1. Introduction
Synthetic analogues of the silicate framework minerals leucite KAlSi
2O
6 [
1] and pollucite CsAlSi
2O
6 [
2] can be prepared with the general formulae
ABSi
2O
6 and
A2CSi
5O
12.
A is an alkali metal cation (K, Rb, Cs),
B is a trivalent cation (Al, B, Fe
3+, Ga) and
C is a divalent cation (Be, Mg, Mn, Fe
2+, Co, Ni, Cu, Zn, Cd). These structures have tetrahedrally coordinated silicate frameworks with
B or
C cations partially substituting for Si on the tetrahedrally coordinated silicon sites (T-sites).
A cations sit in the extra framework channels; these
A cations can be removed by ion exchange which makes them of technological interest as possible storage media for radioactive Cs from nuclear waste [
3].
Leucite analogues with high symmetry structures such as
I4
1/
a tetragonal KGaSi
2O
6 [
4] and
Ia-3
d cubic Rb
2ZnSi
5O
12 [
5] have
B and
C cations disordered over the T-sites. However, lower symmetry leucite structures are known where
C cations are ordered onto separate T-sites. The
P2
1/
c monoclinic K
2MgSi
5O
12 [
6] structure has twelve crystallographically distinct and fully ordered T-sites, ten of these are fully occupied by Si and two are fully occupied by Mg. Three more K
2CSi
5O
12 (
C = Fe
2+, Co, Zn) structures [
7] are known which are isostructural with
P2
1/
c monoclinic K
2MgSi
5O
12 and have fully ordered T-sites. The
Pbca orthorhombic structure of Cs
2CdSi
5O
12 [
8] has six crystallographically distinct and fully ordered T-sites, five of these are fully occupied by Si and one is fully occupied by Cd. Five more structures with the general formula Cs
2CSi
5O
12 (
C = Mg, Mn, Co, Cu, Zn) [
9,
10,
11], four structures with the general formula Rb
2CSi
5O
12 (
C = Mg, Mn, Ni, Cd) [
9,
10,
12] and three structures with the general formula RbCs
CSi
5O
12 (
C = Mg, Ni, Cd) [
13] are all isostructural with the fully T-site cation ordered structure of Cs
2CdSi
5O
12. However, Nuclear Magnetic Resonance spectroscopy [
14] and high-resolution synchrotron X-ray powder diffraction [
10] studies on Cs
2ZnSi
5O
12 described a
Pbca structure where Zn is partially disordered over two of the six T-sites.
High temperature neutron and X-ray powder diffraction studies on KAlSi
2O
6, RbAlSi
2O
6 and KFe
3+Si
2O
6 [
15] and KGaSi
2O
6 [
4] showed first-order phase transitions from
I4
1/
a to
Ia-3
d (isostructural with pollucite CsAlSi
2O
6). High temperature X-ray powder diffraction studies on K
2MgSi
5O
12 [
16] and K
2ZnSi
5O
12 [
17] showed first-order phase transitions from
P2
1/
c to
Pbca (isostructural with Cs
2CdSi
5O
12.)
A high temperature study from 295 to 1173 K [
18] has also been done on three Cs
2CSi
5O
12 (
C = Cu, Cd, Zn) leucite analogues using lower resolution synchrotron X-ray powder diffraction. For Cs
2ZnSi
5O
12, the ambient temperature crystal structure shows (unlike for the high-resolution synchrotron X-ray powder diffraction study) that the
Pbca structure is also isostructural with Cs
2CdSi
5O
12 with complete T-site cation ordering. However, the sample with
C = Zn shows evidence for a transition to a previously unknown
cubic structure, with some T-site cation disorder, at 566 K, on heating. This transition is reversible on cooling to 633 K.
Rb
2CSi
5O
12 leucite structures are known where
C = Cd, Ni, Mg, Mn and Zn [
5,
7,
9,
10,
12] as is the structure of Cs
2CoSi
5O
12 [
9]. A cubic lattice parameter of 13.4(1)Å for Rb
2CoSi
5O
12 has been reported [
19] but there is no published crystal structure for this leucite analogue. Therefore, a sample of Rb
2CoSi
5O
12 leucite analogue has been synthesized. An ambient temperature X-ray powder diffraction study on this analogue has been undertaken to determine the crystal structure for Rb
2CoSi
5O
12. A high temperature X-ray powder diffraction study has also been done to look for any phase transitions.
2. Materials and Methods
A sample of Rb
2CoSi
5O
12 was synthesized from a stoichiometric mixture of Rb
2CO
3 (Alfa Aesar, Heysham, United Kingdom, 99.5%); CoO (Sigma-Aldrich, St. Louis, MO, USA, 99.98%) and SiO
2 (Better Equipped purified white silica sand, Nantwich, United Kingdom). The mixture was then heated overnight at 873 K to decompose the carbonates and melted in a platinum crucible at 1673 K for 1.5 h before quenching to form a glass. The Rb
2CoSi
5O
l2 glass was then dry crystallized at ambient pressure and 1393 K for 5 days. These were the same sample preparation conditions as were used to synthesise Cs
2CoSi
5O
l2 leucite analogue [
9].
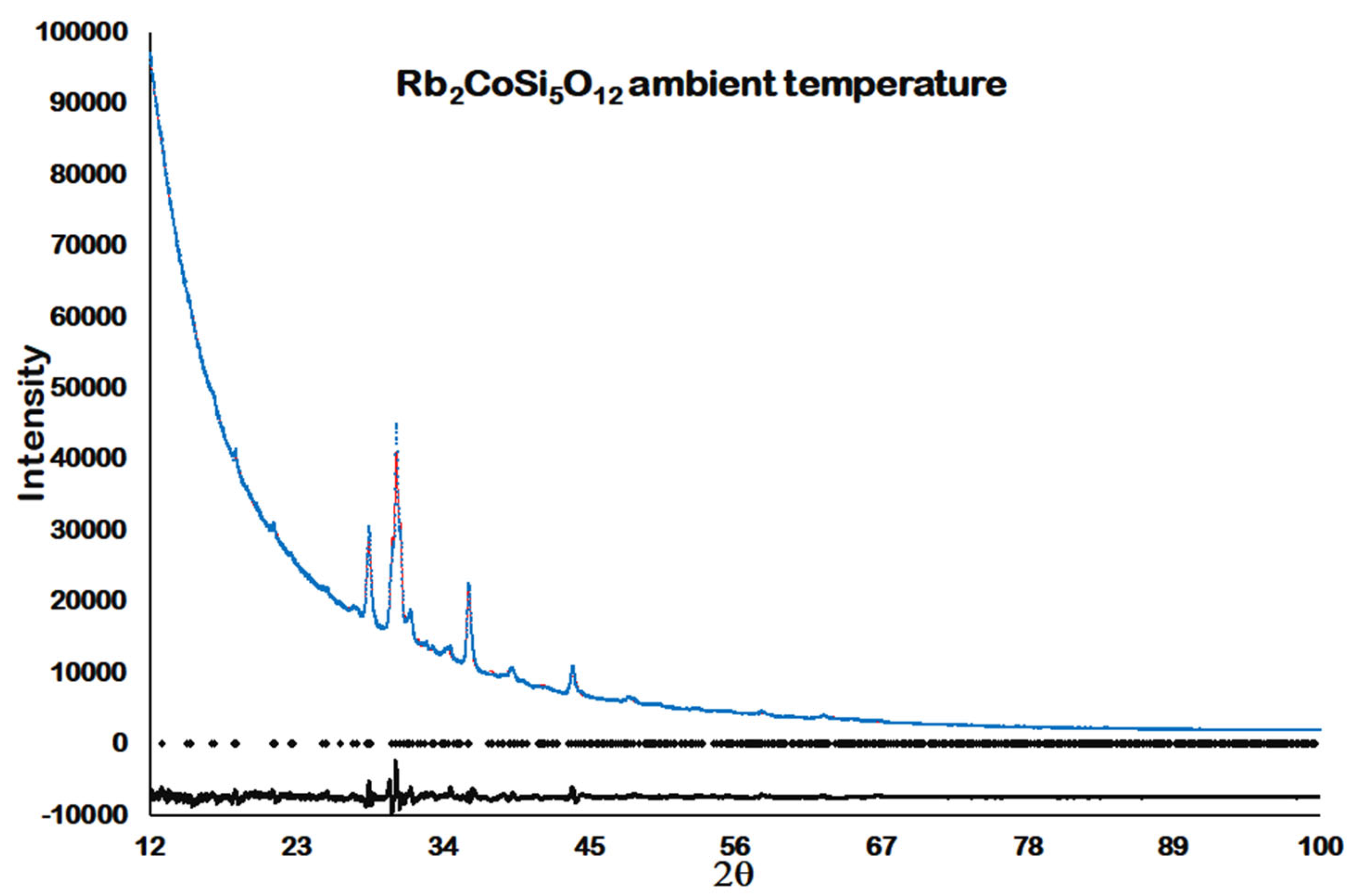
This sample was then mounted on a low-background silicon wafer with a drop of acetone. Ambient temperature X-ray powder diffraction was then collected on this sample using a PANalytical Empyrean X-ray powder diffractometer (PANalytical, Almelo, Netherlands). Data were collected using CoKa X-rays over the range from 12 to 100 °2θ, the scan time was 19 h.
