Understanding Cement Hydration of Cemented Paste Backfill: DFT Study of Water Adsorption on Tricalcium Silicate (111) Surface



Abstract
:
1. Introduction
2. Computational Details
3. Results and Discussion
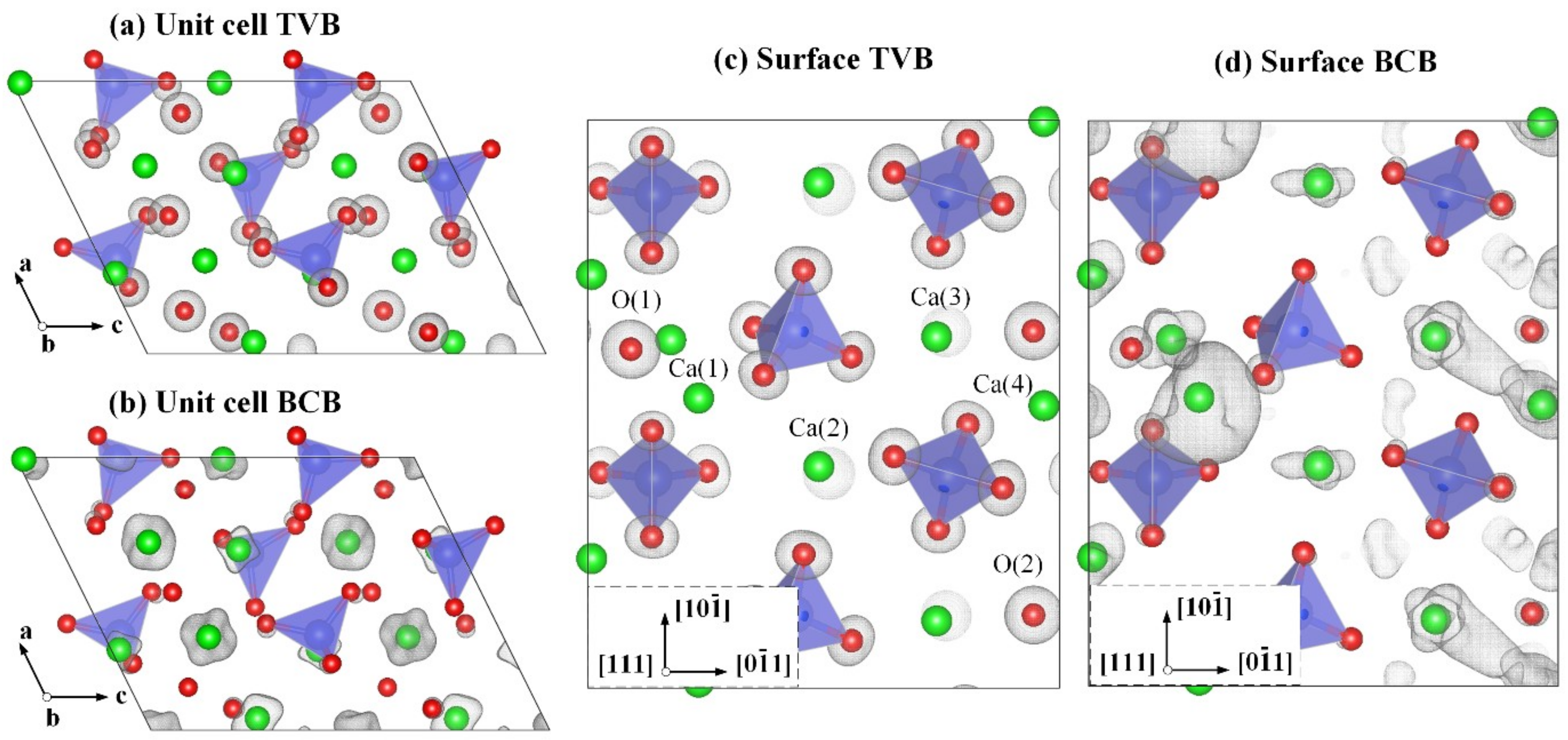
3.1. Electronic Properties of the M3-C3S (111) Surface
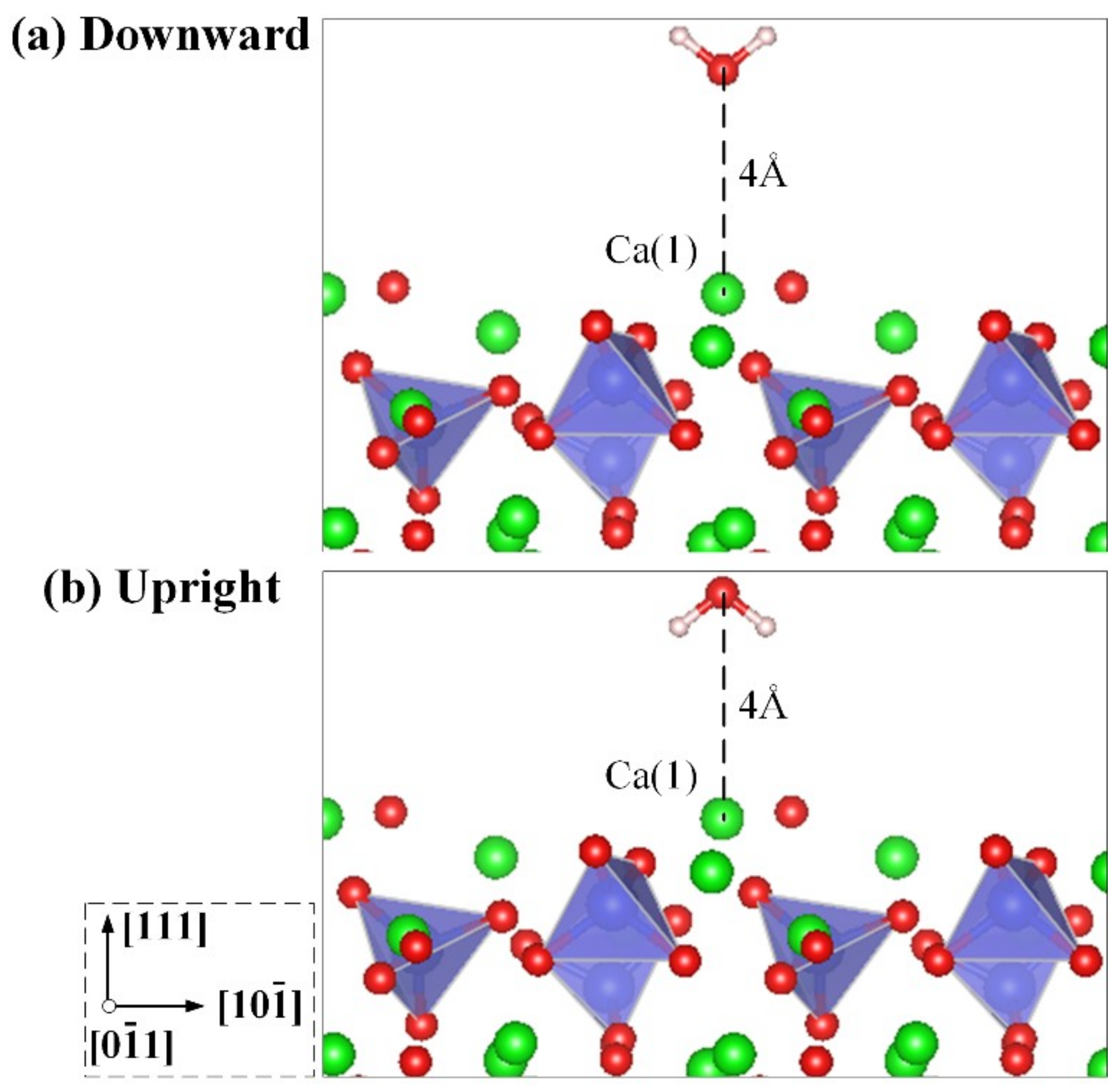
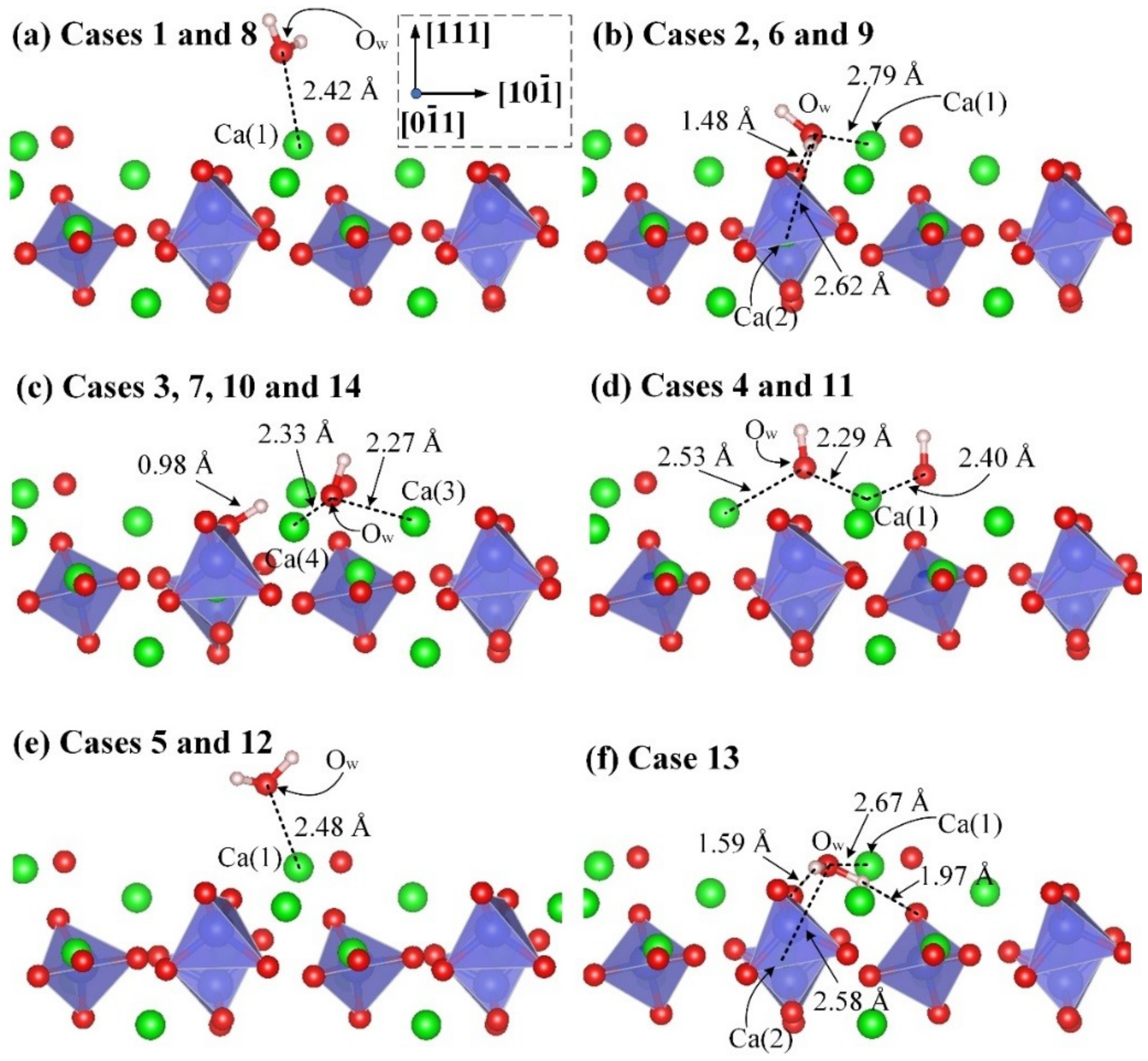
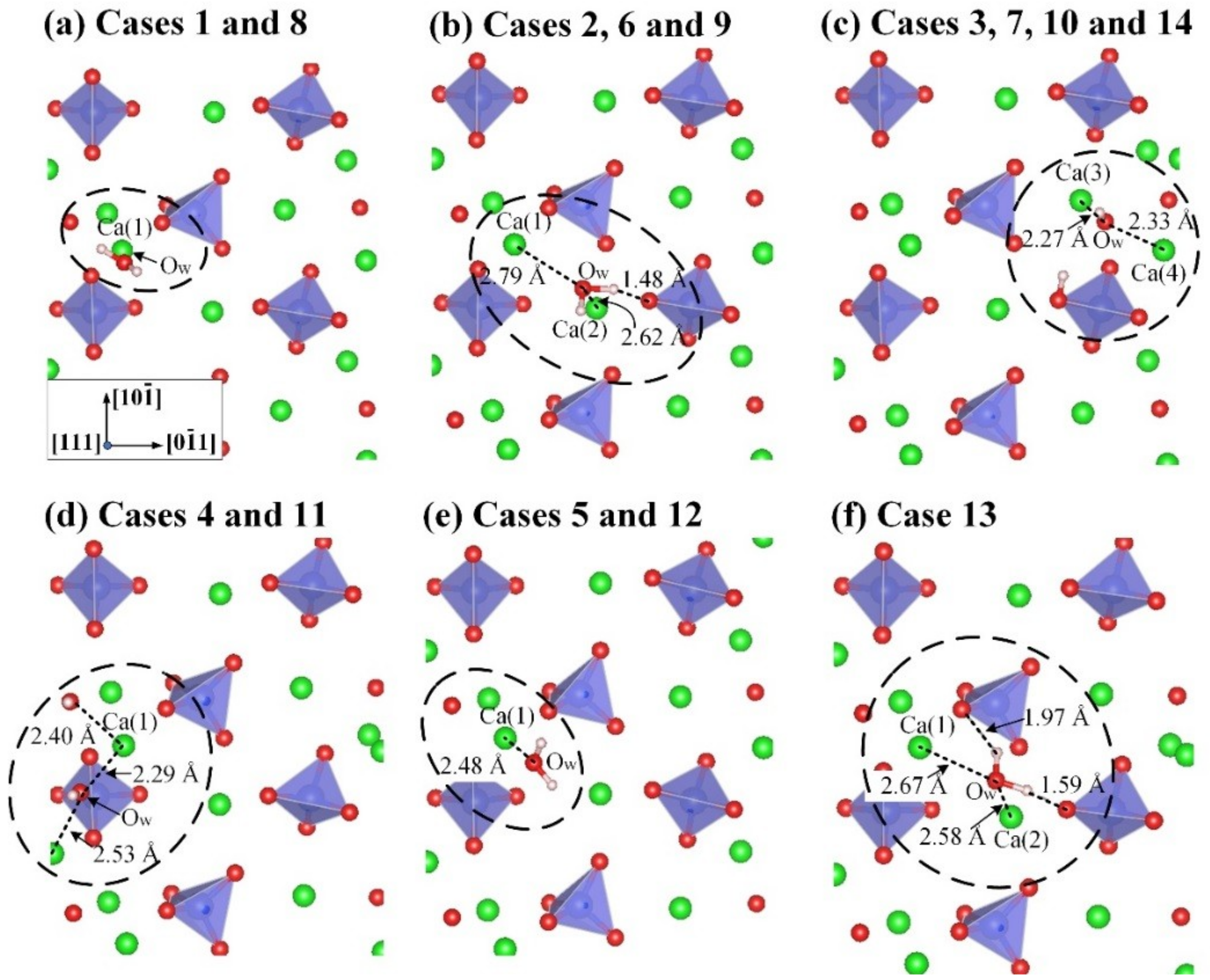
3.2. Adsorption Configuration of a Water Molecule on the M3-C3S (111) Surface
3.3. Electronic Properties of Reactions between a Water Molecule and the M3-C3S (111) Surface
3.3.1. Bader Charge Analysis
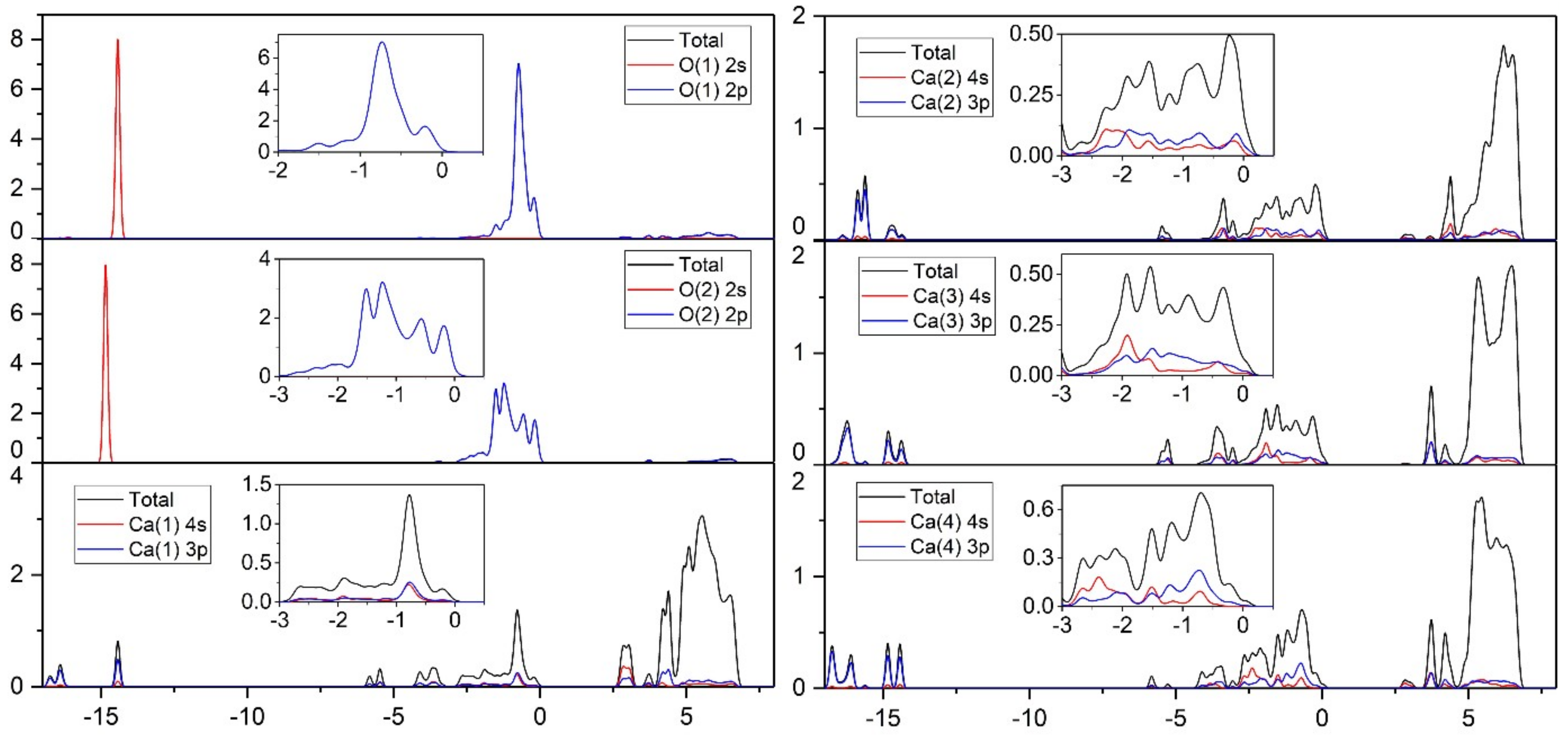
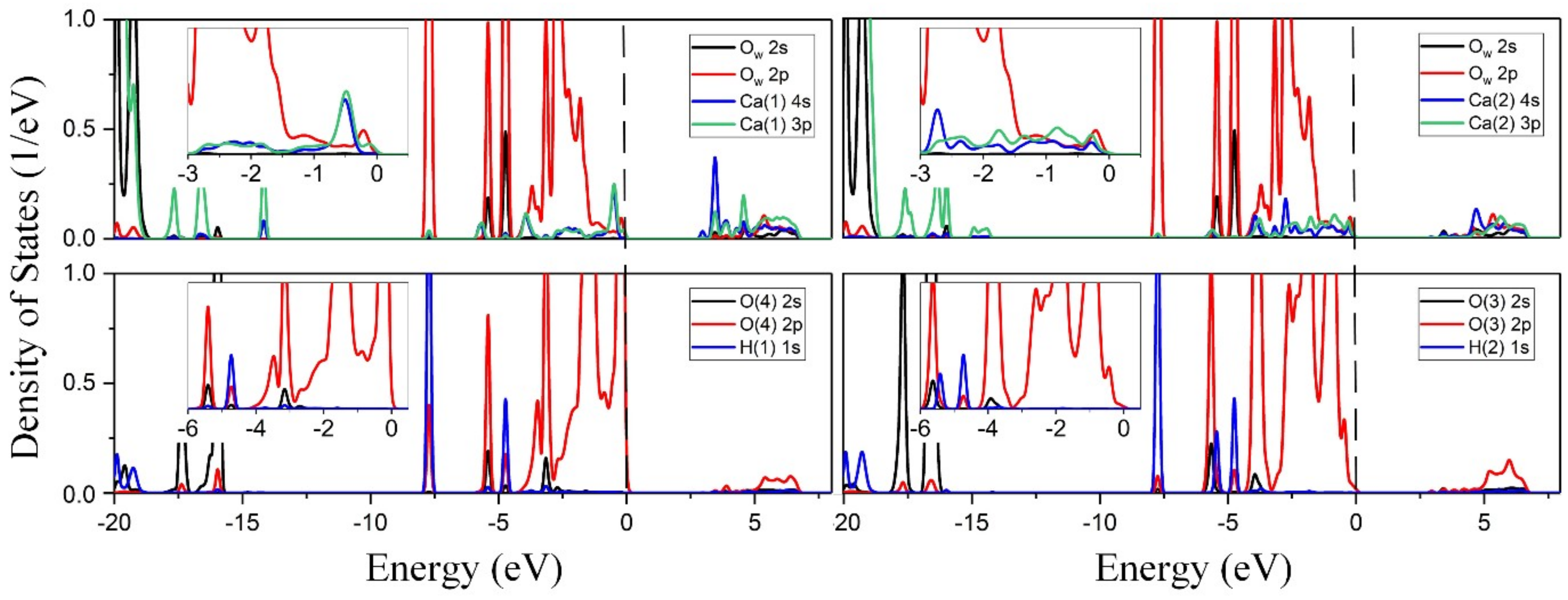
3.3.2. Density of States Analysis
3.4. Limitations and Future Works
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A




References
- Edraki, M.; Baumgartl, T.; Manlapig, E.; Bradshaw, D.; Franks, D.M.; Moran, C.J. Designing mine tailings for better environmental, social and economic outcomes: A review of alternative approaches. J. Clean. Prod. 2014, 84, 411–420. [Google Scholar] [CrossRef]
- Kesimal, A.; Yilmaz, E.; Ercikdi, B.; Alp, I.; Deveci, H. Effect of properties of tailings and binder on the short-and long-term strength and stability of cemented paste backfill. Mater. Lett. 2005, 59, 3703–3709. [Google Scholar] [CrossRef]
- Sun, W.; Wang, C.; Zhang, C. Factor analysis and forecasting of CO2 emissions in Hebei, using extreme learning machine based on particle swarm optimization. J. Clean. Prod. 2017, 162, 1095–1101. [Google Scholar] [CrossRef]
- Yilmaz, E.; Belem, T.; Benzaazoua, M. Effects of curing and stress conditions on hydromechanical, geotechnical and geochemical properties of cemented paste backfill. Eng. Geol. 2014, 168, 23–37. [Google Scholar] [CrossRef]
