Biogeochemical Cycling of Silver in Acidic, Weathering Environments

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
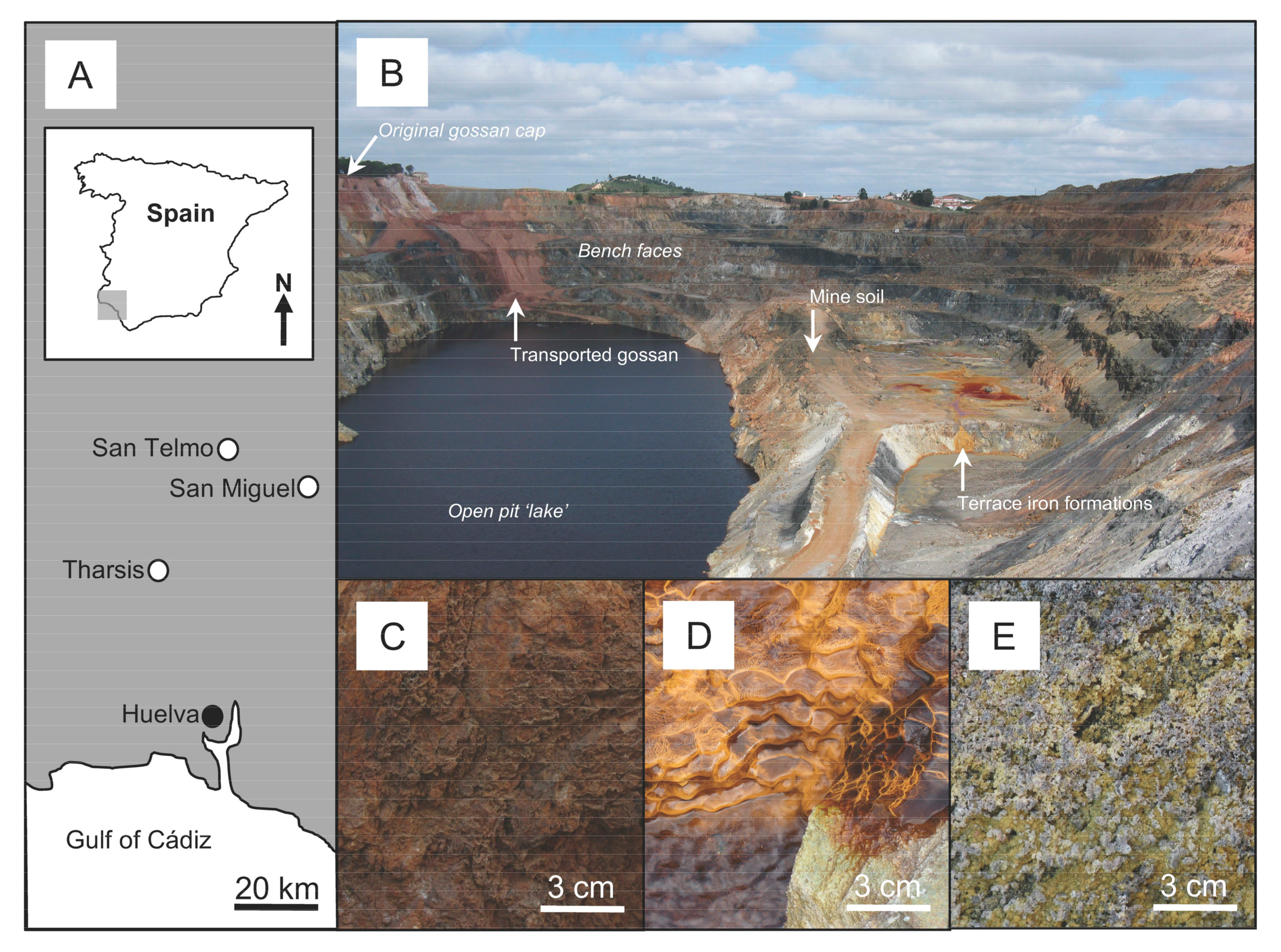
2.1. Study Sites and Sample Acquisition
2.2. Chemical and Structural Characterization of Regolith Materials
2.3. Microbial Enrichments and Enumeration
2.4. Microbial Enrichment-Ag System Experiments
3. Results
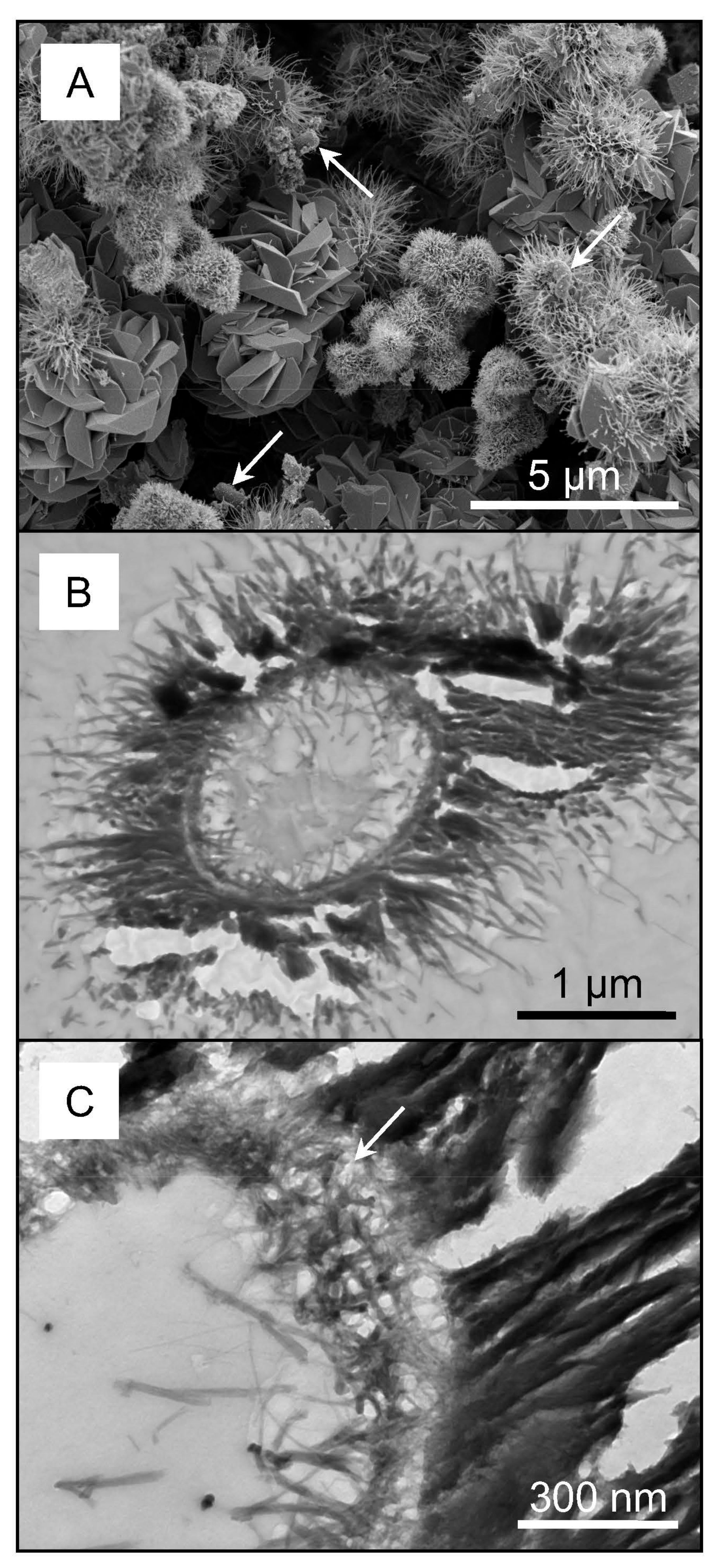
3.1. Chemistry, Mineralogy and Structure of Regolith Materials
3.2. Biogeochemical Reduction of Ag
4. Discussion
4.1. Interpretations of Ag Biogeochemical Cycling within Regolith Materials
4.2. Microbially-Catalyzed Ag Immobilization
4.3. Temporal Estimates of Ag Biogeochemical Cycling
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Boyle, R.W. The geochemistry of silver and its deposits. Geol. Surv. Can. Bull. 1968, 160, 1–282. [Google Scholar]
- Sillitoe, R.H. Supergene silver enrichment reassessed. Soc. Econ. Geol. Spec. Pub. 2009, 14, 15–32. [Google Scholar]
- Steger, H.F.; Desjardins, L.E. Oxidation of sulfide minerals, 4. Pyrite, chalcopyrite and pyrrhotite. Chem. Geol. 1978, 23, 225–237. [Google Scholar] [CrossRef]
- Lowson, R.T. Aqueous oxidation of pyrite by molecular oxygen. Chem. Rev. 1982, 82, 461–496. [Google Scholar] [CrossRef]
- Buckley, A.N.; Walker, G.W. The surface composition of arsenopyrite exposed to oxidising environments. Appl. Surf. Sci. 1988, 35, 227–240. [Google Scholar] [CrossRef]
- Nesbitt, H.W.; Muir, I.J.; Pratt, A.R. Oxidation of arsenopyrite by air and air-saturated, distilled water, and implications for mechanism of oxidation. Geochim. Cosmochim. Acta 1995, 59, 1773–1786. [Google Scholar] [CrossRef]
- Chandra, A.P.; Gerson, A.R. The mechanisms of pyrite oxidation and leaching: A fundamental perspective. Surf. Sci. Rep. 2010, 65, 293–315. [Google Scholar] [CrossRef]
- Nordstrom, D.K. Aqueous pyrite oxidation and the consequent formation of secondary iron minerals. In Acid Sulfate Weathering; Kittrick, J.A., Fanning, D.S., Hosner, L.R., Eds.; Soil Science Society of America: Madison, WI, USA, 1982; pp. 37–56. [Google Scholar]
