The Activation of Prothrombin Seems to Play an Earlier Role than the Complement System in the Progression of Colorectal Cancer: A Mass Spectrometry Evaluation


 , , , ,
, , , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Patient Selection and Ethical Aspects
2.2. Solid Phase Extraction
2.3. API-MALDI/TOF MS Analysis
2.4. Nano-LC Mass Spectrometry
2.5. API-MALDI/TOF MS Data Format Conversion
2.6. Sample Selection
2.7. Spectra Pre-Processing
- Normalize both abundances and adjust m/z values against a reference signal;
- Sum up isotopic peaks of the same molecule;
- Average technical replicas from each sample, thus producing a single spectrum that is representative for the sample;
- Align representative spectra and produce a table of aligned signals with corresponding abundances.
2.8. Statistical Analysis
2.9. Data Validation
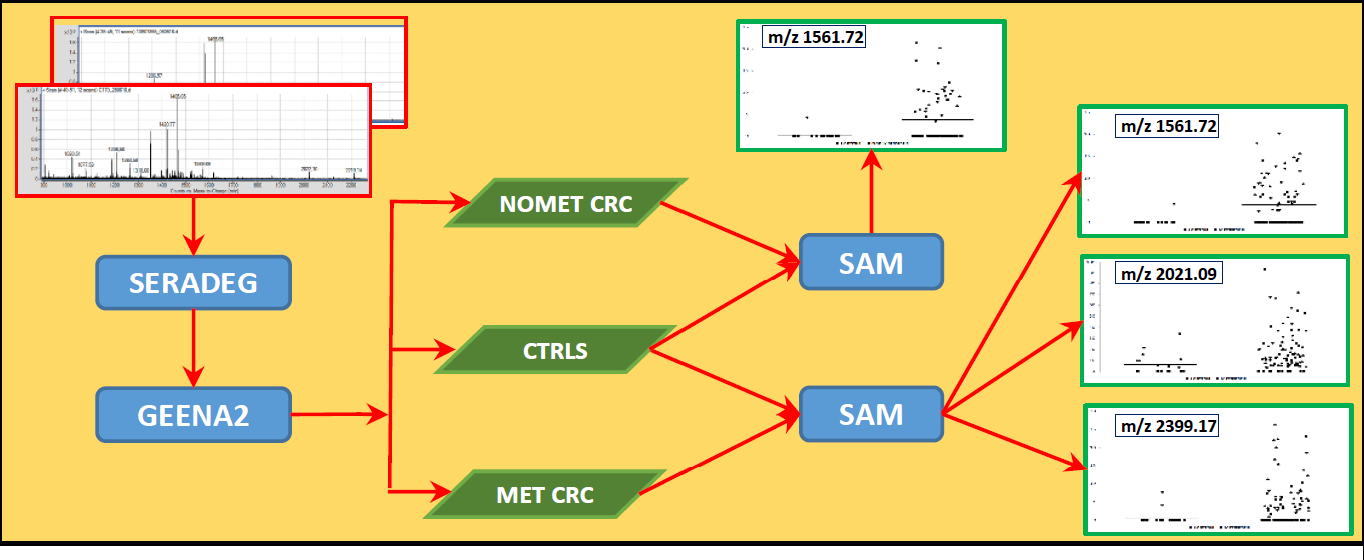
3. Results
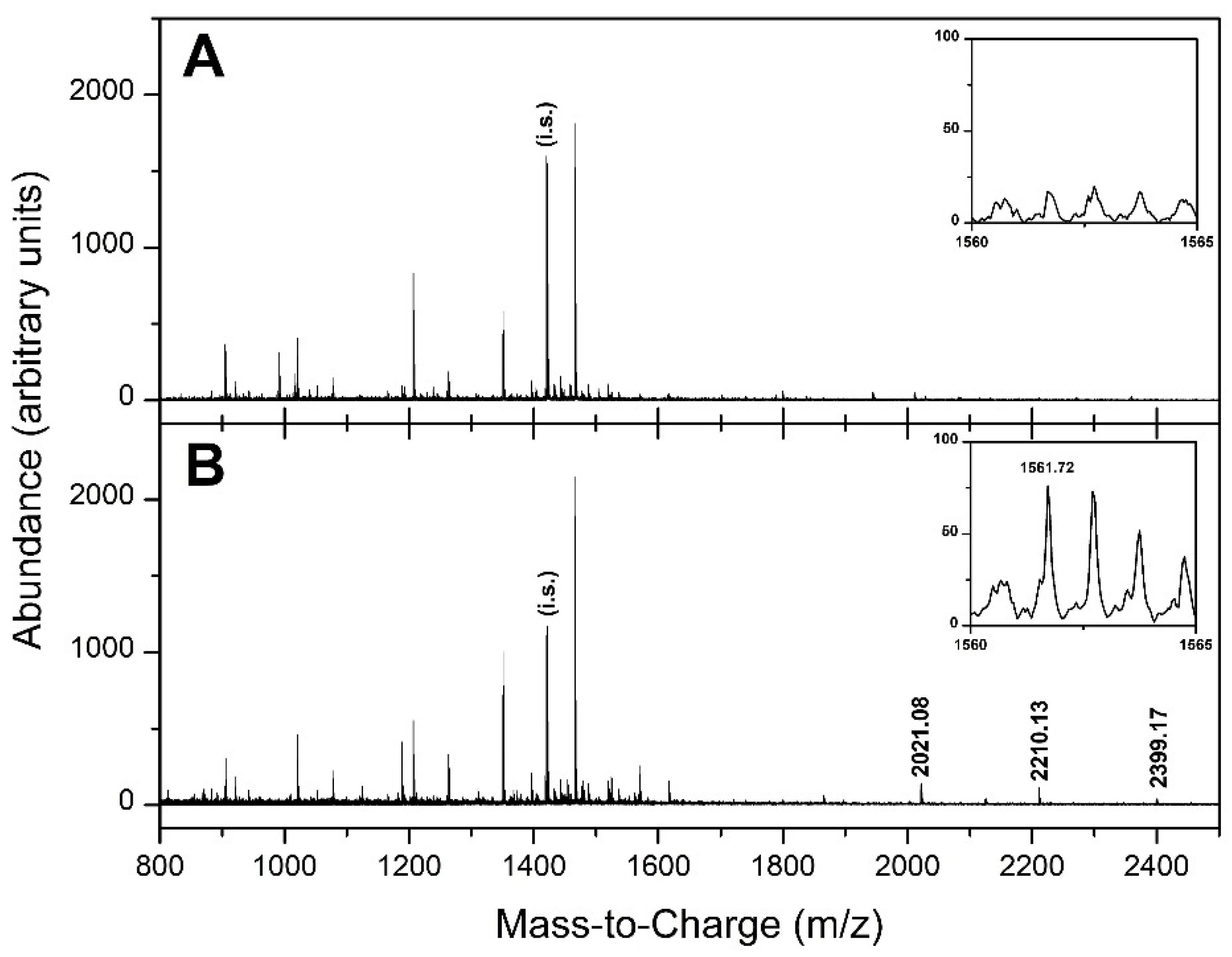
3.1. API-MALDI/TOF MS Analysis
3.2. Selection of Samples
3.3. Selection of Candidate Signals
3.4. Identification of the Selected Serum Peptides by Nano-HPLC-ESI-MS/MS
3.5. Validation by Bootstrap Resampling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| API-MALDI/TOF | atmospheric pressure ionization–matrix-assisted laser desorption/ionization/Time of flight |
| CHCA | alpha-cyano-4-hydroxycinnamic acid |
| CID | collision-induced dissociation |
| CRC | colorectal cancer |
| LC | liquid chromatography |
| FAP | familial adenomatous polyposis |
| FDR | false discovery rate |
| fpA | fibrinopeptide A |
| LMW | low molecular weight |
| MS | mass spectrometry |
| MS/MS | tandem mass spectrometry |
| nano-HPLC-ESI | nanoscale high-pressure liquid chromatography–electron spray ionization |
| SAM | significant analysis of microarrays |
| SAMR | implementation of SAM in the R language |
| SDQS | SeraDeg quality score |
| TFA | trifluoroacetic acid |
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [Green Version]
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics, 2010. CA Cancer J. Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef] [PubMed]
