The Influence of Spectral Interferences on Critical Element Determination with Portable X-Ray Fluorescence (pXRF)

Abstract
:1. Introduction
2. Materials and Methods
2.1. Certified Reference Materials
2.2. Instrument Settings
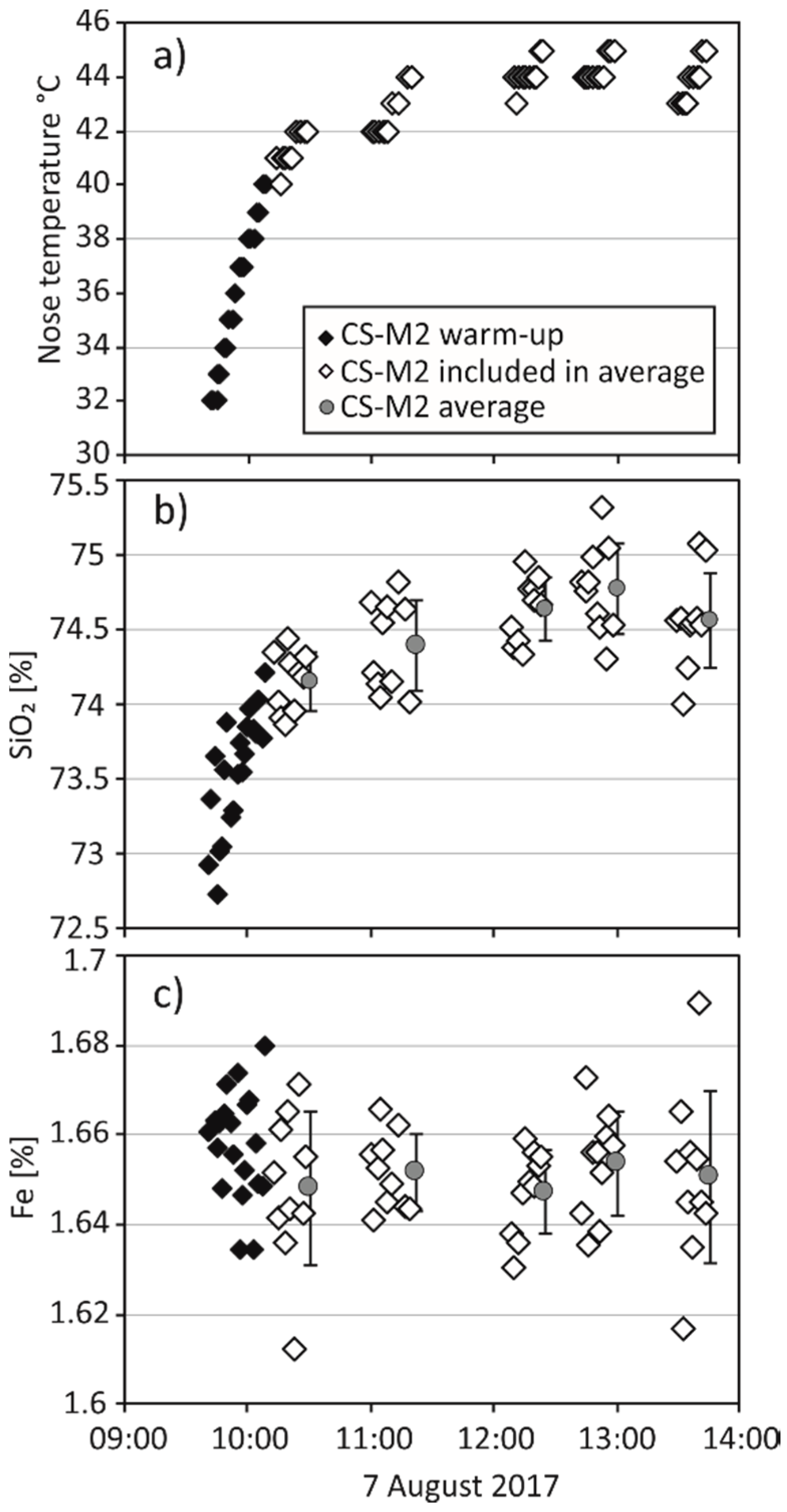
2.3. Monitoring of pXRF Performance
3. Results
3.1. Monitoring of pXRF Performance and General Observations
3.2. Analysis of Certified Reference Materials
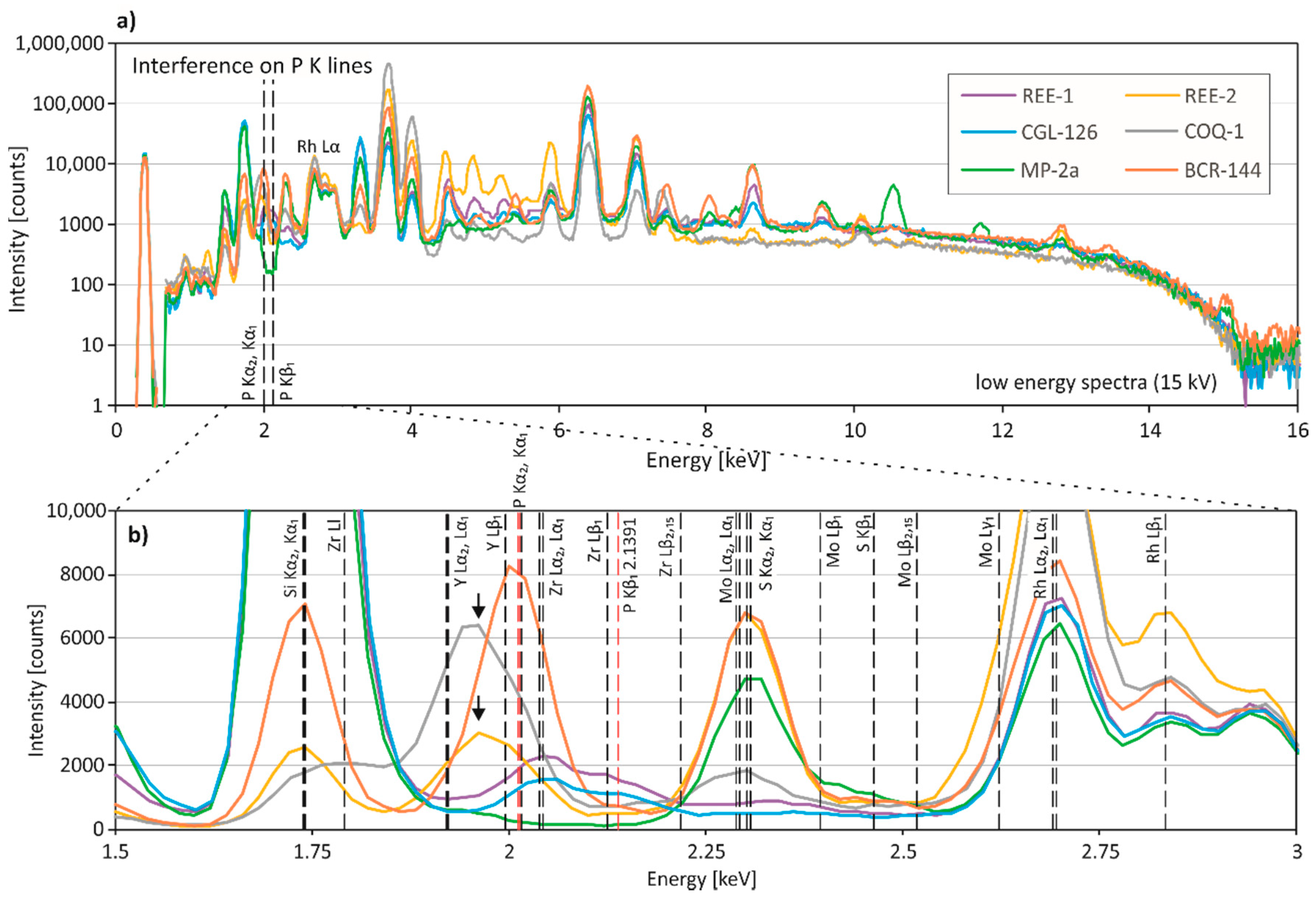
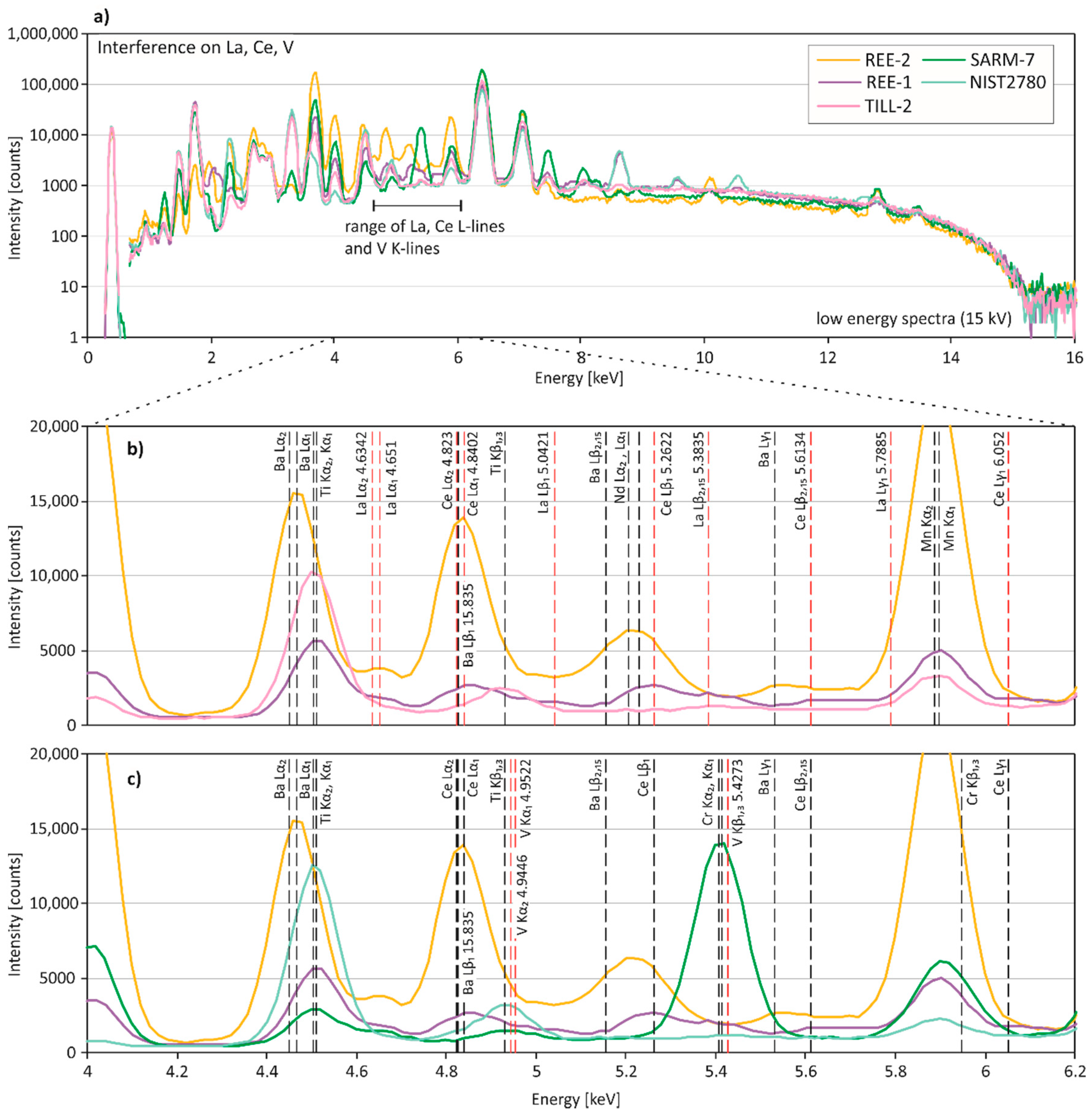
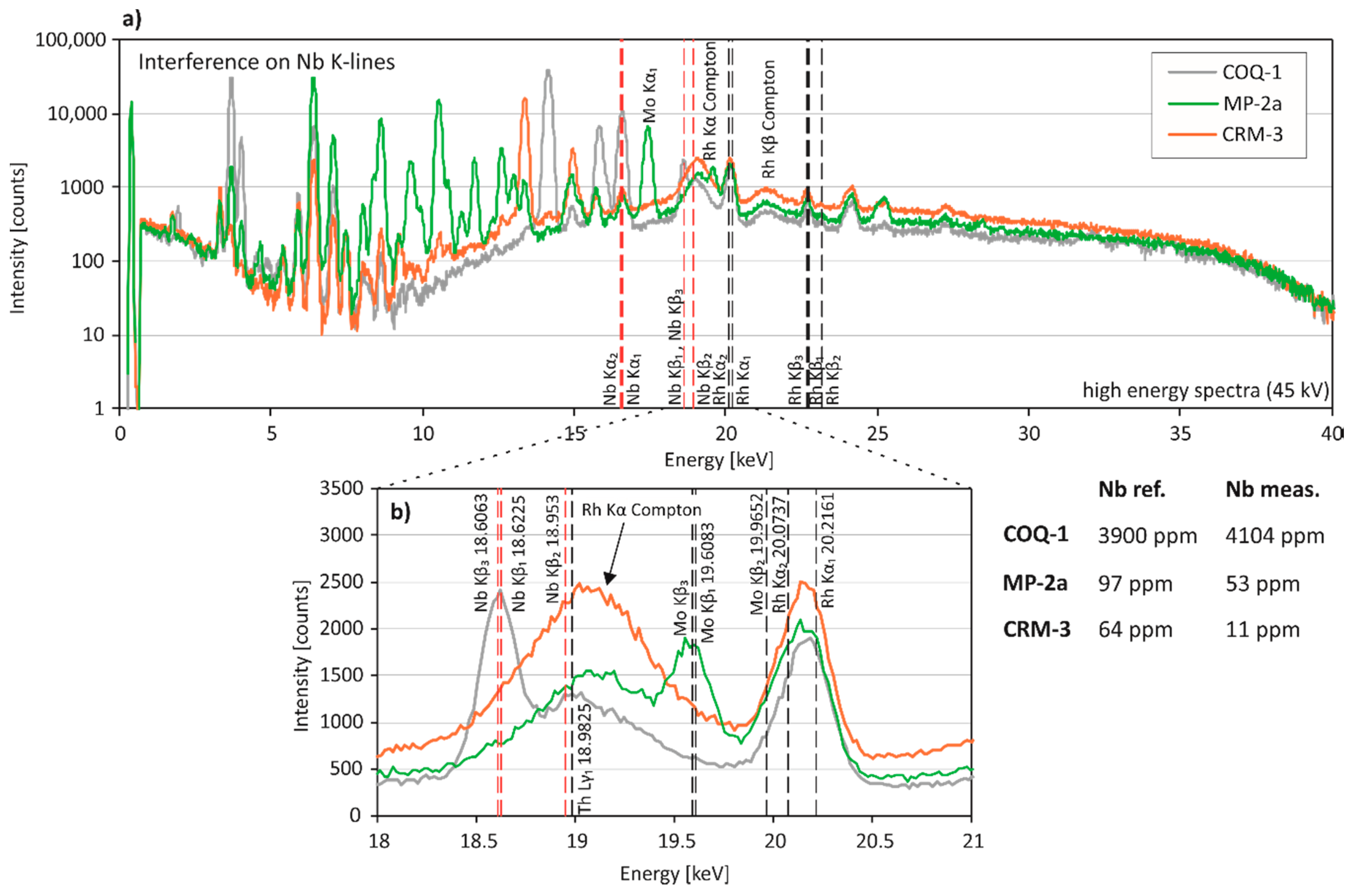
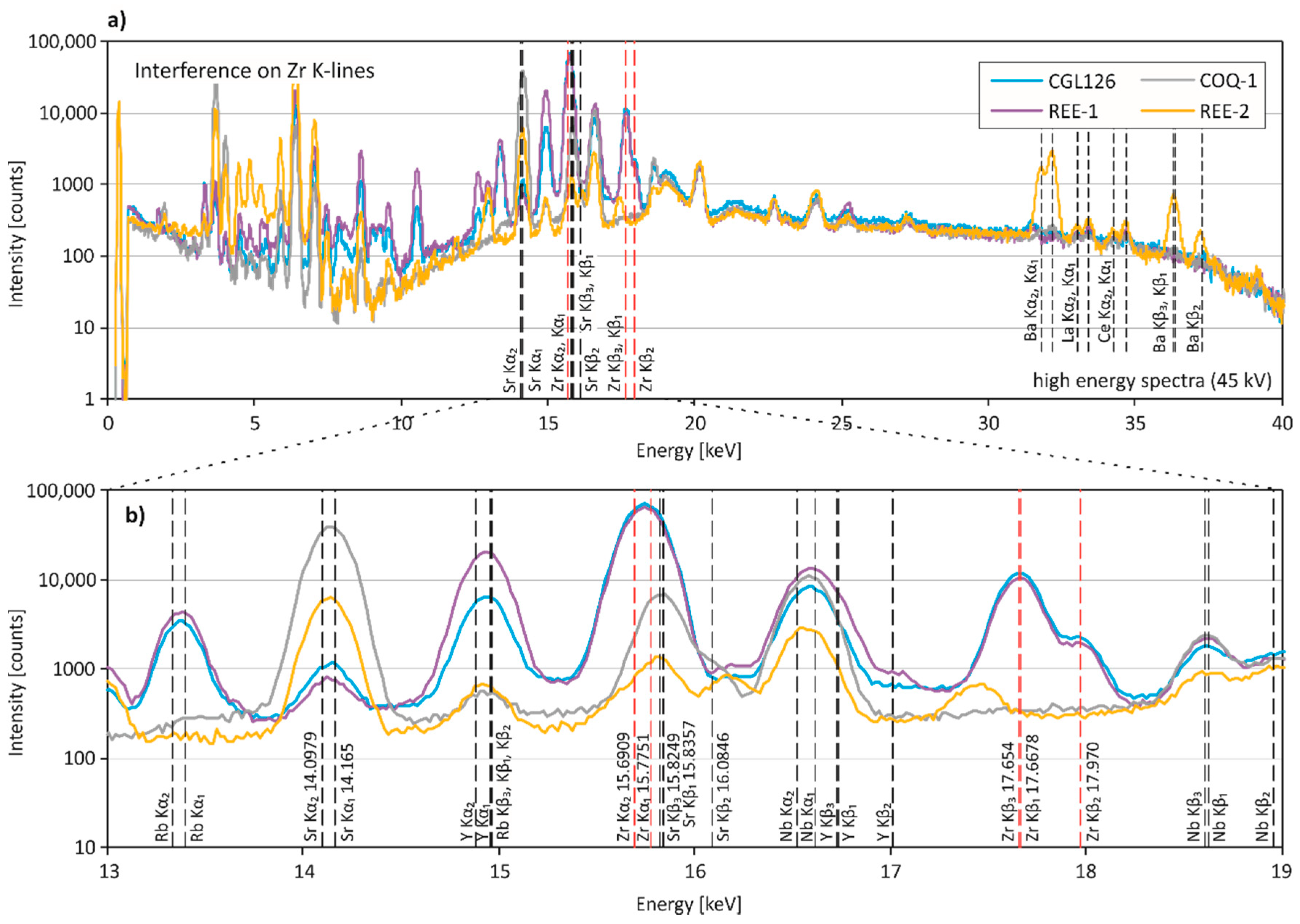
3.3. Spectral Interferences on Critical Elements
3.3.1. Low-Energy Region of the Spectral Data (0–10 keV)
3.3.2. High Energy Region of the Spectral Data (10–20 keV)
4. Discussion
4.1. Analysis of Critical Elements
4.2. Limitations of pXRF
4.3. Implications
5. Conclusions
- Critical elements can be determined in geological materials by pXRF at high concentrations, i.e., when a well-defined element peak is present in the spectra, and/or in samples of little spectral interferences;
- Each matrix type has a distinct combination of spectral interferences;
- Various elements may cause spectral interferences and hamper the quantification of a specific element (e.g., depending on matrix type V may be influenced by Ti, Cr, Ba or Ce);
- Apart from characteristic X-ray lines from interfering elements, Compton scattered peaks (Rh Kα Compton peak on Nb in sediments) and Si escape peaks (Si escape peak for the Ca Kα-lines on P in carbonatites) influence the correct quantification of elements; and
- When re-calibrating pXRF results with standards, matrix-match of standards and samples should be verified by comparing XRF spectra.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Higueras, P.; Oyarzun, R.; Iraizoz, J.M.; Lorenzo, S.; Esbrí, J.M.; Martínez-Coronado, A. Low-cost geochemical surveys for environmental studies in developing countries: Testing a field portable XRF instrument under quasi-realistic conditions. J. Geochem. Explor. 2012, 113, 3–12. [Google Scholar] [CrossRef]