This Rb2CoSi5Ol2 sample was then loaded into a platinum flat plate sample holder which was loaded into an Anton HTK1200N high temperature stage mounted on a PANalytical X’Pert MPD X-ray powder diffractometer (PANalytical, Almelo, Netherlands). Data were collected using CuKa X-rays over the range from 10 to 80 °2θ. Scans were done at ambient temperature (scan time 30 min) and then in 50 K increments (scan time 2.5 h) from 323 to 1373 K. A second series of high temperature scans were done Rb2CoSi5Ol2 on using the HTK1200N over the range from 10 to 80 °2θ. Scans were done in 10 K increments from 423 to 523 K, the scan time at each temperature was 8 h. A final scan on cooling to ambient temperature was also measured over the range from 10 to 80 °2θ, the scan time was 30 min.
High temperature X-ray powder diffraction data were also collected on a sample of MgO (99.99%, Acros Organics, New Jersey, USA) to calibrate the temperature of the HTK1200N. This was loaded into a platinum flat plate sample holder which was loaded onto the same Anton HTK1200N high temperature stage. Data were collected over the range from 33 to 111 °2θ, the scan time at each temperature was 10 min. Scans were done at ambient temperature and then in 50 K increments from 323 to 1473 K.
Thermogravimetric Analysis (TGA) and Differential Scanning Calorimetry (DSC) were collected on a sample of Rb2CoSi5Ol2 from ambient temperature to 1673 K. Data were collected in a TA Instruments SDT 650 simultaneous TGA/DSC (New Castle, DE, USA), under a flow of compressed air. The instrument was suitably calibrated, an empty alumina crucible was used for the sample holder and reference sample, with no background corrections.
