Crystal Chemistry of Sulfates from the Apuan Alps (Tuscany, Italy). V. Scordariite, K8(Fe3+0.67□0.33)[Fe3+3O(SO4)6(H2O)3]2(H2O)11: A New Metavoltine-Related Mineral





Abstract
:1. Introduction
2. Occurrence and Physical Properties
2.1. Physical Properties
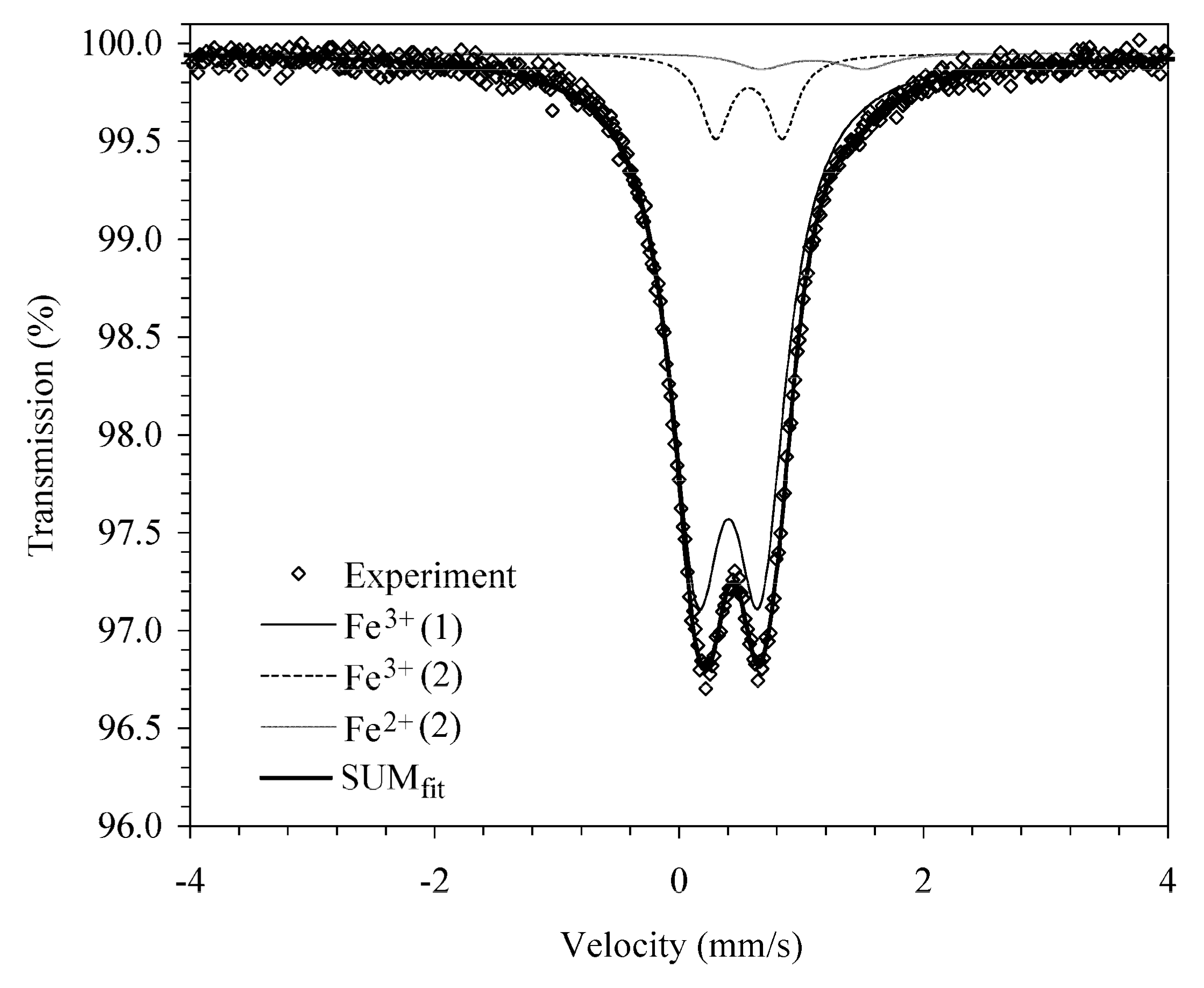
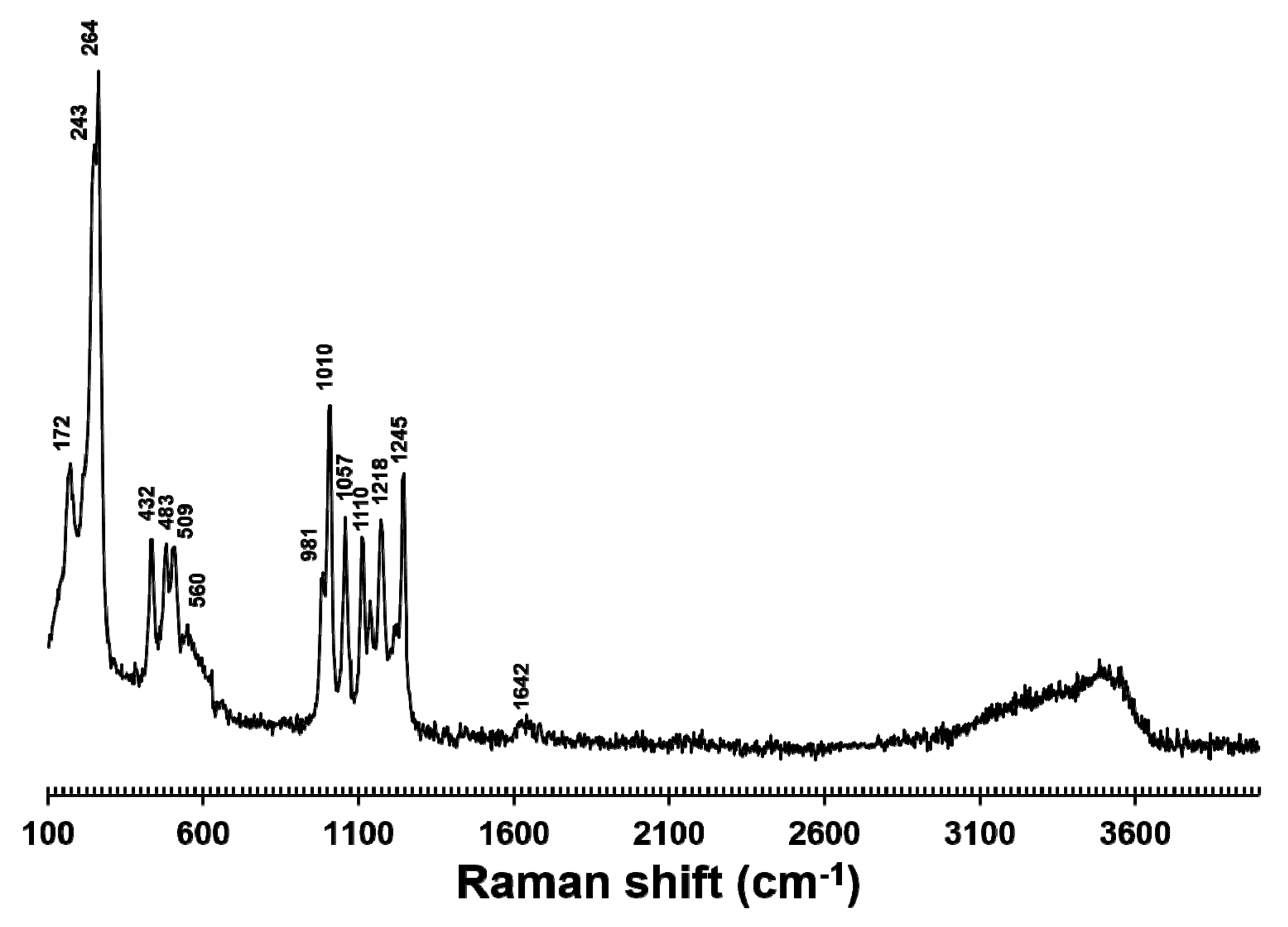
2.2. Chemical and Spectroscopic Data
2.3. X-ray Crystallography
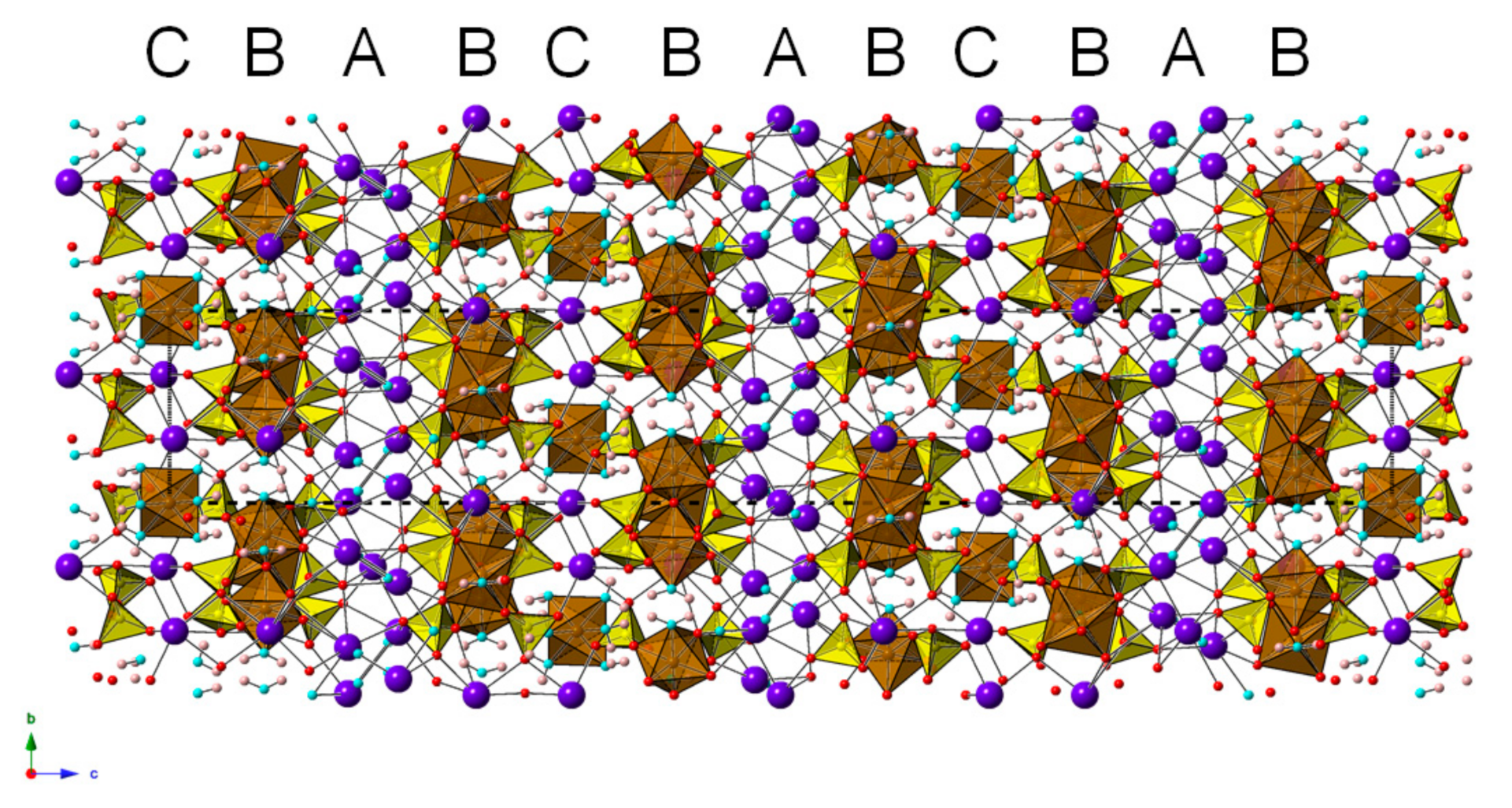
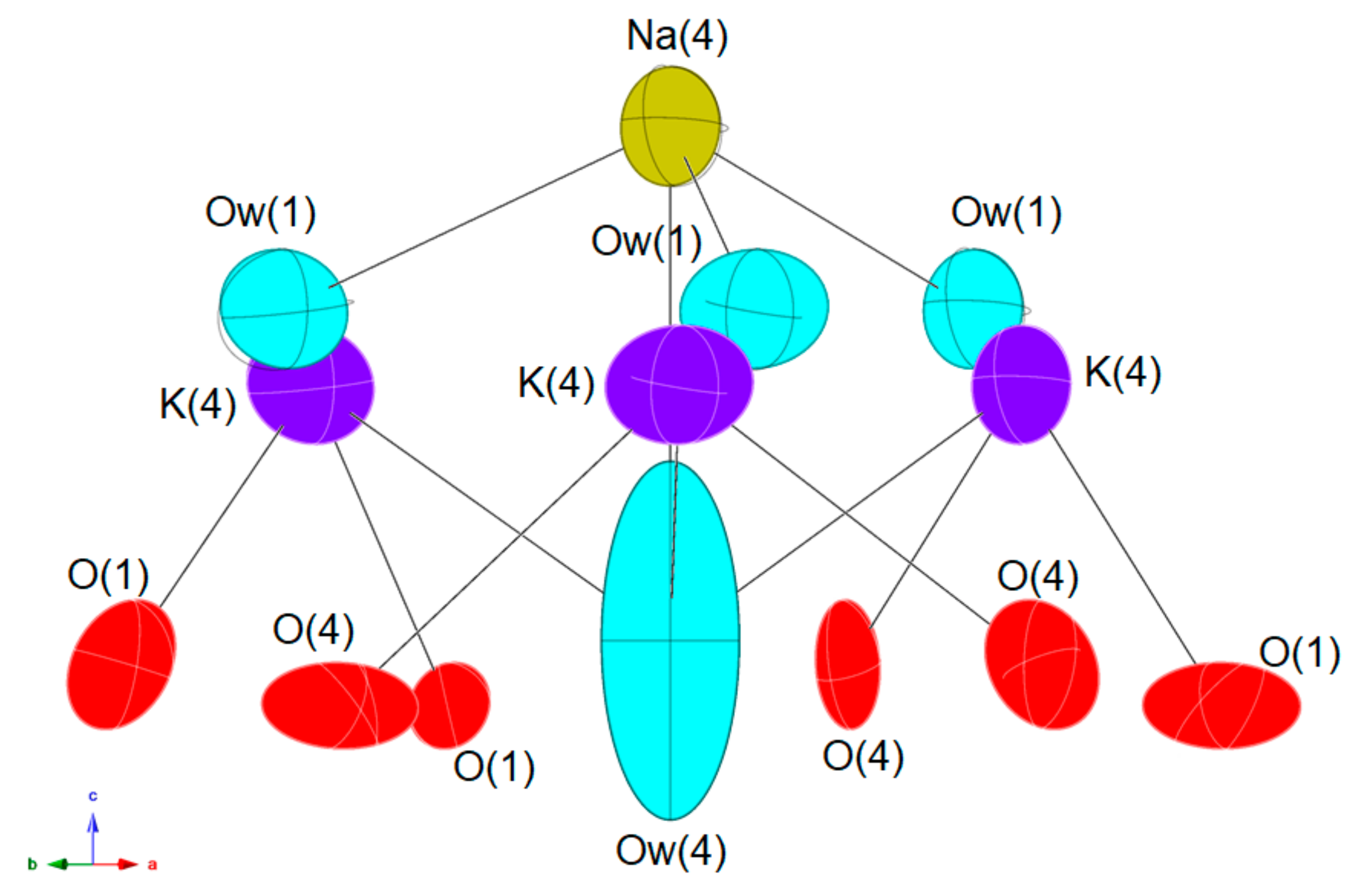
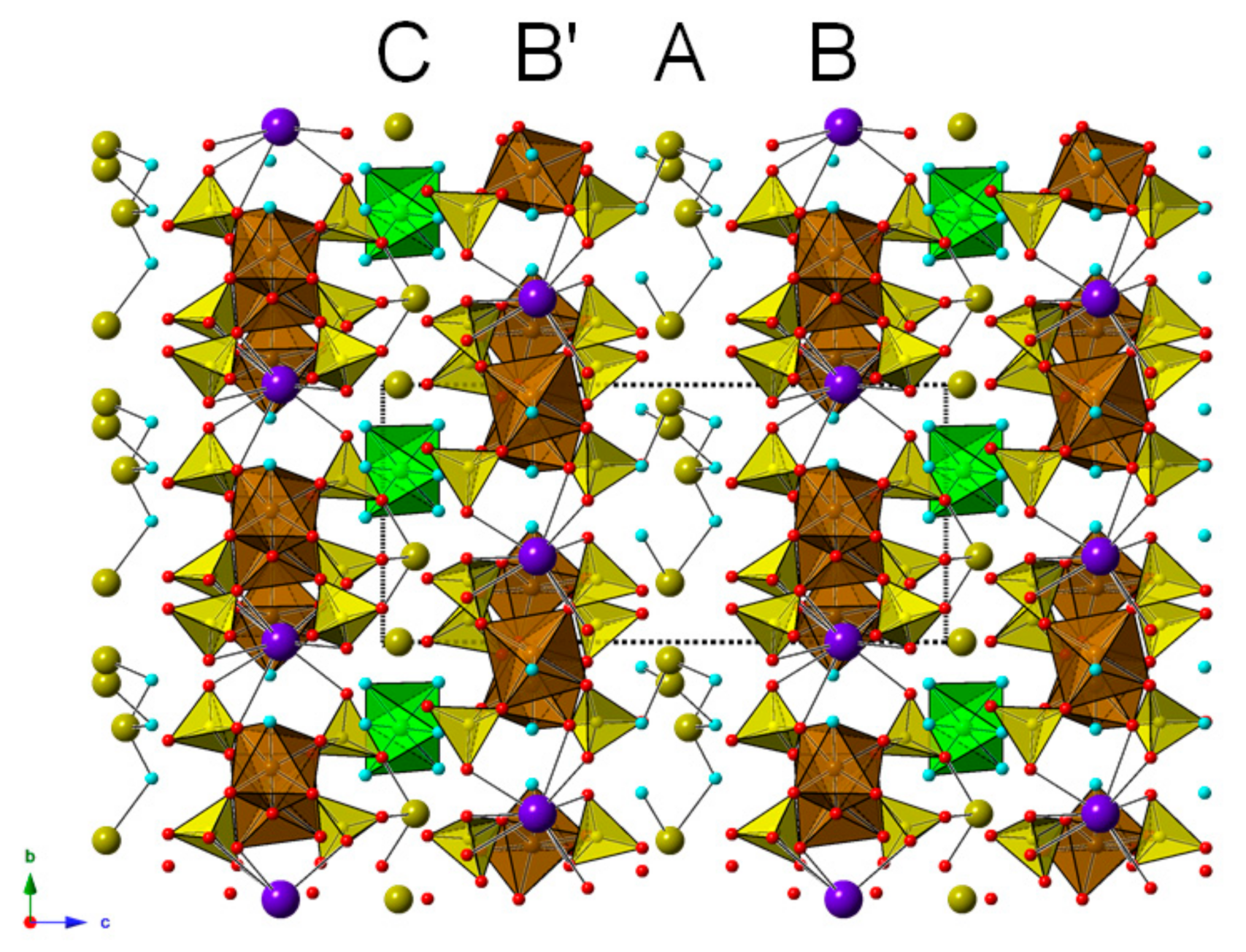
3. Crystal Structure Description
4. Discussion
4.1. The Hydration State of Scordariite
4.2. Scordariite and Metavoltine: A Comparison
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Biagioni, C.; D’Orazio, M.; Vezzoni, S.; Dini, A.; Orlandi, P. Mobilization of Tl-Hg-As-Sb-(Ag,Cu)-Pb sulfosalt melts during low-grade metamorphism in the Alpi Apuane (Tuscany, Italy). Geology 2013, 41, 747–750. [Google Scholar] [CrossRef]
- Biagioni, C.; Orlandi, P.; Pasero, M. Ankangite from the Monte Arsiccio mine (Apuan Alps, Tuscany, Italy): Occurrence, crystal structure and classification problems in cryptomelane group minerals. Period. Mineral. 2009, 78, 3–11. [Google Scholar]
- Biagioni, C.; Orlandi, P.; Pasero, M.; Nestola, F.; Bindi, L. Mapiquiroite, (Sr,Pb)(U,Y)Fe2(Ti,Fe3+)18O38, a new member of the crichtonite group from the Apuan Alps, Tuscany, Italy. Eur. J. Mineral. 2014, 26, 427–437. [Google Scholar] [CrossRef]
- Biagioni, C.; Bindi, L.; Mauro, D.; Pasero, M. Giacovazzoite, IMA 2018-165. CNMNC Newsletter No. 49. Eur. J. Mineral. 2019, 31. [Google Scholar] [CrossRef]
- Biagioni, C.; Bindi, L.; Kampf, A.R. Magnanelliite, IMA 2019-006. CNMNC Newsletter No. 49. Eur. J. Mineral. 2019, 31. [Google Scholar] [CrossRef]