- Lemière, B. A review of pXRF (field portable X-ray fluorescence) applications for applied geochemistry. J. Geochem. Explor. 2018, 188, 350–363. [Google Scholar] [CrossRef]
- Hall, G.E.; Bonham-Carter, G.F.; Buchar, A. Evaluation of portable X-ray fluorescence (pXRF) in exploration and mining: Phase 1, control reference materials. Geochem. Explor. Environ. Anal. 2014, 14, 99–123. [Google Scholar] [CrossRef] [Green Version]
- Conrey, R.M.; Goodman-Elgar, M.; Bettencourt, N.; Seyfarth, A.; van Hoose, A.; Wolff, J.A. Calibration of a portable X-ray fluorescence spectrometer in the analysis of archaeological samples using influence coefficients. Geochem. Explor. Environ. Anal. 2014, 14, 291–301. [Google Scholar] [CrossRef]
- Rouillon, M.; Taylor, M.P. Can field portable X-ray fluorescence (pXRF) produce high quality data for application in environmental contamination research? Environ. Pollut. 2016, 214, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Wiedenbeck, M. The Elements Toolkit: Field-Portable XRF: A Geochemist’s Dream? Elements 2013, 9, 7–8. [Google Scholar]
- Le Vaillant, M.; Barnes, S.J.; Fisher, L.; Fiorentini, M.L.; Caruso, S. Use and calibration of portable X-Ray fluorescence analysers: application to lithogeochemical exploration for komatiite-hosted nickel sulphide deposits. Geochem. Explor. Environ. Anal. 2014, 14, 199–209. [Google Scholar] [CrossRef]
- Quiniou, T.; Laperche, V. An assessment of field-portable X-ray fluorescence analysis for nickel and iron in laterite ore (New Caledonia). Geochem. Explor. Environ. Anal. 2014, 14, 245–255. [Google Scholar] [CrossRef]