4. Discussion
All temperatures referred to in this section are now MgO calibrated temperatures.
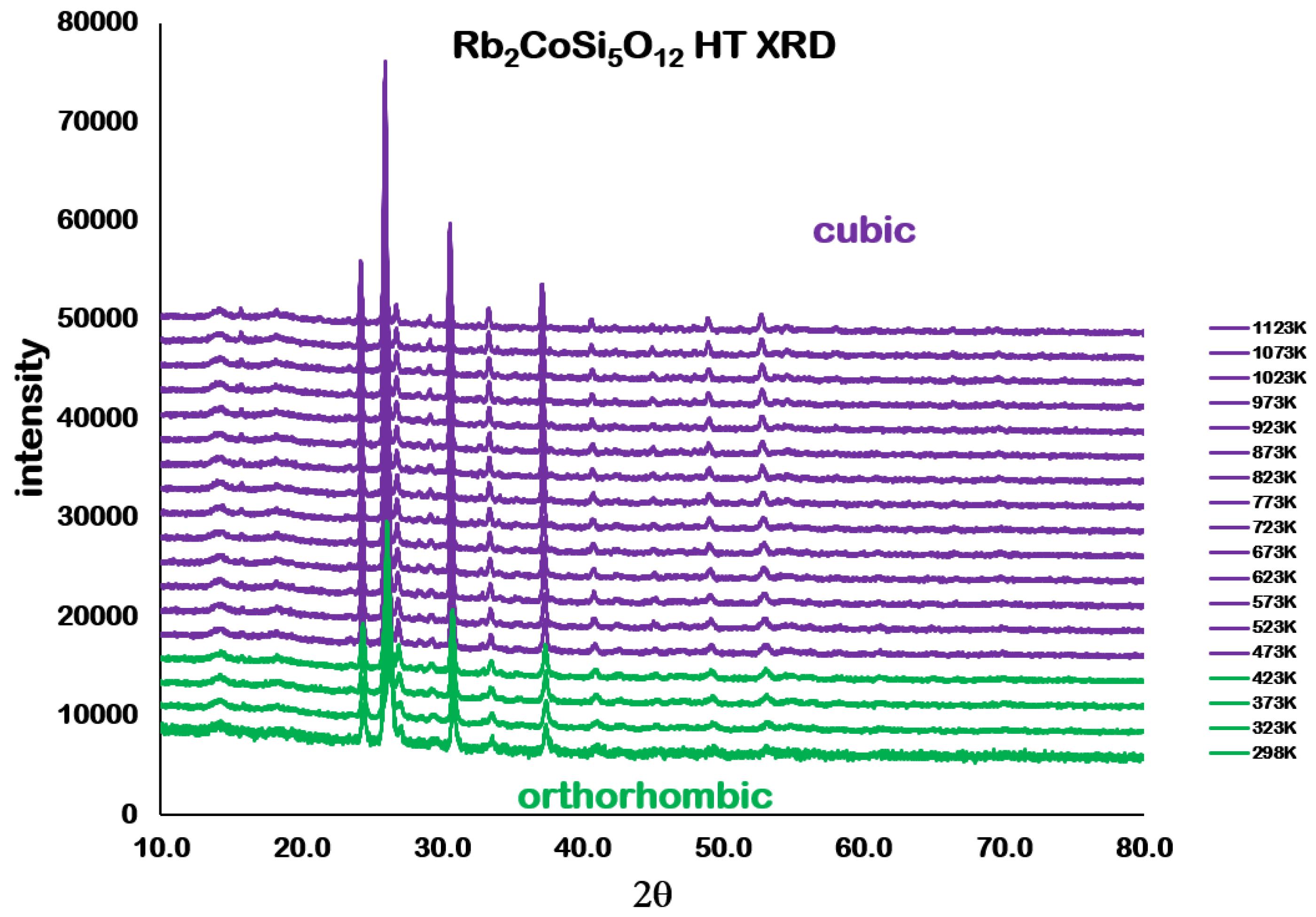
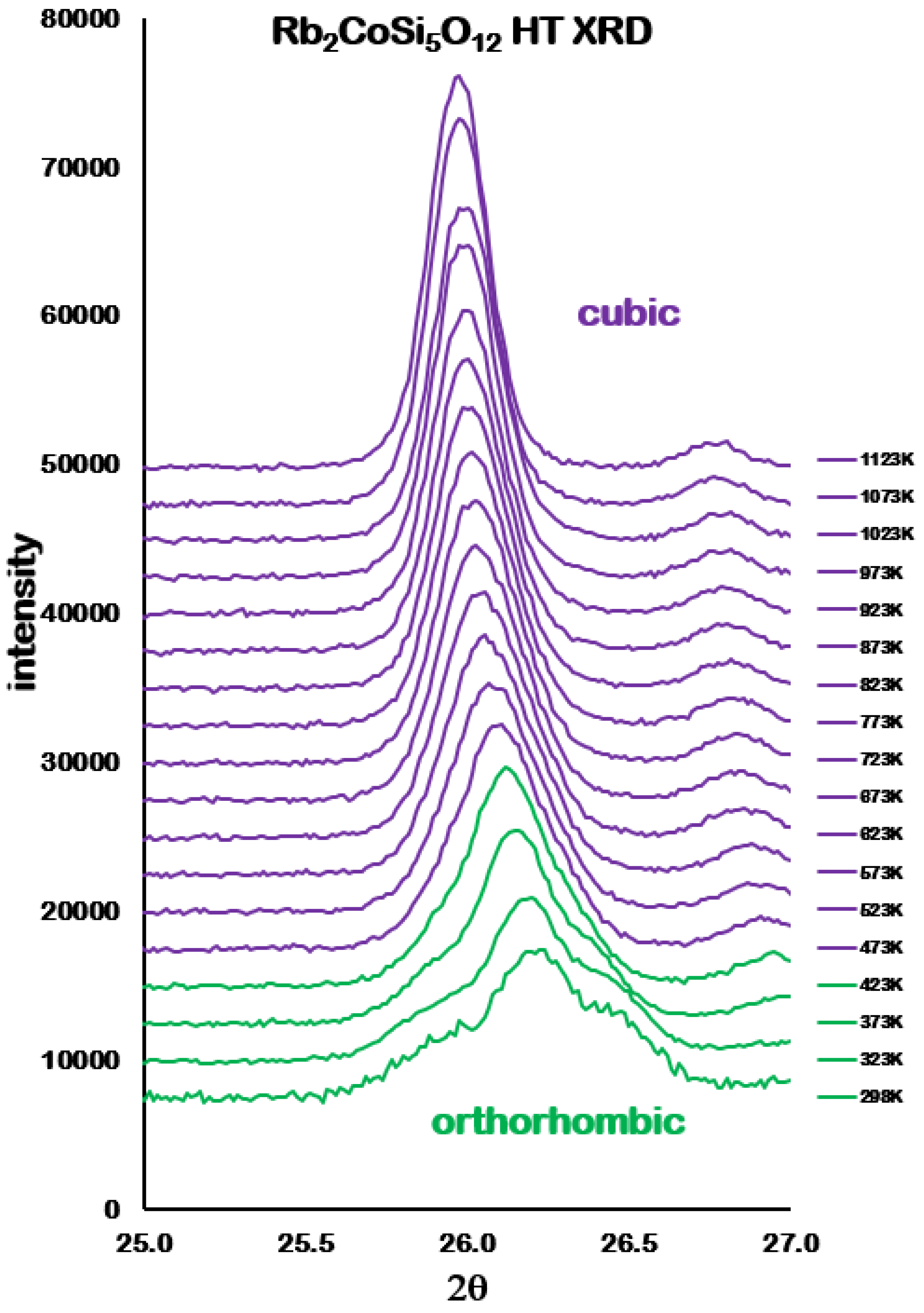
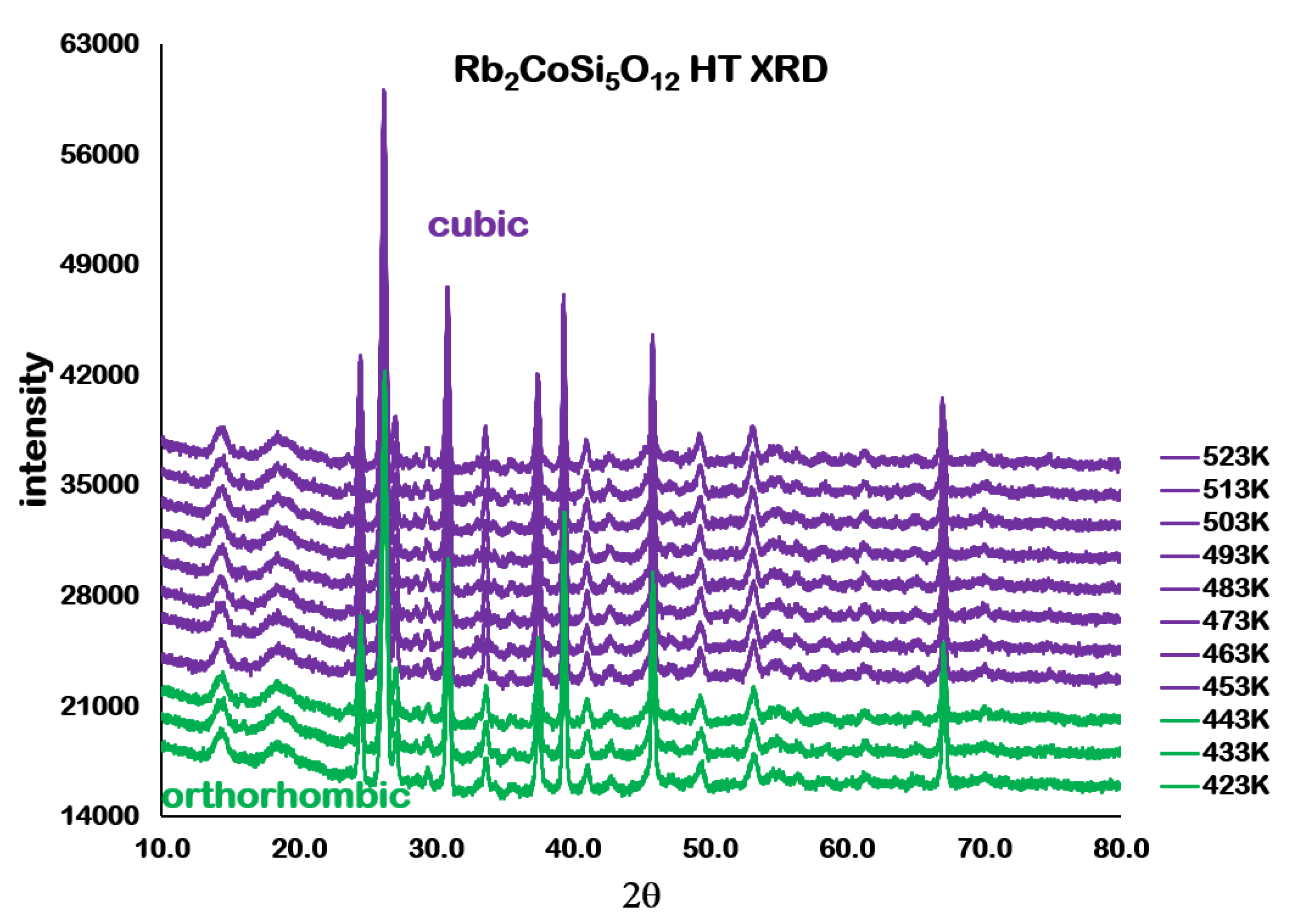
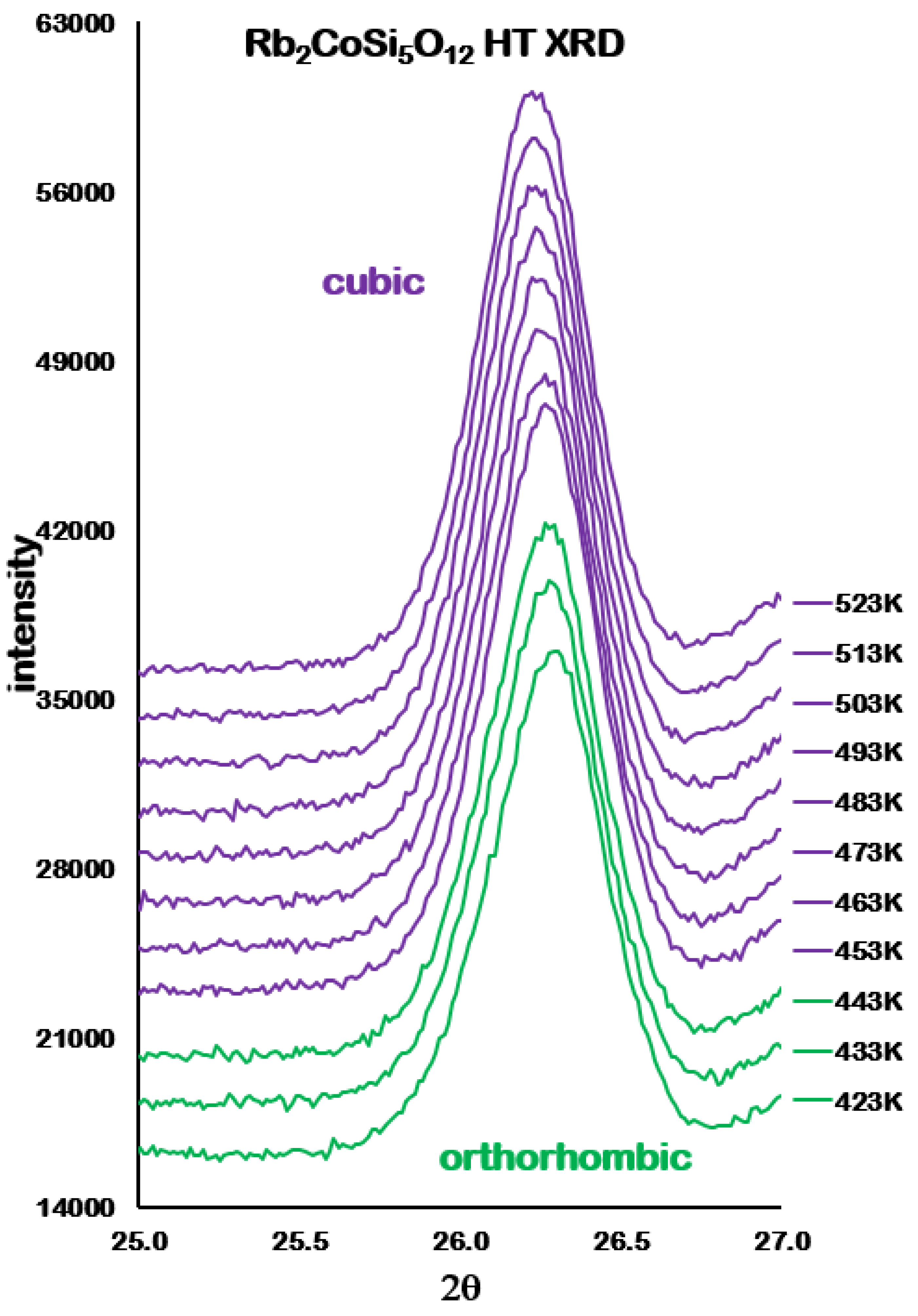
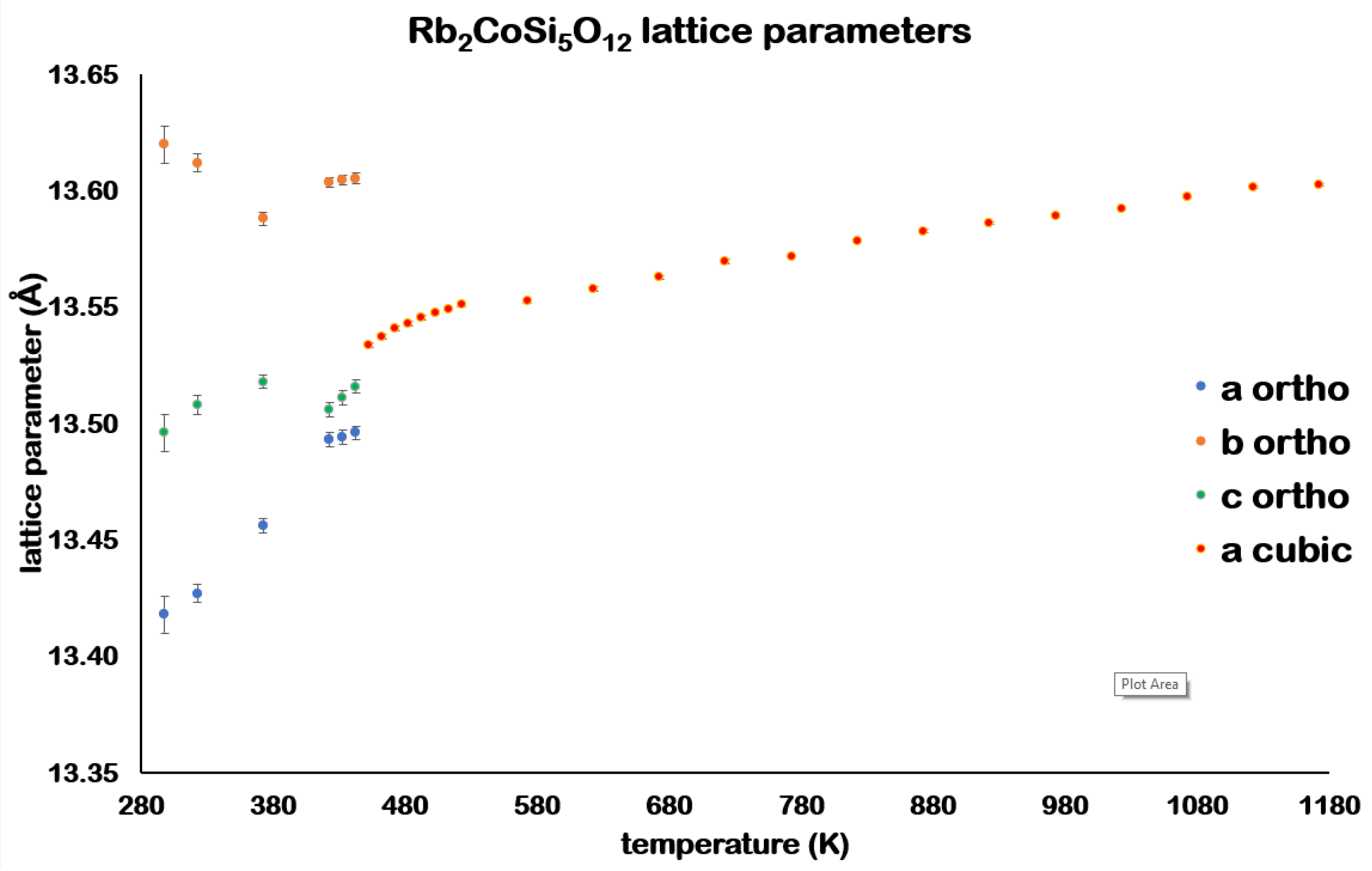
Analysis of the high temperature X-ray powder diffraction data for Rb
2CoSi
5O
l2 showed a first-order phase transition from
Pbca orthorhombic to
cubic at 457 K. However,
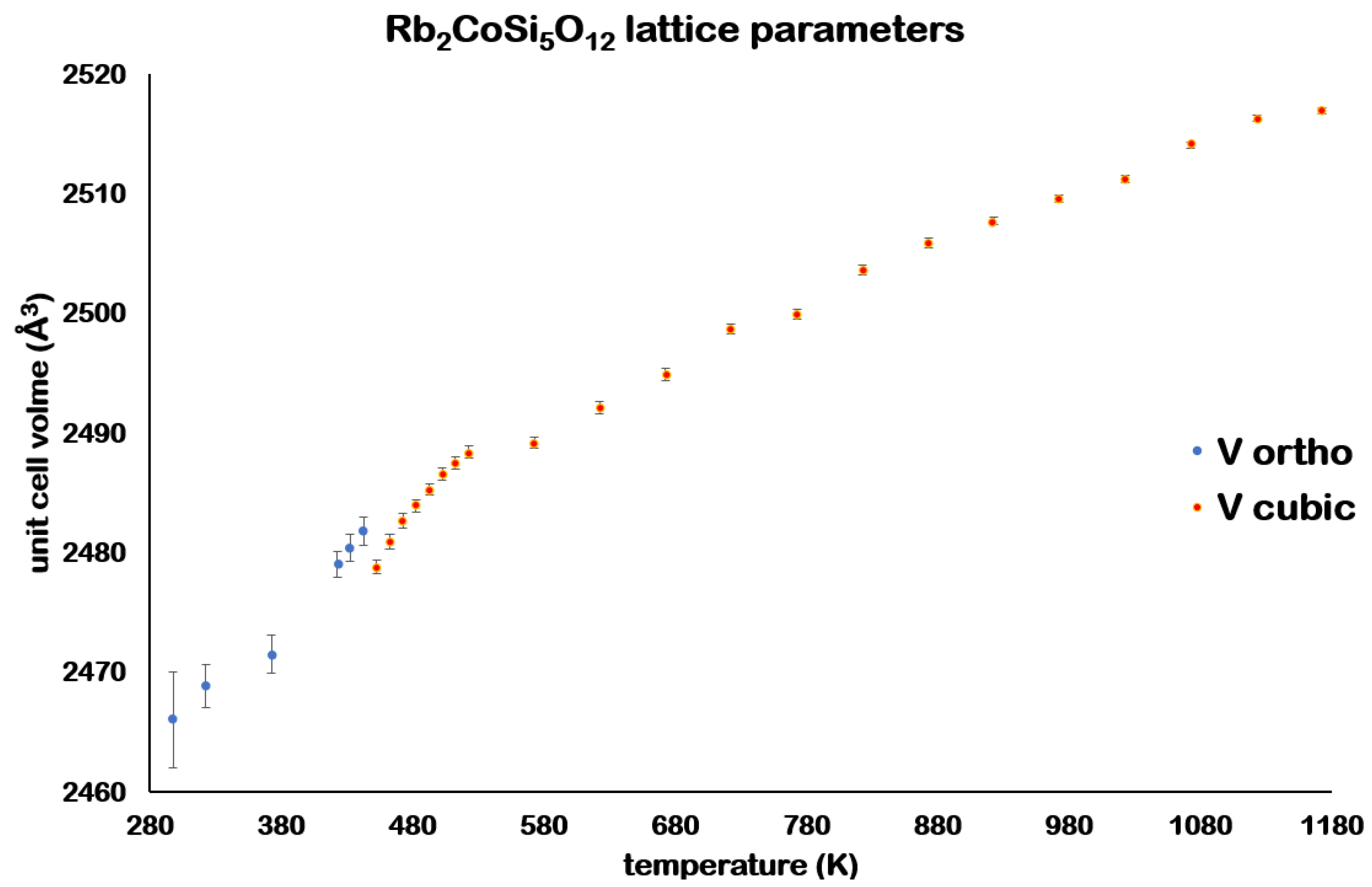
Table 4 and
Figure 7 and
Figure 8 show that the unit cell volume
decreases with increasing temperature after the phase transition. The
Pbca structure for Rb
2CoSi
5O