- Scordari, F.; Stasi, F.; Schingaro, E.; Comunale, G. A survey of (Na,H3O+,K)5Fe3O(SO4)6·H2O compounds: Architectural principles and influence of the Na-K replacement on their structures. Crystal structure, solid-state transformation and its relationship to some analogues. Z. Kristallogr. 1994, 209, 733–737. [Google Scholar]
- Costagliola, P.; Benvenuti, M.; Tanelli, G.; Cortecci, G.; Lattanzi, P. The barite-pyrite-iron oxides deposit of Monte Arsiccio (Apuane Alps). Geological setting, mineralogy, fluid inclusions, stable isotopes and genesis. Boll. Soc. Geol. Ital. 1990, 109, 267–277. [Google Scholar]
- Molli, G.; Vitale-Brovarone, A.; Beyssac, O.; Cinquini, I. RSCM thermometry in the Alpi Apuane (NW Tuscany, Italy): New constraints for the metamorphic and tectonic history of the inner northern Apennines. J. Struct. Geol. 2018, 113, 200–216. [Google Scholar] [CrossRef]
- Anthony, J.W.; Bideaux, R.A.; Bladh, K.W.; Nichols, M.C. Handbook of Mineralogy. Volume V. Borates, Carbonates, Sulfates; Mineralogical Society of America: Chantilly, VA, USA, 2003; p. 791. [Google Scholar]
- Mandarino, J.A. The Gladstone-Dale relationship. Part III. Some general applications. Can. Mineral. 1979, 17, 71–76. [Google Scholar]
- Mandarino, J.A. The Gladstone-Dale relationship. Part IV. The compatibility concept and its application. Can. Mineral. 1981, 19, 441–450. [Google Scholar]
- Prescher, C.; McCammon, C.; Dubrovinsky, L. MossA: A program for analyzing energy-domain Mössbauer spectra from conventional and synchrotron sources. J. Appl. Crystallogr. 2012, 45, 329–331. [Google Scholar] [CrossRef]
- Laugier, J.; Bochu, B. CELREF: Unit Cell Refinement Program from Powder Diffraction Diagram; Laboratoires des Matériaux et du Génie Physique, Ecole Nationale Supériore de Physique de Grenoble (INPG): Grenoble, France, 1999. [Google Scholar]