- Arne, D.C.; Mackie, R.A.; Jones, S.A. The use of property-scale portable X-ray fluorescence data in gold exploration: Advantages and limitations. Geochem. Explor. Environ. Anal. 2014, 14, 233–244. [Google Scholar] [CrossRef]
- Gazley, M.F.; Bonnett, L.C.; Fisher, L.A.; Salama, W.; Price, J.H. A workflow for exploration sampling in regolith-dominated terranes using portable X-ray fluorescence: Comparison with laboratory data and a case study. Aust. J. Earth Sci. 2017, 64, 903–917. [Google Scholar] [CrossRef]
- Ross, P.-S.; Bourke, A.; Fresia, B. Improving lithological discrimination in exploration drill-cores using portable X-ray fluorescence measurements: (2) applications to the Zn-Cu Matagami mining camp, Canada. Geochem. Explor. Environ. Anal. 2014, 14, 187–196. [Google Scholar] [CrossRef]
- Sterk, R.; Gazley, M.F.; Wood, M.P.; Collins, K.S.; Collis, G. Maximising the value of Portable XRF data in exploration: An example from Marirongoe, Mozambique. Geochem. Explor. Environ. Anal. 2018, 18, 142–154. [Google Scholar] [CrossRef]
- Simandl, G.J.; Stone, R.S.; Paradis, S.; Fajber, R.; Reid, H.M.; Grattan, K. An assessment of a handheld X-ray fluorescence instrument for use in exploration and development with an emphasis on REEs and related specialty metals. Miner. Depos. 2014, 49, 999–1012. [Google Scholar] [CrossRef]
- Simandl, G.J.; Paradis, S.; Stone, R.S.; Fajber, R.; Kressall, R.D.; Grattan, K.; Crozier, J.; Simandl, L.J. Applicability of handheld X-Ray fluorescence spectrometry in the exploration and development of carbonatite-related niobium deposits: a case study of the Aley Carbonatite, British Columbia, Canada. Geochem. Explor. Environ. Anal. 2014, 14, 211–221. [Google Scholar] [CrossRef]
- Simandl, G.J.; Fajber, R.; Paradis, S. Portable X-ray fluorescence in the assessment of rare earth element-enriched sedimentary phosphate deposits. Geochem. Explor. Environ. Anal. 2014, 14, 161–169. [Google Scholar] [CrossRef]