- Nordstrom, D.K.; Alpers, C. Negative pH, efflorescent mineralogy, and consequences for environmental restoration at the Iron Mountain Superfund site, California. Proc. Natl. Acad. Sci. USA 1999, 96, 3455–3462. [Google Scholar] [CrossRef] [PubMed]
- Singer, P.C.; Stumm, W. Acidic mine drainage: The rate-determining step. Science 1970, 167, 1121–1123. [Google Scholar] [CrossRef] [PubMed]
- Silverman, M.P.; Lundgren, D.G. Studies on the chemoautotrophic iron bacterium Ferrobacillus ferrooxidans. J. Bacteriol. 1959, 77, 642–647. [Google Scholar] [PubMed]
- Shuster, J.; Lengke, M.F.; Márquez-Zavalía, M.F.; Southam, G. Floating gold grains and nanophase particles produced from the biogeochemical weathering of a gold-bearing ore. Econ. Geol. 2016, 111, 1485–1494. [Google Scholar] [CrossRef]
- Mann, A.W. Mobility of gold and silver in lateritic weathering profiles: Some observations from Western Australia. Econ. Geol. 1984, 79, 38–49. [Google Scholar] [CrossRef]
- Webster, J.G.; Mann, A.W. The Influence of climate, geomorphology and primary geology on the supergene migration of gold and silver. J. Geochem. Explor. 1984, 22, 21–42. [Google Scholar] [CrossRef]
- Craw, D.; MacKenzie, D.J. Near-surface secondary gold mobility and grain-size enhancement, Barewood Mine, east Otago, New Zealand. N. Z. J. Geol. Geophys. 2010, 35, 151–156. [Google Scholar] [CrossRef]
- Craw, D.; Lilly, K. Gold nugget morphology and geochemical environments of nugget formation, southern New Zealand. Ore Geol. Rev. 2016, 79, 301–315. [Google Scholar] [CrossRef]
- Reith, F.; Lengke, M.F.; Falconer, D.; Craw, D.; Southam, G. The geomicrobiology of gold. ISME J. 2007, 1, 567–584. [Google Scholar] [CrossRef] [PubMed]
- Brugger, J.; Etschmann, B.; Grosse, C.; Plumridge, C.; Kaminski, J.; Paterson, D.; Shar, S.S.; Ta, C.; Howard, D.L.; de Jonge, M.D.; et al. Can biological toxicity drive the contrasting behavior of platinum and gold in surface environments? Chem. Geol. 2013, 343, 99–110. [Google Scholar] [CrossRef]
- Greffie, C.; Benedetti, M.F.; Parron, C.; Amouric, M. Gold and iron oxide associations under supergene conditions: An experimental approach. Geochim. Cosmochim. Acta 1996, 60, 1531–1542. [Google Scholar] [CrossRef]
- Webster, J.G. The solubility of gold and silver in the system Au-Ag-S-O2-H2O at 25 °C and 1 atm. Geochim. Cosmochim. Acta 1986, 50, 1837–1845. [Google Scholar] [CrossRef]