- Yılmaz, T.; Ercikdi, B. Predicting the uniaxial compressive strength of cemented paste backfill from ultrasonic pulse velocity test. Nondestruct. Test. Eval. 2016, 31, 247–266. [Google Scholar] [CrossRef]
- Qi, C.; Tang, X.; Dong, X.; Chen, Q.; Fourie, A.; Liu, E. Towards Intelligent Mining for Backfill: A genetic programming-based method for strength forecasting of cemented paste backfill. Miner. Eng. 2019, 133, 69–79. [Google Scholar] [CrossRef]
- Liu, L.; Fang, Z.; Qi, C.; Zhang, B.; Guo, L.; Song, K.I.I.L. Numerical study on the pipe flow characteristics of the cemented paste backfill slurry considering hydration effects. Powder Technol. 2019, 343, 454–464. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, R.; Ji, C.; Huan, C.; Zhang, B.; Liu, L. Parametric study and design of an earth-air heat exchanger using model experiment for memorial heating and cooling. Appl. Therm. Eng. 2019, 148, 838–845. [Google Scholar] [CrossRef]
- Liu, L.; Zhu, C.; Qi, C.; Zhang, B.; Song, K.-I. A microstructural hydration model for cemented paste backfill considering internal sulfate attacks. Constr. Build. Mater. 2019, 211, 99–108. [Google Scholar]
- Qi, C.; Chen, Q.; Fourie, A.; Zhao, J.; Zhang, Q. Pressure drop in pipe flow of cemented paste backfill: Experimental and modeling study. Powder Technol. 2018, 333, 9–18. [Google Scholar] [CrossRef]
- Wang, M.; Liu, L.; Zhang, X.-Y.; Chen, L.; Wang, S.-Q.; Jia, Y.-H. Experimental and numerical investigations of heat transfer and phase change characteristics of cemented paste backfill with pcm. Appl. Therm. Eng. 2019, 150, 121–131. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, Z.; Ji, C.; Liu, L.; Zhang, B.; Huan, C. Characterization of particulate matter from heating and cooling several edible oils. Build. Environ. 2019, 152, 204–213. [Google Scholar] [CrossRef]