- European Commission. Communication from the Commission to the European Parliament, the Council, the European Economic and Social Committee and the Committee of the Regions on the 2017 List of Critical Raw Materials for the EU COM(217) 490 Final; European Commission: Brussels, Belgium, 2017. [Google Scholar]
- Markowicz, A.A. Chapter 2 Quantification and Correction Procedures. In Portable X-ray Fluorescence Spectrometry: Capabilities for In Situ Analysis; The Royal Society of Chemistry: London, UK, 2008; pp. 13–38. [Google Scholar]
- Potts, P.J. Chapter 1 Introduction, Analytical Instrumentation and Application Overview. In Portable X-ray Fluorescence Spectrometry: Capabilities for In Situ Analysis; Potts, P.J., West, M., Eds.; The Royal Society of Chemistry: London, UK, 2008; pp. 1–12. [Google Scholar]
- Potts, P.J.; Webb, P.C. X-ray fluorescence spectrometry. Geoanalysis 1992, 44, 251–296. [Google Scholar] [CrossRef]
- Knesl, I.; Jandova, T.; Rambousek, P.; Breiter, K. Calibration of Portable XRF Spectrometer in Sn-W Ore-Bearing Granites: Application in the Cínovec Deposit (Erzgebirge/Krušné Hory Mts., Czech Republic). Inżynieria Miner. 2015, 16, 67–72. [Google Scholar] [CrossRef]
- Steiner, A.E.; Conrey, R.M.; Wolff, J.A. PXRF calibrations for volcanic rocks and the application of in-field analysis to the geosciences. Chem. Geol. 2017, 453, 35–54. [Google Scholar] [CrossRef]
- Mauriohooho, K.; Barker, S.L.; Rae, A. Mapping lithology and hydrothermal alteration in geothermal systems using portable X-ray fluorescence (pXRF): A case study from the Tauhara geothermal system, Taupo Volcanic Zone. Geothermics 2016, 64, 125–134. [Google Scholar] [CrossRef]
- Gazley, M.F.; Tutt, C.M.; Brisbout, L.I.; Fisher, L.A.; Duclaux, G. Application of portable X-ray fluorescence analysis to characterize dolerite dykes at the Plutonic Gold Mine, Western Australia. Geochem. Explor. Environ. Anal. 2014, 14, 223–231. [Google Scholar] [CrossRef]