- Ciccolallo, L.; Capocaccia, R.; Coleman, M.P.; Berrino, F.; Coebergh, J.W.; Damhuis, R.A.; Faivre, J.; Martinez-Garcia, C.; Møller, H.; Ponz de Leon, M.; et al. Survival differences between European and US patients with colorectal cancer: Role of stage at diagnosis and surgery. Gut 2005, 54, 268–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boccardo, F.; Rubagotti, A.; Nuzzo, P.V.; Argellati, F.; Savarino, G.; Romano, P.; Damonte, G.; Rocco, M.; Profumo, A. Matrix-assisted laser desorption/ionisation (MALDI) TOF analysis identifies serum angiotensin II concentrations as a strong predictor of all-cause and breast cancer (BCa)-specific mortality following breast surgery. Int. J. Cancer 2015, 137, 2394–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opstal-van Winden, A.W.; Krop, E.J.; Kåredal, M.H.; Gast, M.C.; Lindh, C.H.; Jeppsson, M.C.; Jönsson, B.A.; Grobbee, D.E.; Peeters, P.H.; Beijnen, J.H.; et al. Searching for early breast cancer biomarkers by serum protein profiling of pre-diagnostic serum; a nested case–control study. BMC Cancer 2011, 11, 381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, J.; Shaffer, D.R.; Philip, J.; Chaparro, C.A.; Erdjument-Bromage, H.; Olshen, A.B.; Fleisher, M.; Lilja, H.; Brogi, E.; Boyd, J.; et al. Differential exoprotease activities confer tumor-specific serum peptidome patterns. J. Clin. Investig. 2006, 116, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Petricoin, E.F.; Ardekani, S.M.; Hitt, B.A.; Levine, P.J.; Fusaro, V.A.; Steinberg, S.M.; Mills, G.B.; Simone, C.; Fishman, D.A.; Kohn, E.C.; et al. Use of proteomic patterns in serum to identify ovarian cancer. Lancet 2002, 359, 572–577. [Google Scholar] [CrossRef]
- Villanueva, J.; Philip, J.; Entenberg, D.; Chaparro, C.A.; Tanwar, M.K.; Holland, E.C.; Tempst, P. Serum peptide profiling by magnetic particle-assisted, automated sample processing and MALDI–TOF mass spectrometry. Anal. Chem. 2004, 76, 1560–1570. [Google Scholar] [CrossRef]
- Fan, N.J.; Gao, C.F.; Wang, X.L.; Zhao, G.; Liu, Q.Y.; Zhang, Y.Y.; Cheng, B.G. Serum Peptidome Patterns of Colorectal Cancer Based on Magnetic Bead Separation and MALDI-TOF Mass Spectrometry Analysis. J. Biomed. Biotechnol. 2012, 2012, 985020. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Luo, C.; Zhu, S.; Fang, H.; Gao, Q.; Ge, S.; Qu, H.; Ma, Q.; Ren, H.; Wang, Y.; et al. Serum peptidome profiling for the diagnosis of colorectal cancer: Discovery and validation in two independent cohorts. Oncotarget 2017, 8, 59376–59386. [Google Scholar] [CrossRef] [Green Version]
