI) immobilization in hardened cement paste: Influence of experimental conditions on speciation. Radiochimi. Acta 2013, 101, 379–389. [Google Scholar] [CrossRef]
- Häußler, V.; Amayri, S.; Beck, A.; Platte, T.; Stern, T.A.; Vitova, T.; Reich, T. Uptake of actinides by calcium silicate hydrate (C-S-H) phases. Appl. Geochem. 2018, 98, 426–434. [Google Scholar] [CrossRef]
- Ochs, M.; Mallants, D.; Wang, L. Radionuclide and Metal Sorption on Cement and Concrete; Gheorghe, A.V., Ed.; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Guo, X.; Gin, S.; Frankel, G.S. Review of corrosion interactions between different materials relevant to disposal of high-level nuclear waste. npj Mat. Degrad. 2020, 4, 34. [Google Scholar] [CrossRef]
- Duquette, D.J.; Latanision, R.M.; Di Bella, C.A.; Kirstein, B.E. Corrosion issues related to disposal of high-level nuclear waste in the Yucca Mountain repository–Peer reviewers’ perspective. Corrosion 2009, 65, 272–280. [Google Scholar] [CrossRef]
- Tits, J.; Wieland, E. Actinide Sorption by Cementitious Materials; Bericht Nr. 18-02; PSI: Villigen, Switzerland, 2018. [Google Scholar]
- Tits, J.; Gaona, X.; Laube, A.; Wieland, E. Influence of the redox state on the neptunium sorption under alkaline conditions: Batch sorption studies on titanium dioxide and calcium silicate hydrates. Radiochim. Acta 2014, 102, 385–400. [Google Scholar] [CrossRef]
- Fanghänel, T.; Neck, V. Aquatic chemistry and solubility phenomena of actinide oxides/hydroxides. Pure Appl. Chem. 2002, 74, 1895–1907. [Google Scholar] [CrossRef]
- Stockmann, M.; Fritsch, K.; Bok, F.; Fernandes, M.M.; Baeyens, B.; Steudtner, R.; Müller, K.; Nebelung, C.; Brendler, V.; Stumpf, T. New insights into U(VI) sorption onto montmorillonite from batch sorption and spectroscopic studies at increased ionic strength. Sci. Total Environ. 2022, 806, 150653. [Google Scholar] [CrossRef]
- Meleshyn, A. Mechanisms of Transformation of Bentonite Barriers—Testing a New Experimental Concept; Gesellschaft für Anlagen- und Reaktorsicherheit: Cologne, Germany, 2015; GRS-A-3844. [Google Scholar]
- DIN EN 196-3:2005+A1:2008; Prüfverfahren für Zement in Teil 3: Bestimmung der Erstarrungszeiten und der Raumbeständigkeit. Deutsche Fassung EN: Berlin, Germany, 2009.
- Amayri, S.; Fröhlich, D.R.; Kaplan, U.; Trautmann, N.; Reich, T. Distribution coefficients for the sorption of Th, U, Np, Pu, and Am on Opalinus Clay. Radiochim. Acta 2016, 104, 33–40. [Google Scholar] [CrossRef]
- Amayri, S.; Jermolajev, A.; Reich, T. Neptunium(V) sorption on kaolinite. Radiochim. Acta 2011, 99, 349–357. [Google Scholar] [CrossRef]
- Gaona, X.; Wieland, E.; Tits, J.; Scheinost, A.C.; Dähn, R. Np(V/VI) redox chemistry in cementitious systems: XAFS investigations on the speciation under anoxic and oxidizing conditions. Appl. Geochem. 2013, 28, 109–118. [Google Scholar] [CrossRef]
- Tits, J.; Fujita, T.; Tsukamoto, M.; Wieland, E. Uranium(VI) uptake by synthetic calcium silicate hydrates. MRS Online Proc. Libr. 2008, 1107, 467. [Google Scholar] [CrossRef]
- Fanghänel, T.; Neck, V.; Kim, J.I. The ion product of H2O, dissociation constants of H2CO3 and Pitzer parameters in the system Na+/H+/OH−/HCO3−/CO32−/ClO4−/H2O at 25 °C. J. Solut. Chem 1996, 25, 327–343. [Google Scholar] [CrossRef]