- Ross, P.-S.; Bourke, A.; Mercier-Langevin, P.; Lépine, S.; Leclerc, F.; Boulerice, A. High-Resolution Physical Properties, Geochemistry, and Alteration Mineralogy for the Host Rocks of the Archean Lemoine Auriferous Volcanogenic Massive Sulfide Deposit, Canada. Econ. Geol. 2016, 111, 1561–1574. [Google Scholar] [CrossRef] [Green Version]
- Fisher, L.; Gazley, M.F.; Baensch, A.; Barnes, S.J.; Cleverley, J.; Duclaux, G. Resolution of geochemical and lithostratigraphic complexity: A workflow for application of portable X-ray fluorescence to mineral exploration. Geochem. Explor. Environ. Anal. 2014, 14, 149–159. [Google Scholar] [CrossRef]
- Durance, P.; Jowitt, S.M.; Bush, K. An assessment of portable X-ray fluorescence spectroscopy in mineral exploration, Kurnalpi Terrane, Eastern Goldfields Superterrane, Western Australia. Appl. Earth Sci. 2014, 123, 150–163. [Google Scholar] [CrossRef]
- Bourke, A.; Ross, P.-S. Portable X-ray fluorescence measurements on exploration drill-cores: comparing performance on unprepared cores and powders for “whole rock” analysis. Geochem. Explor. Environ. Anal. 2016, 16, 147–157. [Google Scholar] [CrossRef]
- Rowe, H.; Hughes, N.; Robinson, K. The quantification and application of handheld energy-dispersive x-ray fluorescence (ED-XRF) in mudrock chemostratigraphy and geochemistry. Chem. Geol. 2012, 324, 122–131. [Google Scholar] [CrossRef]
- Hall, G.; Buchar, A.; Bonham-Carter, G. Quality Control Assessment of Portable XRF Analysers: Development of Standard Operating Procedures, Performance on Variable Media and Recommended Uses; CAMIRO Project 10E01 Phase 1; Canadian Mining Industry Research Organization (CAMIRO) Exploration Division: Toronto, ON, Canada, 2012; Available online: https://www.appliedgeochemists.org/images/stories/XRF/pXRF%20Report%20Phase%20I%20Report%20rev%20Oct%202013.pdf (accessed on 18 July 2018).