- Nordstrom, D.K.; Southam, G. Geomicrobiology of sulphide mineral oxidation. Rev. Mineral. 1997, 35, 362–390. [Google Scholar]
- Schippers, A.; Sand, W. Bacterial leaching of metal sulfides proceeds by two indirect mechanisms via thiosulfate or via polysulfides and sulfur. Appl. Environ. Microbiol. 1999, 65, 319–321. [Google Scholar] [PubMed]
- Groat, L.A.; Jambor, J.L.; Pemberton, B.C. The crystal structure of argentojarosite, AgFe3(SO4)2(OH)6. Can. Mineral. 2003, 41, 921–928. [Google Scholar] [CrossRef]
- Amorós, J.; Lunar, R.; Tavira, P. Jarosite: A silver bearing mineral of the gossan of Rio Tinto (Huelva) and la Union (Cartagena, Spain). Miner. Depos. 1981, 16, 205–213. [Google Scholar] [CrossRef]
- Sasaki, K.; Tsunekawa, M.; Konno, H. Characterisation of argentojarosite formed from biologically oxidised Fe3+ ions. Can. Mineral. 1995, 33, 1311–1319. [Google Scholar]
- Sasaki, K.; Sakimoto, T.; Endo, M.; Konno, H. FE-SEM study of microbially formed jarosite by Acidithiobacillus ferrooxidans. Mater. Trans. 2006, 47, 1155–1162. [Google Scholar] [CrossRef]
- Sasaki, K.; Konno, H. Morphology of jarosite-group compounds precipitated from biologically and chemically oxidized Fe ions. Can. Mineral. 2000, 38, 45–56. [Google Scholar] [CrossRef]
- Wang, H.; Bigham, J.M.; Tuovinen, O.H. Formation of schwertmannite and its transformation to jarosite in the presence of acidophilic iron-oxidizing microorganisms. Mater. Sci. Eng. C Mater. 2006, 26, 588–592. [Google Scholar] [CrossRef]
- Liao, Y.; Zhou, L.; Liang, J.; Xiong, H. Biosynthesis of schwertmannite by Acidithiobacillus ferrooxidans cell suspensions under different pH condition. Mater. Sci. Eng. C Mater. 2009, 29, 211–215. [Google Scholar] [CrossRef]
- Daoud, J.; Karamanev, D. Formation of jarosite during Fe2+ oxidation by Acidithiobacillus ferrooxidans. Miner. Eng. 2006, 19, 960–967. [Google Scholar] [CrossRef]
- Shuster, J.; Bolin, T.; MacLean, L.C.W.; Southam, G. The effect of iron-oxidising bacteria on the stability of gold (I) thiosulfate complex. Chem. Geol. 2014, 376, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Shuster, J.; Marsden, S.; Maclean, L.; Ball, J.; Bolin, T.; Southam, G. The immobilization of gold from gold (III) chloride by a halophilic sulfate-reducing bacterial consortium. Geol. Soc. Lond. Spec. Publ. 2013, 393, 249–263. [Google Scholar] [CrossRef]