- Willberger, C.; Amayri, S.; Häußler, V.; Scholze, R.; Reich, T. Investigation of the electrophoretic mobility of the actinides Th, U, Np, Pu, and Am in different oxidation states. Anal. Chem. 2019, 91, 11537–11543. [Google Scholar] [CrossRef]
- Scheinost, A.C.; Claussner, J.; Exner, J.; Feig, M.; Findeisen, S.; Hennig, C.; Kvashnina, K.O.; Naudet, D.; Prieur, D.; Rossberg, A. ROBL-II at ESRF: A synchrotron toolbox for actinide research. J. Synchrotron Radiat. 2021, 28, 333–349. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef]
- Schmeide, K.; Reich, T.; Sachs, S.; Bernhard, G. Plutonium(III) complexation by humic substances studied by X-ray absorption fine structure spectroscopy. Inorg. Chim. Acta 2006, 359, 237–242. [Google Scholar] [CrossRef]
- Martin, P.; Grandjean, S.; Valot, C.; Carlot, G.; Ripert, M.; Blanc, P.; Hennig, C. XAS study of (U1−yPuy)O2 solid solutions. J. Alloys Compd. 2007, 444, 410–414. [Google Scholar] [CrossRef]
- George, G.N.; Pickering, I.J. EXAFSPAK—A Suite of Computer Programs for Analysis of X-ray Absortion Spectra; Stanford Synchrotron Radiation Lightsource: Menlo Park, CA, USA, 2000. [Google Scholar]
- Rehr, J.J.; Kas, J.J.; Vila, F.D.; Prange, M.P.; Jorissen, K. Parameter-free calculations of X-ray spectra with FEFF9. Phys. Chem. Chem. Phys. 2010, 12, 5503–5513. [Google Scholar] [CrossRef] [PubMed]
- Chiorescu, I.; Kremleva, A.; Krüger, S. On the sorption mode of U(IV) at calcium silicate hydrate: A comparison of adsorption, absorption in the interlayer, and incorporation by means of density functional calculations. Minerals 2022, 12, 1541. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. PHREEQC (Version 3.3.5)—A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations. Available online: http://wwwbrr.cr.usgs.gov/projects/GWC_coupled/phreeqc/index.html (accessed on 12 October 2023).
- Giffaut, E.; Grivé, M.; Blanc, P.; Vieillard, P.; Colàs, E.; Gailhanou, H.; Gaboreau, S.; Marty, N.; Madé, B.; Duro, L. Andra thermodynamic database for performance assessment: ThermoChimie. Appl. Geochem. 2014, 49, 225–236. [Google Scholar] [CrossRef]
- Pointeau, I.; Landesman, C.; Giffaut, E.; Reiller, P. Reproducibility of the uptake of U(VI) onto degraded cement pastes and calcium silicate hydrate phases. Radiochim. Acta 2004, 92, 645–650. [Google Scholar] [CrossRef]
- Pointeau, I.; Landesman, C.; Coreau, N.; Moisan, C.; Reiller, P. Am(III), Zr(IV), Pu(IV), Nb(V), U(VI) et Tc(IV) par les Matériaux Cimentaires Dégradés; CEA Report; Commissariat à l’énergie atomique: Gif sur Yvette, France, 2004; pp. 3–37. [Google Scholar]
- Wieland, E.; Lothenbach, B.; Glaus, M.A.; Thoenen, T.; Schwyn, B. Influence of superplasticizers on the long-term properties of cement pastes and possible impact on radionuclide uptake in a cement-based repository for radioactive waste. Appl. Geochem. 2014, 49, 126–142. [Google Scholar] [CrossRef]
- Conradson, S.D.; Abney, K.D.; Begg, B.D.; Brady, E.D.; Clark, D.L.; Den Auwer, C.; Ding, M.; Dorhout, P.K.; Espinosa-Faller, F.J.; Gordon, P.L. Higher order speciation effects on plutonium L3 X-ray absorption near edge spectra. Inorg. Chem. 2004, 43, 116–131. [Google Scholar] [CrossRef]