- Fall, M.; Adrien, D.; Célestin, J.C.; Pokharel, M.; Touré, M. Saturated hydraulic conductivity of cemented paste backfill. Miner. Eng. 2009, 22, 1307–1317. [Google Scholar] [CrossRef]
- Qi, C.; Chen, Q.; Fourie, A.; Tang, X.; Zhang, Q.; Dong, X.; Feng, Y. Constitutive modelling of cemented paste backfill: A data-mining approach. Build. Environ. 2019, 197, 262–270. [Google Scholar] [CrossRef]
- Qi, C.; Fourie, A.; Chen, Q.; Tang, X.; Zhang, Q.; Gao, R. Data-driven modelling of the flocculation process on mineral processing tailings treatment. J.Clean. Production 2018, 196, 505–516. [Google Scholar] [CrossRef]
- Scrivener, K. The Concrete Conundrum. Chem. World 2008, 5, 62–66. [Google Scholar]
- Monteiro, P.J.; Miller, S.A.; Horvath, A. Towards sustainable concrete. Nat. Mater. 2017, 16, 698. [Google Scholar] [CrossRef] [PubMed]
- Hou, D.; Zhao, T.; Ma, H.; Li, Z. Reactive molecular simulation on water confined in the nanopores of the calcium silicate hydrate gel: Structure, reactivity, and mechanical properties. J. Phys. Chem. C 2015, 119, 1346–1358. [Google Scholar] [CrossRef]
- Durgun, E.; Manzano, H.; Pellenq, R.; Grossman, J.C. Understanding and controlling the reactivity of the calcium silicate phases from first principles. Chem. Mater. 2012, 24, 1262–1267. [Google Scholar] [CrossRef]
- Durgun, E.; Manzano, H.; Kumar, P.; Grossman, J.C. The characterization, stability, and reactivity of synthetic calcium silicate surfaces from first principles. J. Phys. Chem. C 2014, 118, 15214–15219. [Google Scholar] [CrossRef]