- Bruker AXS Inc. APEX 3. Bruker Advanced X-Ray Solutions; Bruker AXS Inc.: Madison, WI, USA, 2016. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.C. (Ed.) International Tables for Crystallography. Volume C: Mathematical, Physical and Chemical Tables; Kluwer Academic: Dordrecht, The Netherlands, 1992. [Google Scholar]
- Kraus, W.; Nolze, G. PowderCell—A program for the representation and manipulation of crystal structures and calculation of the resulting X-ray powder patterns. J. Appl. Crystallogr. 1996, 29, 301–303. [Google Scholar] [CrossRef]
- Brese, N.E.; O’Keeffe, M. Bond-valence parameters for solids. Acta Crystallogr. 1991, 47, 192–197. [Google Scholar] [CrossRef]
- Giacovazzo, C.; Scordari, F.; Todisco, A.; Menchetti, S. Crystal structure model for metavoltine from Sierra Gorda. Tschermaks Mineral. Petrogr. Mitt. 1976, 23, 155–166. [Google Scholar] [CrossRef]
- Ferraris, G.; Ivaldi, G. Bond valence vs bond length in O…O hydrogen bonds. Acta Crystallogr. 1988, 44, 341–344. [Google Scholar] [CrossRef]
- Kampf, A.R.; Richards, R.P.; Nash, B.P.; Murowchick, J.B.; Rakovan, J.F. Carlsonite, (NH4)5Fe3+3O(SO4)6·7H2O, and huizingite-(Al), (NH4)9Al3(SO4)8(OH)2·4H2O, two new minerals from a natural fire in an oil-bearing shale near Milan, Ohio. Am. Mineral. 2016, 101, 2095–2107. [Google Scholar] [CrossRef]
- Biagioni, C.; Bindi, L.; Mauro, D. Scordariite, IMA 2019-010. CNMNC Newsletter No. 50. Eur. J. Mineral. 2019, 31. [Google Scholar] [CrossRef]
- Blass, J. Beiträge zur Kenntniss natürlicher wasserhaltiger Doppelsulfate. Sitz. Mat. Nat. Cl. Kais. Akad. Wiss. 1883, 87, 141–163. [Google Scholar]
- Scordari, F.; Vurro, F.; Menchetti, S. The metavoltine problem: Relationships between metavoltine and Maus’ salt. Tschermaks Mineral. Petrogr. Mitt. 1975, 22, 88–97. [Google Scholar] [CrossRef]