- Dettmann, S.; Huittinen, N.M.; Jahn, N.; Kretzschmar, J.; Kumke, M.U.; Kutyma, T.; Lohmann, J.; Reich, T.; Schmeide, K.; Shams Aldin Azzam, S.; et al. Influence of gluconate on the retention of Eu(III), Am(III), Th(IV), Pu(IV), and U(VI) by CSH (C/S = 0.8). Front. Nucl. Eng. 2023, 2, 4. [Google Scholar] [CrossRef]
- Reich, T.; Reich, T.Y.; Amayri, S.; Drebert, J.; Banik, N.L.; Buda, R.A.; Kratz, J.V.; Trautmann, N. Application of XAFS spectroscopy to actinide environmental science. AIP Conf. Proc. 2007, 882, 179–183. [Google Scholar] [CrossRef]
- Wedler, G.; Freund, H.-J. Lehrbuch der Physikalischen Chemie; Wiley–VCH: Weinheim, Germany, 2012. [Google Scholar]
- Odler, I. The BET-specific surface area of hydrated Portland cement and related materials. Cem. Concr. Res. 2003, 33, 2049–2056. [Google Scholar] [CrossRef]
- Hong, S.-Y.; Glasser, F.P. Alkali binding in cement pastes: Part I. The C-S-H phase. Cem. Concr. Res. 1999, 29, 1893–1903. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Energy/eV |
|---|---|
| HCP/Pu/VGL (pH 10.4) | 18,062.0 |
| HCP/Pu/VGL (pH 12.7) | 18,062.1 |
| C-S-H (C/S 1.65)/Pu/Milli-Q (pH 12.5) | 18,062.0 |
| Pu(III)(aq) | 18,060.0 |
| Pu(IV)(aq) | 18,063.2 |
| Sample | Shell | N | R/Å | σ2/Å2 |
|---|---|---|---|---|
| HCP/Pu/VGL (pH 10.4) | Pu-O | 6.0 (± 0.2) | 2.24 (± 0.01) | 0.011 (±0.001) |
| (norm. error = 0.2, ΔE0 = 0.46 eV) | Pu-Ca | 2.1 (± 0.4) | 3.41 (± 0.01) | 0.008 (±0.002) |
| HCP/Pu/VGL (pH 12.7) | Pu-O | 7.4 (± 0.4) | 2.27 (± 0.01) | 0.011 (±0.001) |
| (norm. error = 0.4, ΔE0 = 2.11 eV) | Pu-Si | 1.7 (± 0.5) | 3.13 (± 0.01) | 0.008 (±0.003) |
| Pu-Ca | 4.2 (± 1.3) | 4.19 (± 0.01) | 0.009 (±0.003) | |
| C-S-H (C/S 1.65)/Pu/Milli-Q (pH = 12.5) | Pu-O | 6.0 (± 0.3) | 2.27 (± 0.01) | 0.008 (±0.001) |
| (norm. error = 0.3, ΔE0 = 1.31 eV) | Pu-Si | 1.2 (± 0.4) | 3.13 (± 0.01) | 0.006 (±0.002) |
| Pu-Ca | 5.0 (± 1.3) | 4.17 (± 0.01) | 0.011 (±0.002) | |
| C-S-H (C/S 0.8)/Pu/Milli-Q (pH = 10) | Pu-O | 6.8 ± 0.3 | 2.25 (± 0.02) | 0.0119 (± 0.0004) |
| (norm. error = 0.9, ΔE0 = 0.26 eV) | Pu-Si1 | 2.0 ± 0.3 | 3.15 (± 0.02) | 0.0119 * |
| Pu-Si2 | 4.6 ± 0.5 | 3.54 (± 0.02) | 0.0119 * | |
| Pu-Ca | 6.3 ± 1.0 | 4.12 (± 0.02) | 0.0179 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stietz, J.; Amayri, S.; Häußler, V.; Scholze, R.; Reich, T. The Uptake of Actinides by Hardened Cement Paste in High-Salinity Pore Water. Minerals 2023, 13, 1380. https://doi.org/10.3390/min13111380
Stietz J, Amayri S, Häußler V, Scholze R, Reich T. The Uptake of Actinides by Hardened Cement Paste in High-Salinity Pore Water. Minerals. 2023; 13(11):1380. https://doi.org/10.3390/min13111380
Chicago/Turabian StyleStietz, Janina, Samer Amayri, Verena Häußler, Raphael Scholze, and Tobias Reich. 2023. "The Uptake of Actinides by Hardened Cement Paste in High-Salinity Pore Water" Minerals 13, no. 11: 1380. https://doi.org/10.3390/min13111380
APA StyleStietz, J., Amayri, S., Häußler, V., Scholze, R., & Reich, T. (2023). The Uptake of Actinides by Hardened Cement Paste in High-Salinity Pore Water. Minerals, 13(11), 1380. https://doi.org/10.3390/min13111380