l2 has the tetrahedrally coordinated Co and Si atoms ordered onto separate T-sites (1 Co and 5 Si sites). However, the
structure for Rb
2CoSi
5O
l2 has some partial T-site disorder. One T site is fully occupied by Si, the other T site is 2/3 occupied by Si and 1/3 occupied by Co.
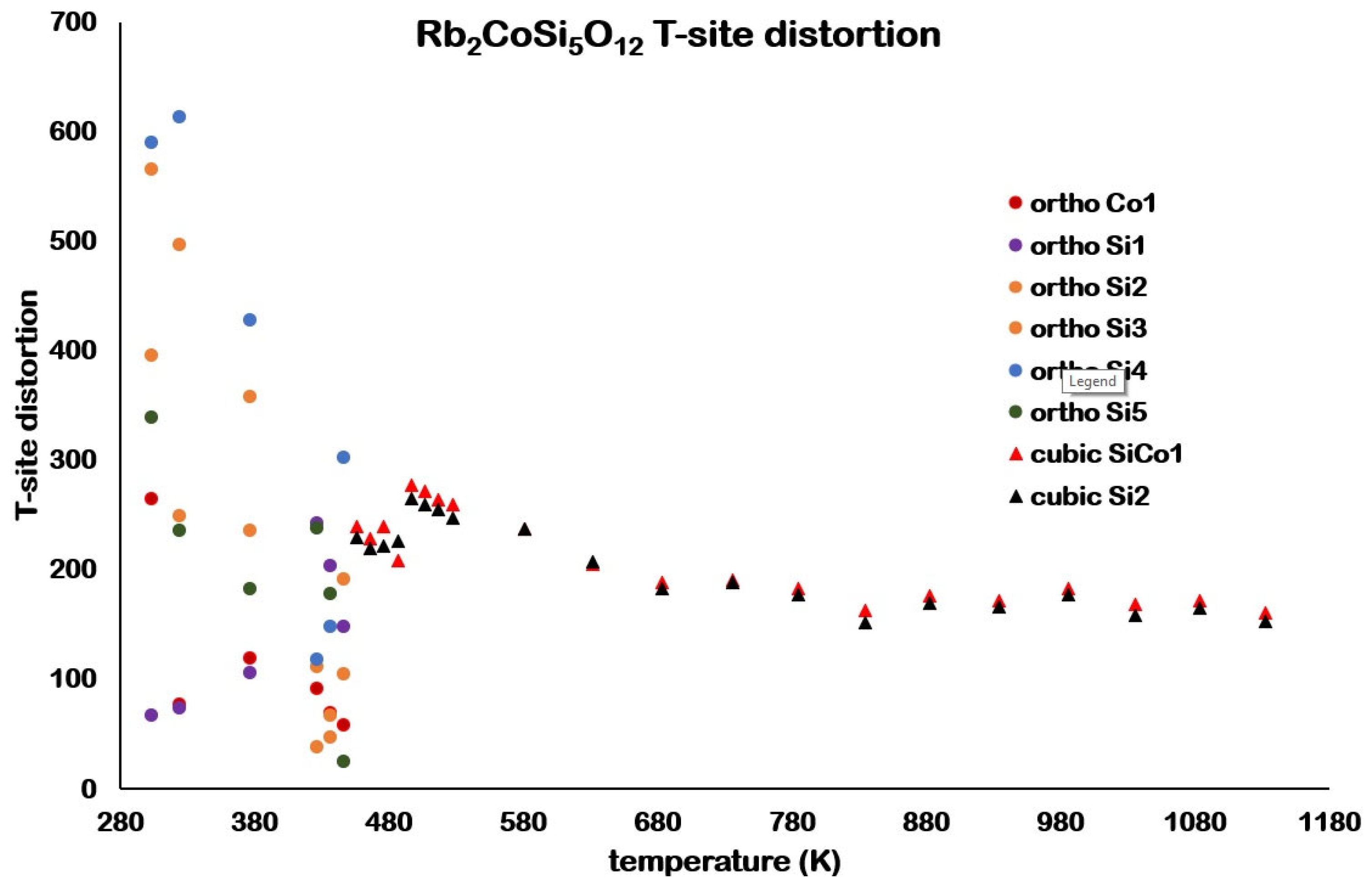
Figure 9 shows how the T-site distortions for Rb
2CoSi
5O
l2 vary with temperature for the
Pbca orthorhombic and
cubic phases. There is a much wider spread of T-site distortions for
Pbca orthorhombic compared with
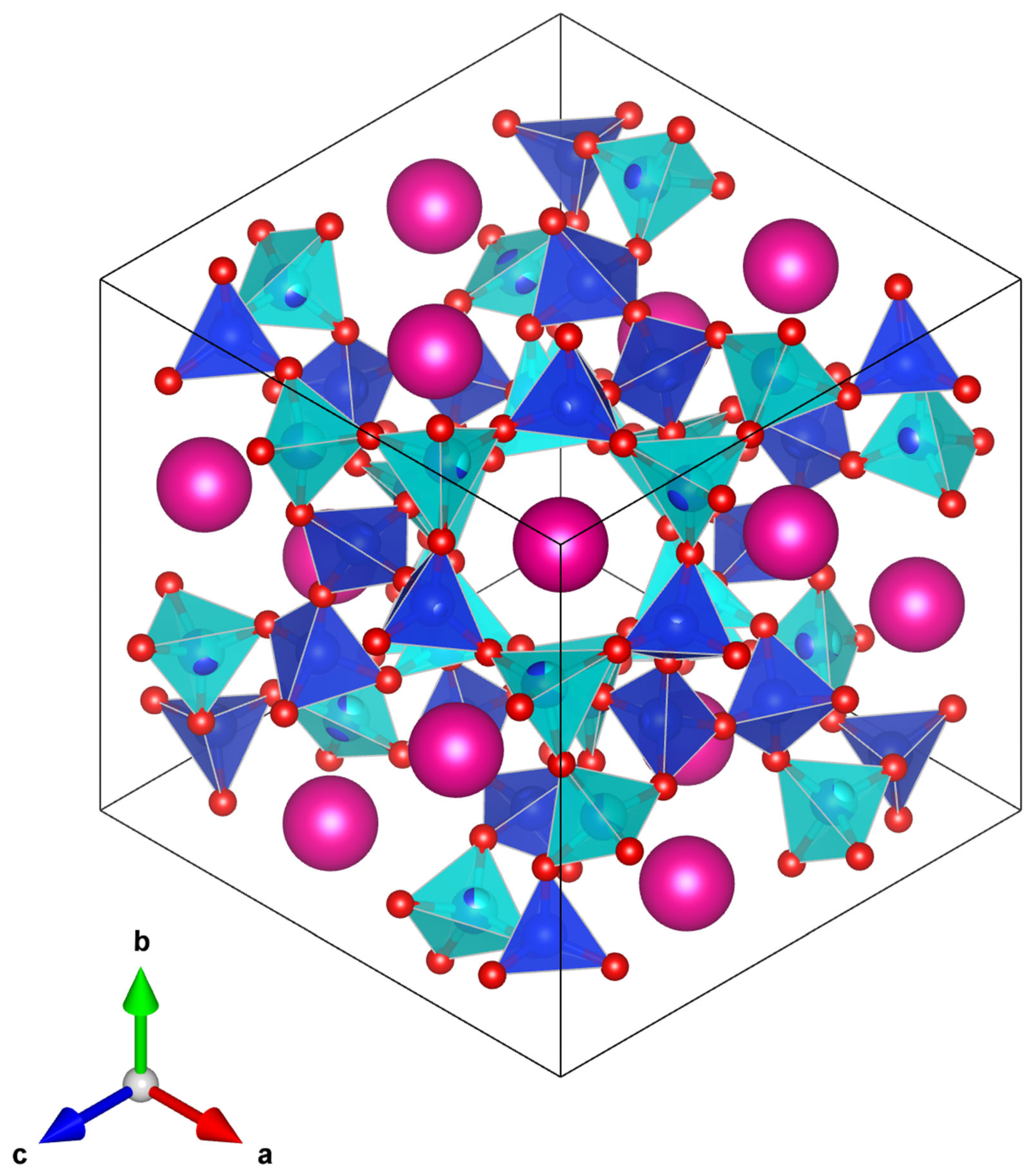
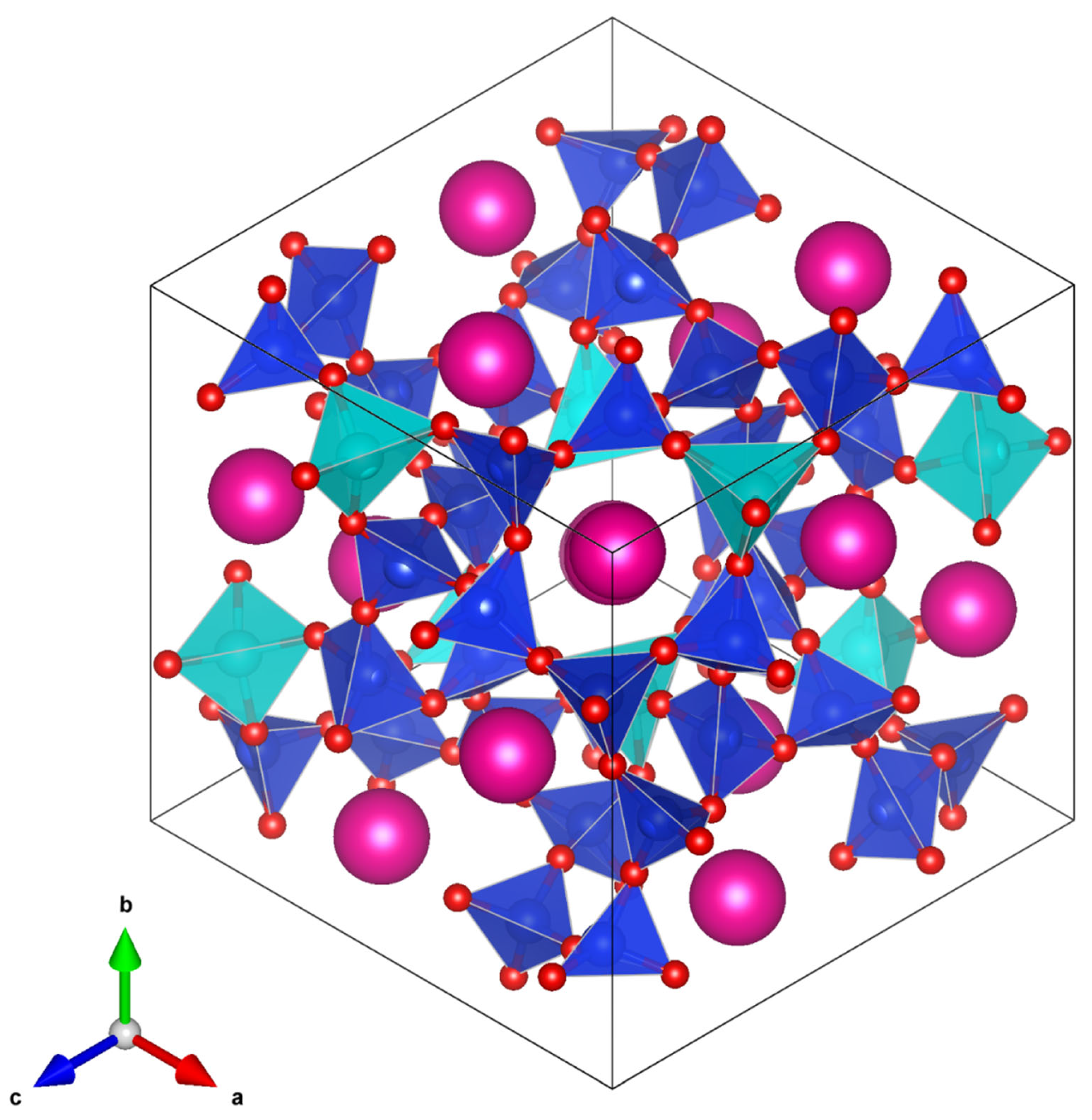
cubic. A plot of the 447 K
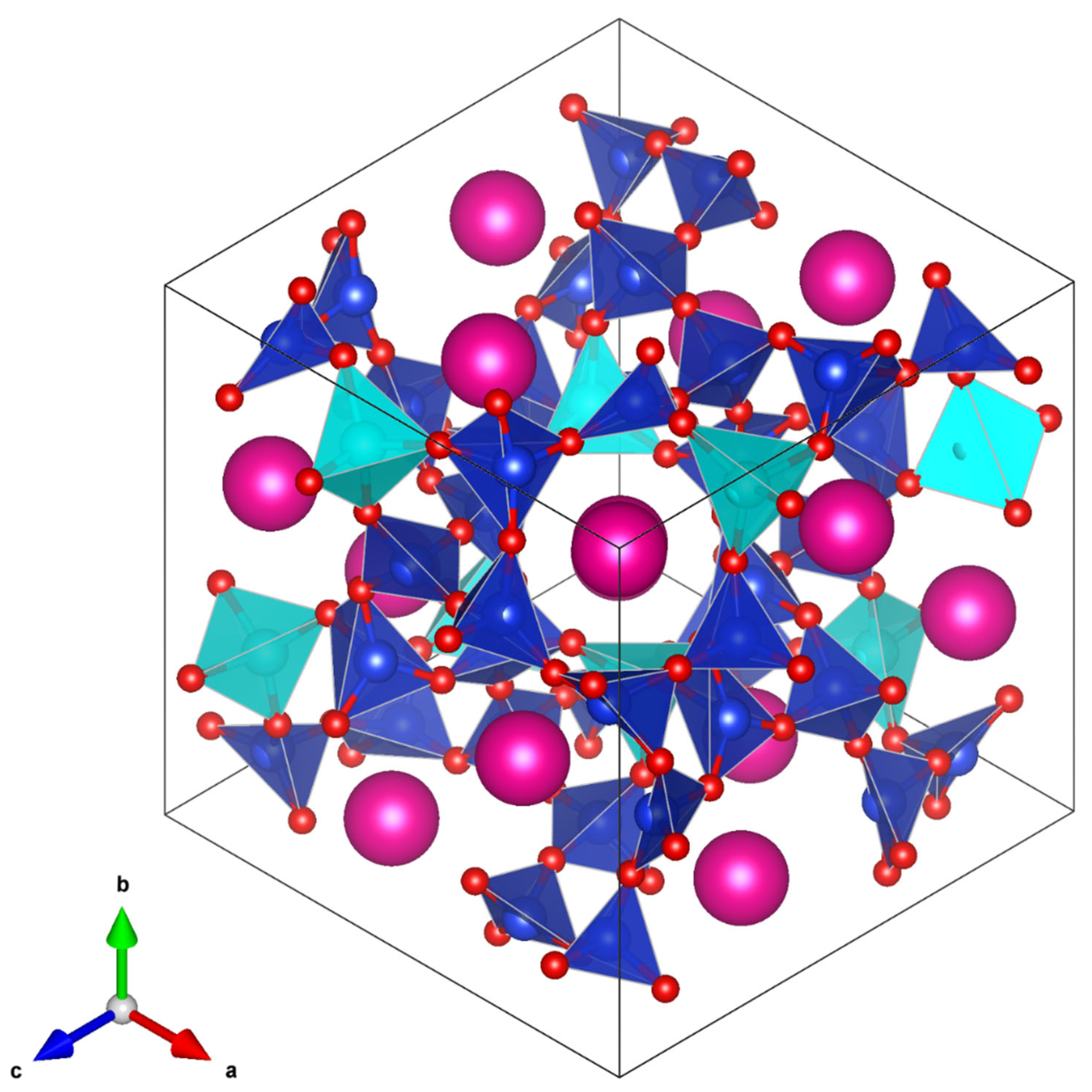
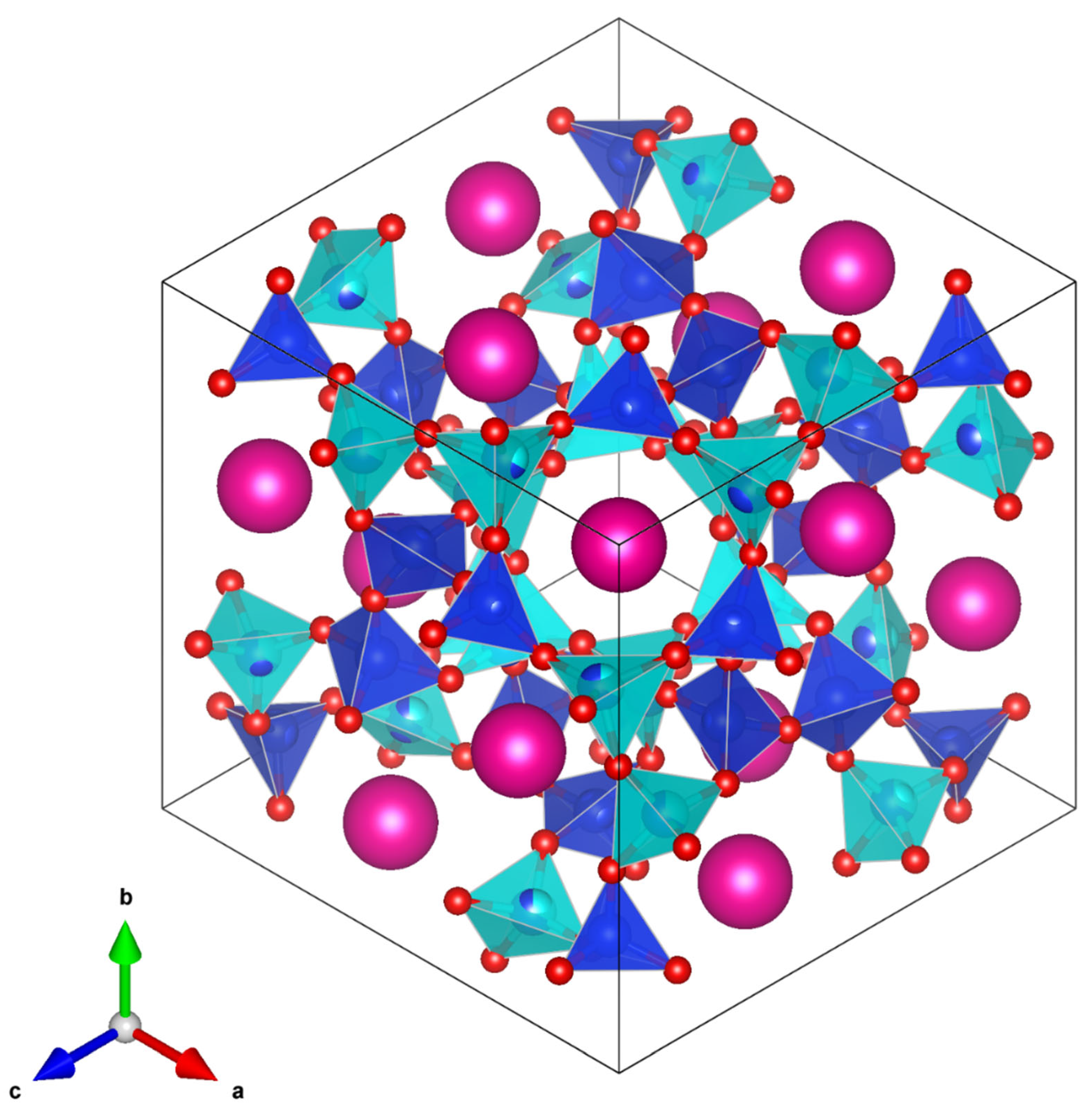
Pbca T-site ordered crystal structure (
Figure 12) shows that the central channel (which contains different sized SiO