- Scordari, F. The metavoltine problem: Metavoltine from Madeni Zakh and Chuquicamata, and a related artificial compound. Mineral. Mag. 1977, 41, 371–374. [Google Scholar] [CrossRef]
- Mills, S.J.; Hatert, F.; Nickel, E.H.; Ferraris, G. The standardization of mineral group hierarchies: Application to recent nomenclature proposals. Eur. J. Mineral. 2009, 21, 1073–1080. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mineral | Chemical Formula | Mineral | Chemical Formula |
|---|---|---|---|
| Alum-(K) | KAl(SO4)2·12H2O | Jarosite | KFe3+3(SO4)2(OH)6 |
| Alunogen | Al2(SO4)3(H2O)12·5H2O | Khademite | Al(SO4)F·5H2O |
| Copiapite group | Krausite | KFe3+(SO4)2·H2O | |
| Coquimbite | AlFe3+3(SO4)6(H2O)12·6H2O | Magnanelliite | K3Fe3+2(SO4)4(OH)(H2O) |
| Giacovazzoite | K5Fe3+3O(SO4)6(H2O)9·H2O | Melanterite | FeSO4·7H2O |
| Goldichite | KFe3+(SO4)2·4H2O | Römerite | Fe2+Fe3+2(SO4)4·14H2O |
| Gypsum | CaSO4·2H2O | Scordariite | K8(Fe3+0.67□0.33)[Fe3+3O(SO4)6(H2O)3]2(H2O)11 |
| Halotrichite | FeAl2(SO4)4·22H2O | Voltaite | K2Fe2+5Fe3+3Al(SO4)12·18H2O |
| Oxide | Wt % (n = 5) | Range | e.s.d. |
|---|---|---|---|
| SO3 | 47.31 | 46.36–48.11 | 1.88 |
| Al2O3 | 0.66 | 0.41–0.83 | 0.12 |
| Fe2O3(tot) | 25.44 | 24.22–26.01 | 1.65 |
| Fe2O3 * | 24.68 | ||
| FeO * | 0.69 | ||
| Na2O | 0.52 | 0.30–0.66 | 0.13 |
| K2O | 17.36 | 16.88–18.01 | 1.32 |
| Sum | 91.22 | ||
| H2Ocalc | 15.06 | ||
| Total | 106.28 |
| Iobs | dobs | Icalc | dcalc | h k l | Iobs | dobs | Icalc | dcalc | h k l | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| mw | 8.8 | 72 | 8.95 | 0 0 6 | w | 2.823 |  | 11 | 2.840 | 2 0 14 | |
| s | 8.3 | 100 | 8.35 | 1 0 1 | 5 | 2.817 | 3 0 0 | ||||