- Hall, G.; Buchar, A.; Bonham-Carter, G. Quality Control Assessment of Portable XRF Analysers: Development of Standard Operating Procedures, Performance on Variable Media and Recommended Uses; CAMIRO Project 10E01 Phase 2; Canadian Mining Industry Research Organization (CAMIRO) Exploration Division: Toronto, ON, Canada, 2013; Available online: https://www.appliedgeochemists.org/images/stories/XRF/pXRF2-Report_Dec_29_2013.pdf (accessed on 18 July 2018).
- Cameron, M. Improving Handheld XRF Performance in Geological Samples. In Proceedings of the Denver X-ray Conference, Big Sky, MT, USA, 31 July–4 August 2017. [Google Scholar]
- Andrew, B.S.; Barker, S.L.L. Determination of carbonate vein chemistry using portable X-ray fluorescence and its application to mineral exploration. Geochem. Explor. Environ. Anal. 2018, 18, 85. [Google Scholar] [CrossRef]
- Haschke, M. XRF-Basics. In Laboratory Micro-X-ray Fluorescence Spectroscopy: Instrumentation and Applications; Haschke, M., Ed.; Springer International Publishing: Cham, Switzerland, 2014; pp. 1–17. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CRM | Type | CRM Group | Certifying Agency | Critical Elements 1 |
|---|---|---|---|---|
| BCR 144 | Sewage sludge | Other CRM | IRMM | P |
| BCR 032 | Moroccan phosphate rock | Other CRM | IRMM | P |
| CGL 126 | Rare earth ore | REE-rich CRM | CGL | Hf, Nb, REE, Ta, W |
| COQ-1 | Carbonatite | REE-rich CRM | USGS | Ba, Nb, P, REE |
| IAG CRM3 | Alkaline Granite | Other CRM | CGL | Bi, Nb, Ta |
| DGPM-1 | Disseminated gold ore | Ore | USGS | Sb,W |
| GTS-2a | Au ore mill tailings | Mine waste | CCRMP | W |
| MAG-1 | Marine sediment | Sediment | USGS | |
| MP-1b | Zn-Sn-Cu-Pb ore | Ore | CCRMP | Bi, Sb, W |
| MP-2a | W-Mo ore | Ore | CCRMP | Nb, REE, Sb, Ta, W |
| NIST 1646a | Estuarine sediment | Sediment | NIST | |
| NIST 2704 | Buffalo River Sediment | Sediment | NIST | |
| NIST 2780 | Hard rock mine waste | Mine waste | NIST | Ba, Sb, V, W |
| REE-1 | REEs, Zr and Nb ore | REE-rich CRM | CCRMP | Hf, Nb, REE, Ta |
| REE-2 | Carbonatite with REEs | REE-rich CRM | CCRMP | Nb, REE, P |
| RTS-3a | Sulphide ore mill tailings | Mine waste | CCRMP | Bi, Co |
| SARM-7 | Platinum ore | Ore | SACCRM | Co, PGE |
| TILL-1 | Soil sample | Sediment | CCRMP | Ba, Hf, Sb |
| TILL-2 | Till sample | Sediment | CCRMP | Hf |
| TILL-3 | Soil sample | Sediment | CCRMP | |
| TILL-4 | Till sample | Sediment | CCRMP | Bi, Hf, W |
| LOD | ||||||
|---|---|---|---|---|---|---|
| Element | SiO2 Matrix 1 | Sediments 2 | REE-Rich CRMs 2 | Ores 2 | Mine Wastes 2 | Other CRMs 2 |
| P2O5 | 90 | 160 | 430 | 350 | 330 | 120 |
| La | 341.54 | 343 | high conc 3 | 373 | 374 | 370 |
| Ce | 85 | 93 | high conc 3 | 107 | 126 | 115 |
| Ba | 188.4 | 168 | 232 | 219 | 241 | 184 |
| V | 8 | 13 | 12 | 9 | 29 | 11 |
| Co | 5 | 5 | 5 | 3 | 4 | 18 |
| Nb | 2 | 5 | high conc 3 | 6 | 9 | 5 |
| Zr | 3 | 7 | 7 | 4 | 7 | 5 |
| Hf | 6 | 22 | 20 | 24 | 22 | 25 |
| Ta | 4 | 9 | 10 | 7 | 13 | 11 |
| W | 6 | 7 | 10 | 8 | 14 | 7 |
| Bi | 12 | 15 | 20 | 17 | 16 | 14 |
| P2O5 (wt %) | La (wt %) | Ce (wt %) | Ba (wt %) | V (wt %) | ||||||||||||||||