- Relvas, J.M.; Tassinari, C.C.; Munhá, J.; Barriga, F.J. Multiple sources for ore-forming fluids in the Neves Corvo VHMS Deposit of the Iberian Pyrite Belt (Portugal): Strontium, neodymium and lead isotope evidence. Miner. Depos. 2001, 36, 416–427. [Google Scholar] [CrossRef]
- Strauss, G.; Rober, G.; Lecolle, M.; Lopera, E. Geochemical and geological study of the volcano-sedimentary sulfide orebody of La Zarza, Huelva Province, Spain. Econ. Geol. 1981, 76, 1975–2000. [Google Scholar] [CrossRef]
- Sáez, R.; Pascual, E.; Toscano, M.; Almodóvar, G. The Iberian type of volcano-sedimentary massive sulfide deposits. Miner. Depos. 1999, 34, 549–570. [Google Scholar]
- Ruiz, C.; Arribas, A.; Arribas, A., Jr. Mineralogy and geochemistry of the Masa Valverde blind massive sulfide deposit, Iberian Pyrite Belt (Spain). Ore Geol. Rev. 2002, 19, 1–22. [Google Scholar] [CrossRef]
- Tornos, F. Environment of formation and styles of volcanogenic massive sulfides: The Iberian Pyrite Belt. Ore Geol. Rev. 2006, 28, 259–307. [Google Scholar] [CrossRef]
- Boyle, D.R. Oxidation of Massive Sulfide Deposits in the Bathurst Mining Camp, New Brunswick. In Environmental Goechemistry of Sulfide Oxidation; Alpers, C., Ed.; American Chemical Society Symposium Series: Washington, DC, USA, 1993; pp. 535–550. [Google Scholar]
- Velasco, F.; Herrero, J.M.; Suárez, S.; Yusta, I.; Alvaro, A.; Tornos, F. Supergene features and evolution of gossans capping massive sulphide deposits in the Iberian Pyrite Belt. Ore Geol. Rev. 2013, 53, 181–203. [Google Scholar] [CrossRef]
- Hutchinson, R.W. Precious metals in massive base metal sulphide deposits. Geol. Rundsch. 1990, 79, 241–262. [Google Scholar] [CrossRef]
- Leistel, J.M.; Marcoux, E.; Deschamps, Y.; Joubert, M. Antithetic behaviour of gold in the volcanogenic massive sulphide deposits of the IPB. Miner. Depos. 1998, 33, 82–97. [Google Scholar] [CrossRef]