| vw | 7.1 | 5 | 7.15 | 1 0 4 | w | 2.746 | 6 | 2.745 | 1 2 −10 | ||
| m | 6.6 | 17 | 6.64 | 1 0 −5 | vw | 2.683 |  | 1 | 2.687 | 3 0 6 | |
| w | 5.7 | 6 | 5.68 | 1 0 7 | 1 | 2.687 | 3 0 −6 | ||||
| vw | 5.3 | 0.1 | 5.26 | 1 0 −8 | vw | 2.621 | 0.2 | 2.628 | 2 0 −16 | ||
| w | 4.89 | 12 | 4.879 | 1 1 0 | w | 2.555 | 5 | 2.547 | 3 0 −9 | ||
| w | 4.72 | 8 | 4.707 | 1 1 3 | w | 2.422 | 9 | 2.417 | 2 2 3 | ||
| vw | 4.50 | 3 | 4.532 | 1 0 10 | w | 2.342 |  | 5 | 2.342 | 1 3 1 | |
| w | 4.29 |  | 1 | 4.284 | 1 1 6 | 9 | 2.335 | 1 3 −2 | |||
| 1 | 4.284 | 1 1 −6 | w | 2.318 | 4 | 2.314 | 2 1 16 | ||||
| mw | 4.22 |  | 8 | 4.226 | 1 0 −11 | w | 2.258 |  | 2 | 2.258 | 2 2 9 |
| 6 | 4.212 | 2 0 −1 | 4 | 2.250 | 1 0 −23 | ||||||
| w | 4.04 | 5 | 4.030 | 2 0 −4 | w | 2.216 |  | 2 | 2.237 | 0 0 24 | |
| vw | 3.930 | 9 | 3.932 | 2 0 5 | 2 | 2.214 | 3 0 15 | ||||
| m | 3.777 |  | 14 | 3.777 | 1 1 −9 | 3 | 2.213 | 1 3 −8 | |||
| 16 | 3.777 | 1 1 9 | vw | 2.151 | 2 | 2.148 | 3 1 −10 | ||||
| w | 3.581 | 9 | 3.576 | 2 0 8 | vw | 2.115 | 2 | 2.116 | 1 2 −19 | ||
| m | 3.299 | 35 | 3.297 | 1 1 12 | vw | 2.040 |  | 2 | 2.048 | 3 0 18 | |
| m | 3.189 |  | 7 | 3.195 | 2 0 11 | 2 | 2.038 | 3 1 −13 | |||
| 12 | 3.189 | 1 2 −1 | vw | 1.988 | 3 | 1.988 | 0 0 27 | ||||
| 13 | 3.172 | 1 2 2 | vw | 1.936 |  | 2 | 1.934 | 2 3 2 | |||
| w | 3.065 | 12 | 3.062 | 1 2 5 | 2 | 1.921 | 1 3 16 | ||||
| w | 2.956 |  | 13 | 2.953 | 2 0 −13 | vw | 1.888 | 3 | 1.888 | 2 2 −18 | |
| 6 | 2.949 | 1 2 −7 | w | 1.847 | 8 | 1.844 | 4 1 0 | ||||
| s | 2.884 |  | 12 | 2.886 | 1 1 15 | vw | 1.812 | ||||
| 24 | 2.886 | 1 1 −15 | vw | 1.790 | |||||||
| 12 | 2.884 | 2 1 −8 | vw | 1.762 | |||||||
| 13 | 2.884 | 1 2 8 | vw | 1.735 |
| Crystal Data | |
|---|---|
| Crystal size (mm) | 0.190 × 0.100 × 0.040 |
| Space group | R-3 |