- Romano, P.; Beitia San Vicente, M.; Profumo, A. A mass spectrometry based method and a software tool to assess degradation status of serum samples to be used in proteomics for biomarker discovery. J. Proteomics 2018, 173, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangerini, R.; Romano, P.; Facchiano, A.; Damonte, G.; Muselli, M.; Rocco, M.; Boccardo, F.; Profumo, A. The application of atmospheric pressure matrix assisted laser desorption/ionization to the analysis of long-term cryopreserved serum peptidome. Anal. Biochem. 2011, 417, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Washington, M.K.; Berlin, J.; Branton, P.; Burgart, L.J.; Carter, D.K.; Fitzgibbons, P.L.; Halling, K.; Frankel, W.; Jessup, J.; Kakar, S.; et al. Protocol for the examination of specimens from patients with primary carcinoma of the colon and rectum. Arch. Pathol. Lab. Med. 2009, 133, 1539–1551. [Google Scholar] [CrossRef]
- Profumo, A.; Mangerini, R.; Rubagotti, A.; Romano, P.; Damonte, G.; Guglielmini, P.; Facchiano, A.; Ferri, F.; Ricci, F.; Rocco, M.; et al. Complement C3f serum levels may predict breast cancer risk in women with gross cystic disease of the breast. J. Proteomics 2013, 85, 44–52. [Google Scholar] [CrossRef]
- Adusumilli, R.; Mallick, P. Data Conversion with ProteoWizard msConvert. Methods Mol. Biol. 2017, 1550, 339–368. [Google Scholar] [CrossRef]
- Gibb, S.; Strimmer, K. MALDIquant: A versatile R package for the analysis of mass spectrometry data. Bioinformatics 2012, 28, 2270–2271. [Google Scholar] [CrossRef]
- Draper, N.; Smith, H. Applied Regression Analysis, 3rd ed.; Wiley: New York, NY, USA, 1998; ISBN 978-0-471-17082-2. [Google Scholar]
- Efron, B.; Tibshirani, R.J. An Introduction to the Bootstrap; Chapman & Hall/CRC: New York, NY, USA, 1993; ISBN 978-0-412-04231-7. [Google Scholar]
- Romano, P.; Profumo, A.; Rocco, M.; Mangerini, R.; Ferri, F.; Facchiano, A. Geena 2, improved automated analysis of MALDI/TOF mass spectra. BMC Bioinform. 2016, 17 (Suppl. 4). [Google Scholar] [CrossRef] [Green Version]
- Sachon, E.; Matheron, L.; Clodic, G.; Blasco, T.; Bolbach, G. MALDI TOF-TOF characterization of a light stabilizer polymer contaminant from polypropylene or polyethylene plastic test tubes. J. Mass Spectrom. 2010, 45, 43–50. [Google Scholar] [CrossRef]
- Eng, J.K.; McCormack, A.L.; Yates, J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 1994, 5, 976–989. [Google Scholar] [CrossRef] [Green Version]
- GitHub. MikeJSeo/SAM: SAM: Significance Analysis of Microarrays. Provide Non-Coding Users to Reproduce as much of the Expressivity and Flexibility of the R Package as Possible without Needing to go beyond the GUI. 2019. Available online: https://github.com/MikeJSeo/SAM (accessed on 18 October 2019).