- Tornos, F.; Solomon, M.; Conde, C.; Spiro, B.F. Formation of the Tharsis massive sulphide deposit, Iberian Pyrite Belt: Geological lithogeochemical, and stable isotope evidence for deposition in a brine pool. Econ. Geol. 2008, 103, 185–214. [Google Scholar] [CrossRef]
- Sánchez España, J.; Santofimia Pastor, E.; López Pamo, E. Iron terraces in acid mine drainage systems: A discussion about the organic and inorganic factors involved in their formation through observations from the Tintillo acidic river (Riotinto mine, Huelva, Spain). Geosphere 2007, 3, 133–151. [Google Scholar] [CrossRef]
- BrukerAXS. DIFFRACplus Evaluation Package Release 2005; Bruker AXS: Karlsruhe, Germany, 2005. [Google Scholar]
- Sánchez España, J.; López Pamo, E.; Santofimia, E.; Aduvire, O.; Reyes, J.; Barettino, D. Acid mine drainage in the Iberian Pyrite Belt (Odiel river watershed, Huelva, SW Spain): Geochemistry, mineralogy and environmental implications. Appl. Geochem. 2005, 20, 1320–1356. [Google Scholar] [CrossRef]
- Cochran, W.G. Estimation of bacterial densities by means of the “Most Probable Number”. Biometrics 1950, 6, 105–116. [Google Scholar] [CrossRef] [PubMed]
- España, J.S.; Pamo, E.L.; Pastor, E.S.; Andrés, J.R.; Rubí, J.A.M. The natural attenuation of two acidic effluents in Tharsis and La Zarza-Perrunal mines (Iberian Pyrite Belt, Huelva, Spain). Environ. Geol. 2005, 49, 253–266. [Google Scholar] [CrossRef]
- Olías, M.; Nieto, J. Background Conditions and Mining Pollution throughout History in the Río Tinto (SW Spain). Environments 2015, 2, 295–316. [Google Scholar] [CrossRef]
- Edwards, K.J.; Bond, P.L.; Druschel, G.K.; McGuire, M.M.; Hamers, R.J.; Banfield, J.F. Geochemical and biological aspects of sulfide mineral dissolution: Lessons from Iron Mountain, California. Chem. Geol. 2000, 169, 383–397. [Google Scholar] [CrossRef]
- Velasco, F.; Tornos, F. Los sulfuros masivos (Cu-Zn-Au) de Lomero Poyatos (Faja Pirítica Iberica): Encuadre geológico, alteración hidrothermal y removilización. Macla 2006, 6, 489–492. [Google Scholar]
- Gabby, K.L.; Eisele, T.C. Selective removal of mercury using zinc sulfide. Miner. Metall. Process. 2013, 30, 91–94. [Google Scholar]
- Colin, F.; Vieillard, P.; Ambrosi, J.P. Quantitative approach to physical and chemical gold mobility in equitorial rainforest lateritic environment. Earth Planet. Sci. Lett. 1993, 114, 269–285. [Google Scholar] [CrossRef]