| a (Å) | 9.7583(12) |
| c (Å) | 53.687(7) |
| V (Å3) | 4427.4(12) |
| Z | 3 |
| Data collection and Refinement | |
| Radiation, wavelength (Å) | MoKα, 0.71073 |
| Temperature (K) | 293 |
| 2θmax (°) | 55.29 |
| Measured reflections | 20268 |
| Unique reflections | 2156 |
| Reflections with Fo > 4σ(Fo) | 1980 |
| Rint | 0.0173 |
| Rσ | 0.0316 |
| Range of h, k, l | −12 ≤ h ≤ 7, −9 ≤ k ≤ 11, −69 ≤ l ≤ 63 |
| R [Fo > 4σ(Fo)] | 0.0571 |
| R (all data) | 0.0624 |
| wR (on Fo2) | 0.1405 |
| Goof | 1.151 |
| Number of least-squares parameters | 165 |
| Maximum and minimum residual peak (e Å−3) | 1.16 [at 0.87 Å from Na(4)] −1.44 [at 0.03 Å from Na(4)] |
| Site | Wyckoff Positions | s.o.f. | x | y | z | Ueq |
|---|---|---|---|---|---|---|
| K(1) | 3b | K1.00 | 0 | 0 | ½ | 0.0431(10) |
| K(2) | 6c | K1.00 | 0 | 0 | 0.67094(5) | 0.0343(6) |
| K(3) | 6c | K1.00 | 0 | 0 | 0.25116(6) | 0.0396(7) |
| K(4) | 18f | K0.4167 | 0.2552(7) | 0.0063(6) | 0.14591(9) | 0.0465(11) |
| Na(4) | 6c | Na0.25 | 0 | 0 | 0.1750(5) | 0.0465(11) |
| Fe(1) | 18f | Fe1.00 | 0.16175(9) | 0.45195(9) | 0.07849(2) | 0.0164(2) |
| Fe(2) | 3a | Fe0.66(1) | 0 | 0 | 0 | 0.0190(11) |
| S(1) | 18f | S1.00 | 0.02166(17) | 0.6090(2) | 0.11696(3) | 0.0204(3) |
| S(2) | 18f | S1.00 | 0.02118(17) | 0.60290(19) | 0.04014(3) | 0.0189(3) |
| O(1) | 18f | O1.00 | 0.2342(6) | 0.3909(6) | 0.10990(10) | 0.0337(12) |
| O(2) | 18f | O1.00 | 0.0132(5) | 0.4877(5) | 0.09921(9) | 0.0262(10) |
| O(3) | 18f | O1.00 | 0.0473(7) | 0.5717(8) | 0.14198(10) | 0.0448(15) |
| O(4) | 18f | O1.00 | 0.3893(7) | 0.2635(6) | 0.11451(10) | 0.0331(12) |
| O(5) | 18f | O1.00 | 0.2636(5) | 0.3570(5) | 0.05845(9) | 0.0270(10) |
| O(6) | 18f | O1.00 | 0.0767(6) | 0.4898(6) | 0.04637(9) | 0.0282(10) |
| O(7) | 18f | O1.00 | 0.4715(6) | 0.3235(6) | 0.04257(11) | 0.0385(13) |