- Gaya da Costa, M.; Poppelaars, F.; van Kooten, C.; Mollnes, T.E.; Tedesco, F.; Würzner, R.; Trouw, L.A.; Truedsson, L.; Daha, M.R.; Roos, A.; et al. Age and Sex-Associated Changes of Complement Activity and Complement Levels in a Healthy Caucasian Population. Front. Immunol. 2018, 9, 2664. [Google Scholar] [CrossRef] [PubMed]
- Rickles, F.R.; Patierno, S.; Fernandez, P.M. Tissue factor, thrombin, and cancer. Chest 2003, 124 (Suppl. 3), 58S–68S. [Google Scholar] [CrossRef] [PubMed]
- Markiewski, M.M.; Lambris, J.D. Is complement good or bad for cancer patients? A new perspective on an old dilemma. Trends Immunol. 2009, 30, 286–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, G.N.; Rosenfeldt, L.; Frederick, M.; Miller, W.; Waltz, D.; Kombrinck, K.; McElhinney, K.E.; Flick, M.J.; Monia, B.P.; Revenko, A.S.; et al. Colon Cancer Growth and Dissemination Relies upon Thrombin, Stromal PAR-1, and Fibrinogen. Cancer Res. 2015, 75, 4235–4243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turpin, B.; Miller, W.; Rosenfeldt, L.; Kombrinck, K.; Flick, M.J.; Steinbrecher, K.A.; Harmel-Laws, E.; Mullins, E.S.; Shaw, M.; Witte, D.P.; et al. Thrombin drives tumorigenesis in colitis-associated colon cancer. Cancer Res. 2014, 74, 3020–3030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, C.; Hirohata, Y.; Kihara, Y.; Nakamura, H.; Abe, S.; Akahane, K.; Okamoto, K.; Itoh, H.; Otsuki, M. Pancreatic cancer complicated by disseminated intravascular coagulation associated with production of tissue factor. J. Gastroenterol. 2001, 36, 848–850. [Google Scholar] [CrossRef]
- Kakkar, A.K.; Lemoine, N.R.; Scully, M.F.; Tebbutt, S.; Williamson, R.C. Tissue factor expression correlates with histological grade in human pancreatic cancer. Br. J. Surg. 1995, 82, 1101–1104. [Google Scholar] [CrossRef]
- Zwicker, J.I.; Furie, B.C.; Furie, B. Cancer-associated thrombosis. Crit. Rev. Oncol. Hematol. 2007, 62, 126–136. [Google Scholar] [CrossRef]
- Kikkawa, S.; Sogawa, K.; Satoh, M.; Umemura, H.; Kodera, Y.; Matsushita, K.; Tomonaga, T.; Miyazaki, M.; Yokosuka, O.; Nomura, F. Identification of a Novel Biomarker for Biliary Tract Cancer Using Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry. Int. J. Proteomics 2012, 2012, 108609. [Google Scholar] [CrossRef] [Green Version]
- Zain, J.; Huang, Y.-Q.; Feng, X.-S.; Nierodzik, M.L.; Li, J.-J.; Karpatkin, S. Concentration-dependent dual effect of thrombin on impaired growth/apoptosis or mitogenesis in tumor cells. Blood 2000, 95, 3133–3138. [Google Scholar] [CrossRef]
- Huang, Y.Q.; Li, J.J.; Karpatkin, S. Thrombin inhibits tumor cell growth in association with up regulation of p21 and caspases via a p53-independent STAT-1 dependent pathway. J. Biol. Chem. 2000, 275, 6462–6468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remiker, A.S.; Palumbo, J.S. Mechanisms coupling thrombin to metastasis and tumorigenesis. Thromb. Res. 2018, 164 (Suppl. 1), S29–S33. [Google Scholar] [CrossRef] [PubMed]