- Golubic, S.; Friedmann, I.; Schneider, J. The lithobiontic ecological niche, with special reference to microorganisms. J. Sediment. Petrol. 1981, 51, 475–478. [Google Scholar]
- Friedmann, I. Endolithic microorganisms in the Antarctic cold desert. Science 1982, 215, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Westall, F. The nature of fossil bacteria: A guide to the search for extraterrestrial life. J. Geophys. Res. Planet. 1999, 104, 16437–16451. [Google Scholar] [CrossRef]
- Fernández-Remolar, D.C.; Knoll, A.H. Fossilization potential of iron-bearing minerals in acidic environments of Rio Tinto, Spain: Implications for Mars exploration. Icarus 2008, 194, 72–85. [Google Scholar] [CrossRef]
- Levett, A.; Gagen, E.; Shuster, J.; Rintoul, L.; Tobin, M.; Vongsvivut, J.; Bambery, K.; Vasconcelos, P.; Southam, G. Evidence of biogeochemical processes in iron duricrust formation. J. S. Am. Earth Sci. 2016, 71, 131–142. [Google Scholar] [CrossRef]
- Tsezos, M.; Remoudaki, E.; Angelatou, V. Biosorption sites of selected metals using eletron microscopy. Comp. Biochem. Physiol. 1997, 118, 481–487. [Google Scholar] [CrossRef]
- Lin, Z.; Zhou, C.; Wu, J.; Zhou, J.; Wang, L. A further insight into the mechanism of Ag+ biosorption by Lactobacillus sp. strain A09. Spectrochim. Acta A 2005, 61, 1195–1200. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Hu, Q.; Zeng, J.; Qi, H.; Zhuang, G. Resistance and biosorption mechanism of silver ions by Bacillus cereus biomass. J. Environ. Sci. 2011, 23, 108–111. [Google Scholar] [CrossRef]
- Levard, C.; Hotze, E.M.; Lowry, G.V.; Brown, G.E., Jr. Environmental transformations of silver nanoparticles: Impact on stability and toxicity. Environ. Sci. Technol. 2012, 46, 6900–6914. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lenhart, J.J.; Walker, H.W. Dissolution-accompanied aggregation kinetics of silver nanoparticles. Langmuir 2010, 26, 16690–16698. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hurt, R.H. Ion release kinetics and particle persistence in aqueous nano-silver colloids. Environ. Sci. Technol. 