| O(8) | 18f | O1.00 | 0.0767(8) | 0.6665(8) | 0.01534(10) | 0.0495(16) |
| O(9) | 6c | O1.00 | 0 | 0 | 0.58722(13) | 0.0154(13) |
| Ow(1) | 18f | O0.50 | 0.274(2) | 0.0594(16) | 0.1544(2) | 0.0465(11) |
| Ow(2) | 18f | O1.00 | 0.2494(7) | 0.0312(6) | 0.07739(11) | 0.0389(13) |
| Ow(3) | 18f | O1.00 | 0.1826(6) | 0.0096(6) | 0.02341(10) | 0.0290(11) |
| Ow(4) | 6c | O1.00 | 0 | 0 | 0.1172(6) | 0.142(9)* |
| H(21) | 18f | H1.00 | 0.160(17) | 0.038(16) | 0.0387(14) | 0.10(5)* |
| H(22) | 18f | H1.00 | 0.172(15) | −0.086(8) | 0.028(2) | 0.08(4)* |
| H(31) | 18f | H1.00 | 0.260(13) | 0.074(12) | 0.0614(10) | 0.06(3)* |
| H(32) | 18f | H1.00 | 0.257(14) | 0.085(12) | 0.0920(12) | 0.07(3)* |
| K(1) | –O(3) | 2.796(6) ×6 | Fe(1) | –O(9) | 1.9223(9) |
| –O(5) | 1.984(4) | ||||
| K(2) | –O(8) | 2.716(7) ×3 | –O(2) | 1.988(5) | |
| –O(7) | 2.772(6) ×3 | –O(6) | 2.027(5) | ||
| average | 2.744 | –O(1) | 2.028(5) | ||
| –Ow(2) | 2.110(5) | ||||
| K(3) | –O(7) | 2.824(6) ×3 | average | 2.010 | |
| –O(4) | 2.993(6) ×3 | ||||
| –O(2) | 3.073(5) ×3 | Fe(2) | –Ow(3) | 2.144(5) ×6 | |
| average | 2.963 | ||||
| S(1) | –O(3) | 1.446(5) | |||
| K(4) | –O(4) | 2.751(7) | –O(4) | 1.454(5) | |
| –O(3) | 2.800(7) | –O(1) | 1.486(5) | ||
| –O(1) | 2.813(7) | –O(2) | 1.489(5) | ||
| –Ow(1) | 2.873(16) | average | 1.469 | ||
| –Ow(4) | 2.903(16) | ||||
| –O(3) | 2.931(9) | S(2) | –O(7) | 1.439(5) | |
| –O(2) | 3.363(7) | –O(8) | 1.455(6) | ||
| average | 2.942 | –O(6) | 1.491(5) | ||
| –O(5) | 1.497(5) | ||||
| Na(4) | –Ow(1) | 2.670(19) ×3 | average | 1.470 | |
| –Ow(4) | 3.10(4) |
| K(1) | K(2) | K(3) | K(4) | Na(4) | Fe(1) | Fe(2) | S(1) | S(2) | Σanions | |
|---|---|---|---|---|---|---|---|---|---|---|
| O(1) | 0.07 | 0.48 | 1.45 | 2.00 | ||||||