- Saritas, K.; Ataca, C.; Grossman, J.C. Predicting electronic structure in tricalcium silicate phases with impurities using first-principles. J. Phys. Chem. C 2015, 119, 5074–5079. [Google Scholar] [CrossRef]
- Manzano, H.; Dolado, J.S.; Ayuela, A. Structural, mechanical, and reactivity properties of tricalcium aluminate using first-principles calculations. J. Am. Ceram. Soc. 2009, 92, 897–902. [Google Scholar] [CrossRef]
- Huang, J.; Wang, B.; Yu, Y.; Valenzano, L.; Bauchy, M.; Sant, G. Electronic origin of doping-induced enhancements of reactivity: Case study of tricalcium silicate. J. Phys. Chem. C 2015, 119, 25991–25999. [Google Scholar] [CrossRef]
- Sanna, S.; Schmidt, W.G.; Thissen, P. Formation of Hydroxyl Groups at Calcium-Silicate-Hydrate (C-S-H): Coexistence of Ca–OH and Si–OH on Wollastonite(001). J. Phys. Chem. C 2014, 118, 8007–8013. [Google Scholar] [CrossRef]
- Thissen, P.; Natzeck, C.; Giraudo, N.; Weidler, P.; Wöll, C. Hydration of Concrete: The First Steps. Chem. A Eur. J. 2018, 24, 8603–8608. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Fourie, A.; Chen, Q.; Liu, P. Application of first-principles theory in ferrite phases of cemented paste backfill. Miner. Eng. 2019, 133, 47–51. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, X.; Song, D.; Liu, S. The adsorption of a single water molecule on low-index C3S surfaces: A DFT approach. Appl. Surf. Sci. 2019, 471, 658–663. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Viñes, F.; Sousa, C.; Liu, P.; Rodriguez, J.A.; Illas, F. A systematic density functional theory study of the electronic structure of bulk and (001) surface of transition-metals carbides. J. Chem. Phys. 2005, 122, 174709. [Google Scholar] [CrossRef]
- Yu, B.D.; Scheffler, M. Physical origin of exchange diffusion on fcc(100) metal surfaces. Phys. Rev. B 1997, 56, R15569–R15572. [Google Scholar] [CrossRef]