2010, 44, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- Lombi, E.; Donner, E.; Taheri, S.; Tavakkoli, E.; Jamting, A.K.; McClure, S.; Naidu, R.; Miller, B.W.; Scheckel, K.G.; Vasilev, K. Transformation of four silver/silver chloride nanoparticles during anaerobic treatment of wastewater and post-processing of sewage sludge. Environ. Pollut. 2013, 176, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, S.; Beveridge, T. Pseudomonas aeruginosa biofilms react with and precipitate toxic soluble gold. Environ. Microbiol. 2002, 4, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, L.E.; Hendrix, J.L. Inhibition of Thiobacillus ferrooxidans by soluble silver. Biotechnol. Bioeng. 1976, 18, 1161–1165. [Google Scholar] [CrossRef] [PubMed]
- Zipperian, D.; Raghavan, S. Gold and silver extraction by ammoniacal thiosulfate leaching from a rhyolite ore. Hydrometallurgy 1988, 19, 361–375. [Google Scholar] [CrossRef]
- Lawrence, R.W.; Gunn, J.D. Biological preoxidation of pyrite gold concentrate. In Frontier Technology in Mineral Processing; Spisak, J.F., Jergensen, G.V., Eds.; Society of Mining Engineers of AIME: New York, NY, USA, 1985; pp. 13–17. [Google Scholar]
- Benedetti, M.; Boulegue, J. Mechanism of gold transfer and deposition in a supergene environment. Geochim. Cosmochim. Acta 1991, 55, 1539–1547. [Google Scholar] [CrossRef]
- Xu, Y.; Schoonen, M. The stability of thiosulfate in the presence of pyrite in low-temperature aqueous solutions. Geochim. Cosmochim. Acta 1995, 59, 4605–4622. [Google Scholar] [CrossRef]
- Aylmore, M.G.; Muir, D.M. Thiosulfate leaching of gold—A review. Miner. Eng. 2001, 14, 135–174. [Google Scholar] [CrossRef]
- Jeffrey, M.I. Kinetic aspects of gold and silver leaching in ammonia-thiosulphate solutions. Hydrometallurgy 2001, 60, 7–16. [Google Scholar] [CrossRef]
- Temple, K.; Colmer, A. The autotrophic oxidation of iron by a new bacterium—Thiobacillus ferrooxidans. J. Bacteriol. 1951, 62, 605–611. [Google Scholar] [PubMed]
- Lyalikova, N.N. A study of chemosynthesis in Thiobacillus ferrooxidans. Mikrobiologiya 1958, 27, 556–559. [Google Scholar]
- Portal, C.C.K. The World Bank Group: Climate Change Knowledge Portal Website. Available online: http://sdwebx.worldbank.org/climateportal/index.cfm (accessed on 05 May 2017).