4 and CoO
4 tetrahedra) is much more distorted than the central channel of the partially T-site disordered 457 K
structure (
Figure 13). This agrees with the plot of T-site distortions shown in
Figure 9. In the
structure, there is a smaller size difference between the SiO
4 and (Si
2/3Co
1/3)O
4 tetrahedra compared with the size difference between the
Pbca SiO
4 and CoO
4 tetrahedra. The smaller size difference between the TO
4 (T = Si or Co) tetrahedra in the
structure compared with the
Pbca structure means that the
structure is less distorted than the
Pbca structure. This decrease in distortion of the crystal structure through the phase transition is why the central channel
contracts through the phase transition. This is reflected in the decrease in unit cell volume.
Such a phase transition with a unit cell contraction has not been observed before in a leucite mineral analogue. However, similar transitions have been observed in other materials such as BiNiO
3 [
26] and Hf
0.86Ta
0.14Fe
2 [
27].
Thermal expansion due to the increase in temperature means that the cubic unit cell volume eventually becomes greater than the orthorhombic unit cell volume at 497 K, 40 K higher than the phase transition temperature. The
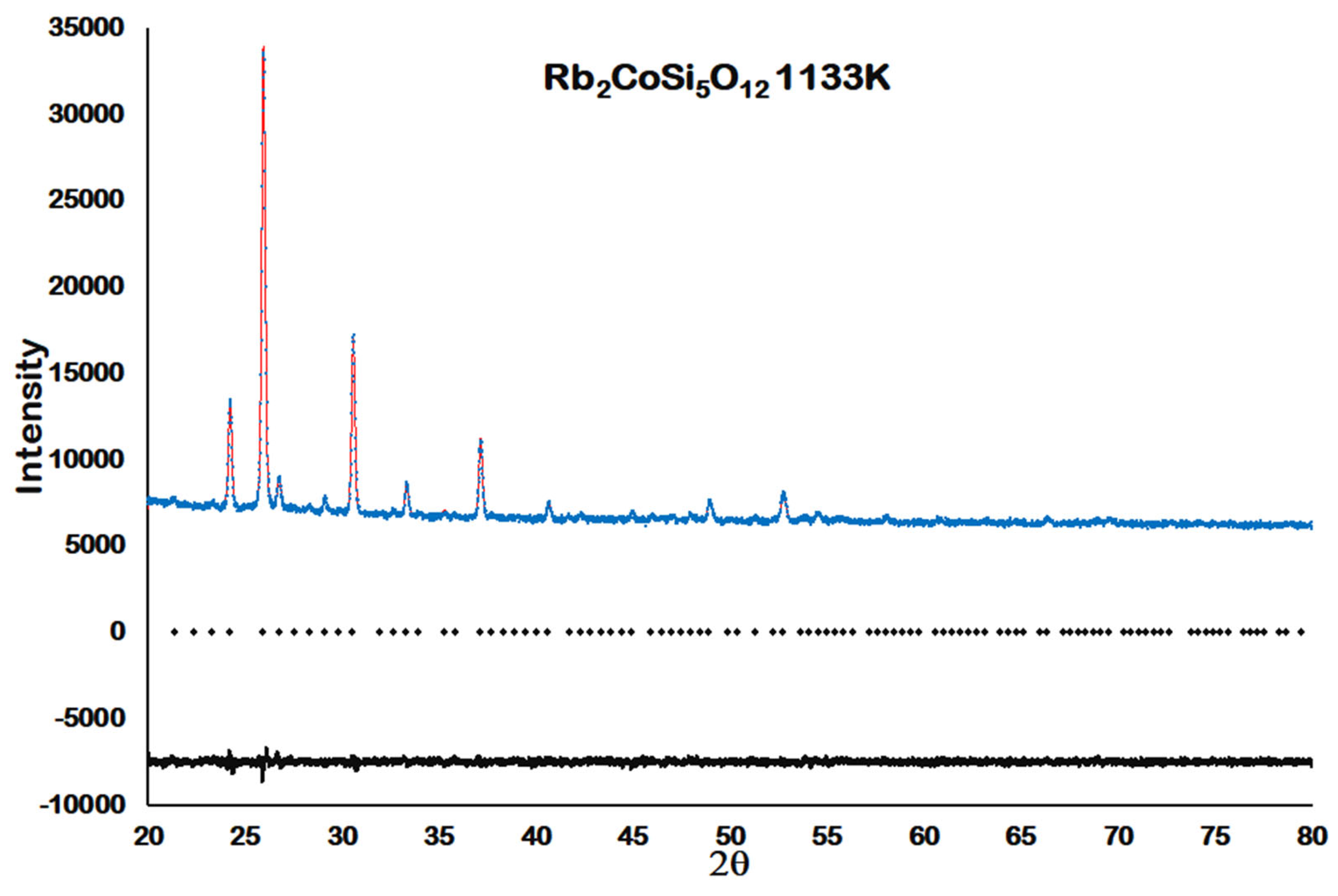
cubic structure is retained up to 1133 K. Above this temperature the Rb
2CoSi
5O
l2 sample starts to decompose before partially melting at 1323 K.
Table 8 shows the thermal expansion coefficients (TEC) for the two Rb
2CoSi
5O
l2 phases. TEC for other leucite phases are given as comparison. It can be seen that the TEC for Rb
2CoSi
5O
l2 are in the same order of magnitude as those reported for other leucites.
The
Pbca orthorhombic to
cubic leucite phase transition was first observed in Cs
2ZnSi
5O
12 [
18]. There was no unit cell contraction noticed through the phase transition in Cs
2ZnSi
5O
12 and this was assumed to be a second-order phase transition. However, the high temperature X-ray powder diffraction data for Cs
2ZnSi
5O
12 were collected every 30 K, a larger temperature increment than was used for Rb
2CoSi
5O
l2. It would be interesting to repeat the high temperature XRD study on Cs
2ZnSi
5O
12 with a smaller temperature increment to see if a unit cell contraction could be observed for this phase transition.


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}