- Shi, X.-R.; Wang, S.-G.; Wang, H.; Deng, C.-M.; Qin, Z.; Wang, J. Structure and stability of β-Mo2C bulk and surfaces: A density functional theory study. Surf. Sci. 2009, 603, 852–859. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Qu, Z.W.; Zhu, H.; Kroes, G.J.; Nørskov, J.K. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 2007, 607, 83–89. [Google Scholar] [CrossRef]
- Guo, P.; Guo, X.; Zheng, C. Roles of γ-Fe2O3 in fly ash for mercury removal: Results of density functional theory study. Appl. Surf. Sci. 2010, 256, 6991–6996. [Google Scholar] [CrossRef]
- De Noirfontaine, M.N.; Dunstetter, F.; Courtial, M.; Gasecki, G.; Signes-Frehel, M. Polymorphism of tricalcium silicate, the major compound of Portland cement clinker: 2. Modelling alite for Rietveld analysis, an industrial challenge. Cement Concr. Res. 2006, 36, 54–64. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, X.; He, Z.; Song, D. Molecular and dissociative adsorption of a single water molecule on a β-dicalcium silicate (100) surface explored by a DFT approach. J. Am. Ceram. Soc. 2018, 101, 2428–2437. [Google Scholar] [CrossRef]
- Schiros, T.; Haq, S.; Ogasawara, H.; Takahashi, O.; Öström, H.; Andersson, K.; Pettersson, L.G.M.; Hodgson, A.; Nilsson, A. Structure of water adsorbed on the open Cu(110) surface: H-up, H-down, or both? Chem. Phys. Lett. 2006, 429, 415–419. [Google Scholar] [CrossRef]
- Peng, C.; Min, F.; Liu, L.; Chen, J. A periodic DFT study of adsorption of water on sodium-montmorillonite (001) basal and (010) edge surface. Appl. Surf. Sci. 2016, 387, 308–316. [Google Scholar] [CrossRef]