| CRM | Mean | REF | RSD% | %diff | Mean | REF | RSD% | %diff | Mean | REF | RSD% | %diff | Mean | REF | RSD% | %diff | Mean | REF | RSD% | %diff |
| DGPM-1 | 0.08 | 13.6 | 0.1144 | 3.6 | 0.0049 | 4.3 | ||||||||||||||
| MP-1b | 0.06 | 0.05 | 23.6 | 35.6 | ||||||||||||||||
| MP-2a | 0.06 | 0.02 | 18.2 | 213.3 | 0.0157 | 0.0244 | 0.0357 | 9.4 | −31.5 | 0.0012 | 0.0032 | 9.6 | ||||||||
| SARM-7 | 0.06 | 0.10 | 20.7 | −43.5 | 0.0005 | 0.0050 | 0.0184 | 0.0060 | 3.5 | 206.7 | ||||||||||
| BCR 032 | 40.46 | 32.98 | 0.5 | 22.7 | 0.0152 | 0.0153 | 8.3 | −0.9 | ||||||||||||
| BCR 144 | 5.93 | 5.08 | 0.7 | 16.7 | 0.0246 | 27.9 | 0.0195 | 14.1 | 0.0065 | 18.3 | ||||||||||
| IAG CRM3 | 0.04 | 0.03 | 9.2 | 43.1 | 0.0008 | 0.0027 | 0.0012 | 0.0016 | 4.6 | |||||||||||
| CGL 126 | 0.03 | 0.0419 | 0.0434 | 8.5 | −3.5 | 0.0862 | 0.1000 | 4.8 | −13.8 | 0.0095 | 0.0050 | 11.1 | ||||||||
| COQ-1 | 2.28 | 2.60 | 3.3 | −12.1 | 0.0561 | 0.0750 | 6.8 | −25.2 | 0.0493 | 0.1700 | 12.9 | −71.0 | 0.0776 | 0.1000 | 5.6 | −22.4 | 0.0191 | 0.0110 | 12.5 | 74.0 |
| REE-1 | 0.37 | 0.06 | 5.0 | 525.3 | 0.0747 | 0.1661 | 14.1 | −55.0 | 0.2925 | 0.3960 | 4.0 | −26.1 | 0.0100 | 0.0141 | 0.0010 | 3.0 | 1320.2 | |||
| REE-2 | 1.39 | 1.06 | 2.3 | 31.8 | 0.3477 | 0.5130 | 4.6 | −32.2 | 1.6934 | 0.9610 | 2.1 | 76.2 | 3.0911 | 5.0200 | 1.6 | −38.4 | 0.0509 | 0.0079 | 7.9 | 544.6 |
| MAG-1 | 0.11 | 0.16 | 11.5 | −31.7 | 0.0043 | 0.0088 | 0.0577 | 0.0480 | 9.1 | 20.3 | 0.0061 | 0.0140 | 27.4 | −56.7 | ||||||
| NIST 1646a | 0.06 | 0.06 | 8.2 | 2.9 | 0.0017 | 0.0034 | 0.0015 | 0.0045 | 27.4 | −65.7 | ||||||||||
| NIST 2704 | 0.22 | 0.23 | 3.6 | −5.4 | 0.0029 | 0.0072 | 0.0335 | 0.0414 | 15.0 | −19.1 | 0.0070 | 0.0095 | 4.6 | −25.9 | ||||||
| TILL-1 | 0.27 | 0.22 | 2.6 | 21.9 | 0.0028 | 0.0186 | 0.0071 | 27.5 | 161.6 | 0.0646 | 0.0702 | 4.0 | −7.9 | 0.0056 | 0.0099 | 24.1 | −43.0 | |||
| TILL-2 | 0.15 | 0.17 | 6.8 | −12.7 | 0.0044 | 0.0169 | 0.0098 | 20.2 | 72.4 | 0.0393 | 0.0540 | 6.6 | −27.3 | 0.0052 | 0.0077 | 10.7 | −32.7 | |||
| TILL-3 | 0.12 | 0.11 | 5.1 | 5.4 | 0.0021 | 0.0042 | 0.0426 | 0.0489 | 18.0 | −13.0 | 0.0040 | 0.0062 | 14.5 | −34.8 | ||||||
| TILL-4 | 0.21 | 0.20 | 3.9 | 2.7 | 0.0041 | 0.0078 | 0.0353 | 0.0395 | 21.3 | −10.5 | 0.0045 | 0.0067 | 20.7 | −32.8 | ||||||
| GTS-2a | 0.19 | 0.20 | 2.9 | −9.0 | 0.0009 | 0.0024 | 0.0186 | 0.0120 | 0.0166 | 11.9 | −27.6 | |||||||||
| NIST 2780 | 0.09 | 0.10 | 11.9 | −6.0 | 0.0038 | 0.0064 | 0.0829 | 0.0993 | 9.8 | −16.6 | 0.0161 | 0.0268 | 4.0 | −40.1 | ||||||
| RTS-3a | 0.10 | 0.10 | 17.2 | −2.0 | 0.001 | 0.0030 | 0.0106 | 0.0089 | 0.0120 | 10.8 | −25.8 | |||||||||
| Co (wt %) | Nb (wt %) | Zr (wt %) | W (wt %) | Bi (wt %) | ||||||||||||||||
| CRM | Mean | REF | RSD% | %diff | Mean | REF | RSD% | %diff | Mean | REF | RSD% | %diff | Mean | REF | RSD% | %diff | Mean | REF | RSD% | %diff |
| DGPM-1 | 0.0005 | 12.8 | 0.0327 | 2.9 | 0.0028 | 0.0076 | 7.1 | −63.2 | ||||||||||||
| MP-1b | 0.0142 | 0.0004 | 15.1 | 3460.0 | 0.0038 | 17.2 | 0.0190 | 0.0150 | 9.3 | 26.8 | 0.0915 | 0.1100 | 2.0 | −16.8 | 0.1023 | 0.0954 | 4.7 | 7.2 | ||