- Kelley, K.; Hudson, T. Natural versus anthropogenic dispersion of metals to the environment in the Wulik River area, western Brooks Range, northern Alaska. Geochem. Explor. Environ. Anal. 2007, 7, 87–96. [Google Scholar] [CrossRef]
- Ratte, H. Bioaccumulation and toxicity of silver compounds: A review. Environ. Toxicol. Chem. 1999, 18, 89–108. [Google Scholar] [CrossRef]
- Bridge, G. CONTESTED TERRAIN: Mining and the Environment. Annu. Rev. Environ. Resour. 2004, 29, 205–259. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Regolith Material Sample Location | Major Minerals (ca. 25–100 vol %) | Minor Minerals (ca. 5–20 vol %) |
|---|---|---|
| Gossans | ||
| San Telmo | goethite hematite quartz galena | argentojarosite plumbojarosite jarosite |
| San Miguel | goethite hematite pyrite | barite quartz |
| Tharsis | goethite quartz pyrite | hematite jarosite |
| Terrace iron formations | ||
| San Telmo | schwertmannite hydroniumjarosite | quartz gypsum |
| San Miguel | schwertmannite | hydroniumjarosite gypsum |
| Tharsis | schwertmannite hydroniumjarosite | gypsum pyrite quartz |
| Mine soils | ||
| San Telmo | bernalite copiapite goethite | melanterite |
| San Miguel | copiapite goethite hydroniumjarosite quartz | kaolinite |
| Tharsis | goethite hydroniumjarosite kaolinite | coquimbite |
| Type of Regolith Material | Ag | Hg | Pb |
|---|---|---|---|
| Sample Location | (µg·g−1) | (µg·g−1) | (µg·g−1) |
| Gossans | |||
| San Telmo | 4.01 × 102 | 7.04 × 102 | 1.22 × 104 |
| San Miguel | 8.50 | 2.14 × 102 | 2.0 × 103 |
| Tharsis | 56.7 | 53.4 | 2.52 × 103 |
| Terrace iron formations | |||
| San Telmo | 2.46 | <30 | 1.29 × 103 |
| San Miguel | 4.60 | 31.7 | 2.59 × 102 |
| Tharsis | 27.6 | 58.4 | 8.14 × 103 |
| Mine soils | |||
| San Telmo | 3.64 | <30 | 2.28 × 102 |
| San Miguel | 1.86 | <30 | 1.23 × 102 |
| Tharsis | 1.69 | <30 | 95.1 |
| Type of Regolith Material | Sample Mass | Ag Mass | Time |
|---|---|---|---|
| Sample Location | (g) | (µg) A | (Years) B |
| Gossans | |||
| San Telmo | 5.06 | 2.03 × 103 | 1.46 × 103 |
| San Miguel | 1.03 | 8.78 | 0.63 |
| Tharsis | 11.4 | 6.44 × 102 | 46.4 |
| Terrace iron formations | |||
| San Telmo | 10.8 | 26.5 | 1.91 |
| San Miguel | 29.9 | 1.37 × 102 | 9.9 |
| Tharsis | 21.5 | 5.93 × 102 | 42.7 |
| Mine soils | |||
| San Telmo | 6.25 | 6.25 | 1.64 |
| San Miguel | 6.62 | 6.62 | 0.89 |
| Tharsis | 5.58 | 5.58 | 0.68 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shuster, J.; Reith, F.; Izawa, M.R.M.; Flemming, R.L.; Banerjee, N.R.; Southam, G. Biogeochemical Cycling of Silver in Acidic, Weathering Environments. Minerals 2017, 7, 218. https://doi.org/10.3390/min7110218
Shuster J, Reith F, Izawa MRM, Flemming RL, Banerjee NR, Southam G. Biogeochemical Cycling of Silver in Acidic, Weathering Environments. Minerals. 2017; 7(11):218. https://doi.org/10.3390/min7110218
Chicago/Turabian StyleShuster, Jeremiah, Frank Reith, Matthew R. M. Izawa, Roberta L. Flemming, Neil R. Banerjee, and Gordon Southam. 2017. "Biogeochemical Cycling of Silver in Acidic, Weathering Environments" Minerals 7, no. 11: 218. https://doi.org/10.3390/min7110218
APA StyleShuster, J., Reith, F., Izawa, M. R. M., Flemming, R. L., Banerjee, N. R., & Southam, G. (2017). Biogeochemical Cycling of Silver in Acidic, Weathering Environments. Minerals, 7(11), 218. https://doi.org/10.3390/min7110218