- Xin, Y.; Hou, S.C.; Xiang, L.; Yu, Y.-X. Adsorption and substitution effects of Mg on the growth of calcium sulfate hemihydrate: An ab initio DFT study. Appl. Surf. Sci. 2015, 357, 1552–1557. [Google Scholar] [CrossRef]
- Liu, J.; Wen, S.; Deng, J.; Chen, X.; Feng, Q. DFT study of ethyl xanthate interaction with sphalerite (110) surface in the absence and presence of copper. Appl. Surf. Sci. 2014, 311, 258–263. [Google Scholar] [CrossRef]
- Sholl, D.; Steckel, J.A. Density Functional Theory: A Practical Introduction; John Wiley Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Fan, X.; Chang, C.H.; Zheng, W.T.; Kuo, J.-L.; Singh, D.J. The Electronic Properties of Single-Layer and Multilayer MoS2 under High Pressure. J. Phys. Chem. C 2015, 119, 10189–10196. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Astala, R.; Stott, M. First-principles study of hydroxyapatite surfaces and water adsorption. Phys. Rev. B 2008, 78, 075427. [Google Scholar] [CrossRef]
- Bader, R.F. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chen, J.; Li, Y.; Huang, W.D.; Li, W. DFT study of interactions between calcium hydroxyl ions and pyrite, marcasite, pyrrhotite surfaces. Appl. Surf. Sci. 2015, 355, 577–581. [Google Scholar] [CrossRef]
- Odelius, M. Mixed Molecular and Dissociative Water Adsorption on MgO[100]. Phys. Rev. Lett. 1999, 82, 3919–3922. [Google Scholar] [CrossRef]
- Schaub, R.; Thostrup, P.; Lopez, N.; Lægsgaard, E.; Stensgaard, I.; Nørskov, J.K.; Besenbacher, F. Oxygen Vacancies as Active Sites for Water Dissociation on Rutile TiO2(110). Phys. Rev. Lett. 2001, 87, 266104. [Google Scholar] [CrossRef]
- Hass, K.C.; Schneider, W.F.; Curioni, A.; Andreoni, W. The chemistry of water on alumina surfaces: Reaction dynamics from first principles. Science 1998, 282, 265–268. [Google Scholar] [CrossRef]