| MP-2a | 0.0007 | 0.0006 | 14.3 | 27.3 | 0.0053 | 0.0097 | 5.1 | −45.6 | 0.0123 | 0.0134 | 1.1 | −8.1 | 0.1091 | 0.3380 | 2.1 | −67.7 | 0.0944 | 0.0989 | 2.2 | −4.5 |
| SARM-7 | 0.0101 | 0.0036 | 7.8 | 180.6 | 0.0001 | 0.0048 | 0.0010 | 11.2 | 376.0 | |||||||||||
| BCR 032 | 0.0001 | 0.0126 | 1.8 | |||||||||||||||||
| BCR 144 | 0.0009 | 0.0040 | 7.0 | 0.0030 | 7.6 | |||||||||||||||
| IAG CRM3 | 0.0011 | 0.0064 | 14.4 | −82.8 | 0.0031 | 0.0040 | 5.1 | −22.7 | 0.0132 | 6.9 | ||||||||||
| CGL 126 | 0.1297 | 0.1700 | 1.6 | −23.7 | 1.3885 | 1.5800 | 1.5 | −12.1 | 0.0088 | 0.0093 | 25.9 | |||||||||
| COQ-1 | 0.0005 | 0.4104 | 0.3900 | 1.2 | 5.2 | 0.1234 | 0.0065 | 0.9 | 1798.8 | |||||||||||
| REE-1 | 0.0010 | 0.0002 | 16.1 | 545.6 | 0.2692 | 0.4050 | 2.2 | −33.5 | 1.5646 | 1.9100 | 1.6 | −18.1 | 0.0028 | 0.0010 | 12.9 | 175.0 | 0.0413 | 0.0001 | 7.2 | 63304.9 |
| REE-2 | 0.1290 | 0.1060 | 2.1 | 21.7 | 0.0298 | 0.0032 | 0.8 | 826.1 | 0.0010 | 0.0267 | 0.0002 | 6.3 | 13230.0 | |||||||
| MAG-1 | 0.0040 | 0.0020 | 7.6 | 99.0 | 0.0008 | 0.0012 | 8.8 | −33.3 | 0.0107 | 0.0130 | 2.2 | −17.4 | 0.0001 | 0.0021 | 0.00003 | 6.7 | 6076.5 | |||
| NIST 1646a | 0.0004 | 12.4 | 0.0361 | 0.9 | ||||||||||||||||
| NIST 2704 | 0.0009 | 0.0014 | 10.4 | −38.6 | 0.0007 | 7.9 | 0.0226 | 0.0300 | 4.4 | −24.7 | ||||||||||
| TILL-1 | 0.0012 | 0.0018 | 27.1 | −34.4 | 0.0005 | 0.0010 | 0.0 | −50.0 | 0.0390 | 0.0502 | 1.9 | −22.4 | 0.0001 | 0.0005 | ||||||
| TILL-2 | 0.0008 | 0.0015 | 22.9 | −48.0 | 0.0008 | 0.0020 | 21.6 | −58.0 | 0.0279 | 0.0390 | 1.9 | −28.5 | 0.0005 | 0.0022 | 0.0005 | 13.2 | 348.0 | |||
| TILL-3 | 0.0189 | 0.0230 | 1.5 | −17.8 | 0.0001 | 0.0005 | ||||||||||||||
| TILL-4 | 0.0006 | 0.0008 | 9.8 | −30.0 | 0.0007 | 0.0015 | 17.3 | −56.0 | 0.0291 | 0.0385 | 1.9 | −24.4 | 0.0072 | 0.0204 | 2.1 | −64.9 | 0.0056 | 0.0040 | 7.7 | 41.0 |
| GTS-2a | 0.0062 | 0.0022 | 7.0 | 179.6 | 0.0110 | 0.0114 | 1.2 | −3.7 | 0.0011 | 0.0026 | 9.5 | −59.3 | 0.0000 | |||||||
| NIST 2780 | 0.0002 | 0.0002 | 72.4 | −18.2 | 0.0018 | 0.0167 | 0.0176 | 2.4 | −5.1 | 0.0024 | 0.0047 | 21.5 | ||||||||
| RTS-3a | 0.0143 | 0.0004 | 0.0072 | 0.0078 | 3.0 | −7.9 | 0.0030 | 0.0002 | 8.8 | 1400.0 | 0.0048 | 0.0031 | 14.7 | 52.1 | ||||||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallhofer, D.; Lottermoser, B.G. The Influence of Spectral Interferences on Critical Element Determination with Portable X-Ray Fluorescence (pXRF). Minerals 2018, 8, 320. https://doi.org/10.3390/min8080320
Gallhofer D, Lottermoser BG. The Influence of Spectral Interferences on Critical Element Determination with Portable X-Ray Fluorescence (pXRF). Minerals. 2018; 8(8):320. https://doi.org/10.3390/min8080320
Chicago/Turabian StyleGallhofer, Daniela, and Bernd G. Lottermoser. 2018. "The Influence of Spectral Interferences on Critical Element Determination with Portable X-Ray Fluorescence (pXRF)" Minerals 8, no. 8: 320. https://doi.org/10.3390/min8080320
APA StyleGallhofer, D., & Lottermoser, B. G. (2018). The Influence of Spectral Interferences on Critical Element Determination with Portable X-Ray Fluorescence (pXRF). Minerals, 8(8), 320. https://doi.org/10.3390/min8080320