| O(2) | 0.08↓×3 | 0.01 | 0.54 | 1.44 | 2.07 | |||||
| O(3) | 0.17↓×6 | 0.07 0.05 | 1.62 | 1.91 | ||||||
| O(4) | 0.10↓×3 | 0.08 | 1.58 | 1.76 | ||||||
| O(5) | 0.54 | 1.43 | 1.97 | |||||||
| O(6) | 0.48 | 1.41 | 1.89 | |||||||
| O(7) | 0.18↓×3 | 0.15↓×3 | 1.65 | 1.98 | ||||||
| O(8) | 0.21↓×3 | 1.58 | 1.79 | |||||||
| O(9) | →×30.64 | 1.92 | ||||||||
| Ow(1) | 0.06 | 0.02 | 0.08 | |||||||
| Ow(2) | 0.39 | 0.39 | ||||||||
| Ow(3) | 0.24↓×6 | 0.24 | ||||||||
| Ow(4) | →×30.05 | 0.01 | 0.16 | |||||||
| Σcations | 1.02 | 1.17 | 0.99 | 0.39 | 0.03 | 3.07 | 1.44 | 6.09 | 6.07 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biagioni, C.; Bindi, L.; Mauro, D.; Hålenius, U. Crystal Chemistry of Sulfates from the Apuan Alps (Tuscany, Italy). V. Scordariite, K8(Fe3+0.67□0.33)[Fe3+3O(SO4)6(H2O)3]2(H2O)11: A New Metavoltine-Related Mineral. Minerals 2019, 9, 702. https://doi.org/10.3390/min9110702
Biagioni C, Bindi L, Mauro D, Hålenius U. Crystal Chemistry of Sulfates from the Apuan Alps (Tuscany, Italy). V. Scordariite, K8(Fe3+0.67□0.33)[Fe3+3O(SO4)6(H2O)3]2(H2O)11: A New Metavoltine-Related Mineral. Minerals. 2019; 9(11):702. https://doi.org/10.3390/min9110702
Chicago/Turabian StyleBiagioni, Cristian, Luca Bindi, Daniela Mauro, and Ulf Hålenius. 2019. "Crystal Chemistry of Sulfates from the Apuan Alps (Tuscany, Italy). V. Scordariite, K8(Fe3+0.67□0.33)[Fe3+3O(SO4)6(H2O)3]2(H2O)11: A New Metavoltine-Related Mineral" Minerals 9, no. 11: 702. https://doi.org/10.3390/min9110702
APA StyleBiagioni, C., Bindi, L., Mauro, D., & Hålenius, U. (2019). Crystal Chemistry of Sulfates from the Apuan Alps (Tuscany, Italy). V. Scordariite, K8(Fe3+0.67□0.33)[Fe3+3O(SO4)6(H2O)3]2(H2O)11: A New Metavoltine-Related Mineral. Minerals, 9(11), 702. https://doi.org/10.3390/min9110702