- Tilocca, A.; Selloni, A. Reaction pathway and free energy barrier for defect-induced water dissociation on the (101) surface of TiO2-anatase. J. Chem. Phys. 2003, 119, 7445–7450. [Google Scholar] [CrossRef]
- Liu, X.; Sui, Y.; Meng, C.; Han, Y. Tuning the reactivity of Ru nanoparticles by defect engineering of the reduced graphene oxide support. RSC Adv. 2014, 4, 22230–22240. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, Y.; Liu, Y.; Li, H. DFT study of single water molecule adsorption on the (100) and (101) surfaces of KH2PO4. RSC Adv. 2017, 7, 26170–26178. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| References | a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | |
|---|---|---|---|---|---|---|---|
| Experimental | [29] | 9.30 | 7.08 | 12.22 | 90 | 116.08 | 90 |
| DFT calculations | This work | 9.36 | 7.15 | 12.25 | 90 | 116.44 | 90 |
| [13] | 9.38 | 7.20 | 12.46 | 90 | 116.23 | 90 | |
| [20] | 9.33 | 7.14 | 12.35 | 90 | 116.04 | 90 | |
| [14] | 9.23 | 7.11 | 12.21 | 90 | 116.08 | 90 |
| Case No. | Position a | Configuration | Case No. | Position | Configuration |
|---|---|---|---|---|---|
| 1 | Ca(1) | Downward | 8 | Ca(1) | Upright |
| 2 | Ca(2) | Downward | 9 | Ca(2) | Upright |
| 3 | Ca(3) | Downward | 10 | Ca(3) | Upright |
| 4 | Ca(4) | Downward | 11 | Ca(4) | Upright |
| 5 | Ca(1)–Ca(2) | Downward | 12 | Ca(1)–Ca(2) | Upright |
| 6 | Ca(2)–Ca(3) | Downward | 13 | Ca(2)–Ca(3) | Upright |
| 7 | Ca(3)–Ca(4) | Downward | 14 | Ca(3)–Ca(4) | Upright |
| AC No. | Adsorption Mechanism | Adsorption Energy (eV) a | N b |
|---|---|---|---|
| AC1 | Molecular | −1.10 | 1 |
| AC2 | Molecular | −1.74 | 3 |
| AC3 | Dissociative | −2.28 | 2 |
| AC4 | Dissociative | −2.48 | 2 |
| AC5 | Molecular | −0.93 | 1 |
| AC6 | Molecular | −1.81 | 4 |
| Bader Charge (eV) | O(1) | Ow | H(1) | H(2) | Ca(1) | Ca(5) |
|---|---|---|---|---|---|---|
| Before adsorption | −1.438 | −1.184 | +0.592 | +0.592 | +1.490 | +1.505 |
| After adsorption | −1.367 | −1.349 | +0.548 | +0.569 | +1.564 | +1.542 |
| Charge transfer | +0.071 | −0.165 | −0.044 | −0.023 | +0.074 | +0.037 |
| Bader Charge (eV) | O(3) | O(4) | Ow | H(1) | H(2) | Ca(1) | Ca(2) |
|---|---|---|---|---|---|---|---|
| Before adsorption | −1.560 | −1.558 | −1.184 | +0.592 | +0.592 | +1.490 | +1.554 |
| After adsorption | −1.543 | −1.539 | −1.311 | +0.648 | +0.585 | +1.529 | +1.568 |
| Charge transfer | +0.017 | +0.019 | −0.127 | +0.056 | −0.007 | +0.039 | +0.014 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, C.; Liu, L.; He, J.; Chen, Q.; Yu, L.-J.; Liu, P. Understanding Cement Hydration of Cemented Paste Backfill: DFT Study of Water Adsorption on Tricalcium Silicate (111) Surface. Minerals 2019, 9, 202. https://doi.org/10.3390/min9040202
Qi C, Liu L, He J, Chen Q, Yu L-J, Liu P. Understanding Cement Hydration of Cemented Paste Backfill: DFT Study of Water Adsorption on Tricalcium Silicate (111) Surface. Minerals. 2019; 9(4):202. https://doi.org/10.3390/min9040202
Chicago/Turabian StyleQi, Chongchong, Lang Liu, Jianyong He, Qiusong Chen, Li-Juan Yu, and Pengfei Liu. 2019. "Understanding Cement Hydration of Cemented Paste Backfill: DFT Study of Water Adsorption on Tricalcium Silicate (111) Surface" Minerals 9, no. 4: 202. https://doi.org/10.3390/min9040202
APA StyleQi, C., Liu, L., He, J., Chen, Q., Yu, L. -J., & Liu, P. (2019). Understanding Cement Hydration of Cemented Paste Backfill: DFT Study of Water Adsorption on Tricalcium Silicate (111) Surface. Minerals, 9(4), 202. https://doi.org/10.3390/min9040202