- Janssen, B.J.; Huizinga, E.G.; Raaijmakers, H.C.; Roos, A.; Daha, M.R.; Nilsson-Ekdahl, K.; Nilsson, B.; Gros, P. Structures of complement component C3 provide insights into the function and evolution of immunity. Nature 2005, 437, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Levin, B.; Brooks, D.; Smith, R.A.; Stone, A. Emerging technologies in screening for colorectal cancer: CT colonography, immunochemical fecal occult blood tests, and stool screening using molecular markers. CA Cancer J. Clin. 2003, 53, 44–55. [Google Scholar] [CrossRef] [Green Version]
- Ishida, Y.; Yamashita, K.; Sasaki, H.; Takajou, I.; Kubuki, Y.; Morishita, K.; Tsubouchi, H.; Okayama, A. Activation of complement system in adult T-cell leukemia occurs mainly through lectin pathway: A serum proteomic approach using mass spectrometry. Cancer Lett. 2008, 271, 167–177. [Google Scholar] [CrossRef]
- Markiewski, M.M.; Lambris, J.D. Unwelcome complement. Cancer Res. 2009, 69, 6367–6370. [Google Scholar] [CrossRef] [Green Version]
- Ning, C.; Li, Y.-Y.; Wang, Y.; Han, G.-C.; Wang, R.-X.; Xiao, H.; Li, X.-Y.; Hou, C.-M.; Ma, Y.-F.; Sheng, D.-S.; et al. Complement activation promotes colitis-associated carcinogenesis through activating intestinal IL-1 beta/IL-17A axis. Mucosal Immunol. 2015, 8, 1275–1284. [Google Scholar] [CrossRef] [Green Version]
- Piao, C.; Zhang, W.M.; Li, T.T.; Zhang, C.C.; Qiu, S.; Liu, Y.; Liu, S.; Jin, M.; Jia, L.X.; Song, W.C.; et al. Complement 5a stimulates macrophage polarization and contributes to tumor metastases of colon cancer. Exp. Cell Res. 2018, 366, 127–138. [Google Scholar] [CrossRef]
- Pio, R.; Ajona, D.; Lambris, J.D. Complement inhibition: A promising concept for cancer treatment. Semin. Immunol. 2013, 25, 54–64. [Google Scholar] [CrossRef] [Green Version]
- Liang, T.; Wang, N.; Li, W.; Li, A.; Wang, J.; Cui, J.; Liu, N.; Li, Y.; Li, L.; Yang, G.; et al. Identification of complement C3f-desArg and its derivative for acute leukemia diagnosis and minimal residual disease assessment. Proteomics 2010, 10, 90–98. [Google Scholar] [CrossRef]
- Chang, J.T.; Chen, L.C.; Wei, S.Y.; Chen, Y.J.; Wang, H.M.; Liao, C.T.; Chen, I.H.; Cheng, A.J. Increase diagnostic efficacy by combined use of fingerprint markers in mass spectrometry—Plasma peptidomes from nasopharyngeal cancer patients for example. Clin. Biochem. 2006, 39, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Bedin, C.; Sara Crotti, S.; Ragazzi, E.; Pucciarelli, S.; Agatea, L.; Tasciotti, E.; Ferrari, M.; Traldi, P.; Rizzolio, F.; Giordano, A.; et al. Alterations of the plasma peptidome profiling in colorectal cancer progression. J. Cell. Physiol. 2016, 231, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Agatea, L.; Crotti, S.; Ragazzi, E.; Bedin, C.; Urso, E.; Mammi, I.; Traldi, P.; Pucciarelli, S.; Nitti, D.; Agostini, M. Peptide Patterns as Discriminating Biomarkers in Plasma of Patients With Familial Adenomatous Polyposis. Clin. Colorectal Cancer 2015, 15, e75–e92. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Sample Size | Gender | Mean Age | Age Range |
|---|---|---|---|---|
| Metastatic CRC | 92 | 28F/64M | 71 | 34–93 |
| Non-Metastatic CRC | 80 | 26F/54M | 71 | 44–89 |
| Healthy controls | 19 | 10F/9M | 54 | 40–67 |
| Controls vs. Non Metastatic | |||||
| Signal | Controls (n = 19) Mean (SE) | Cases (n = 80) Mean (SE) | Score | Fold Change | q-Value |
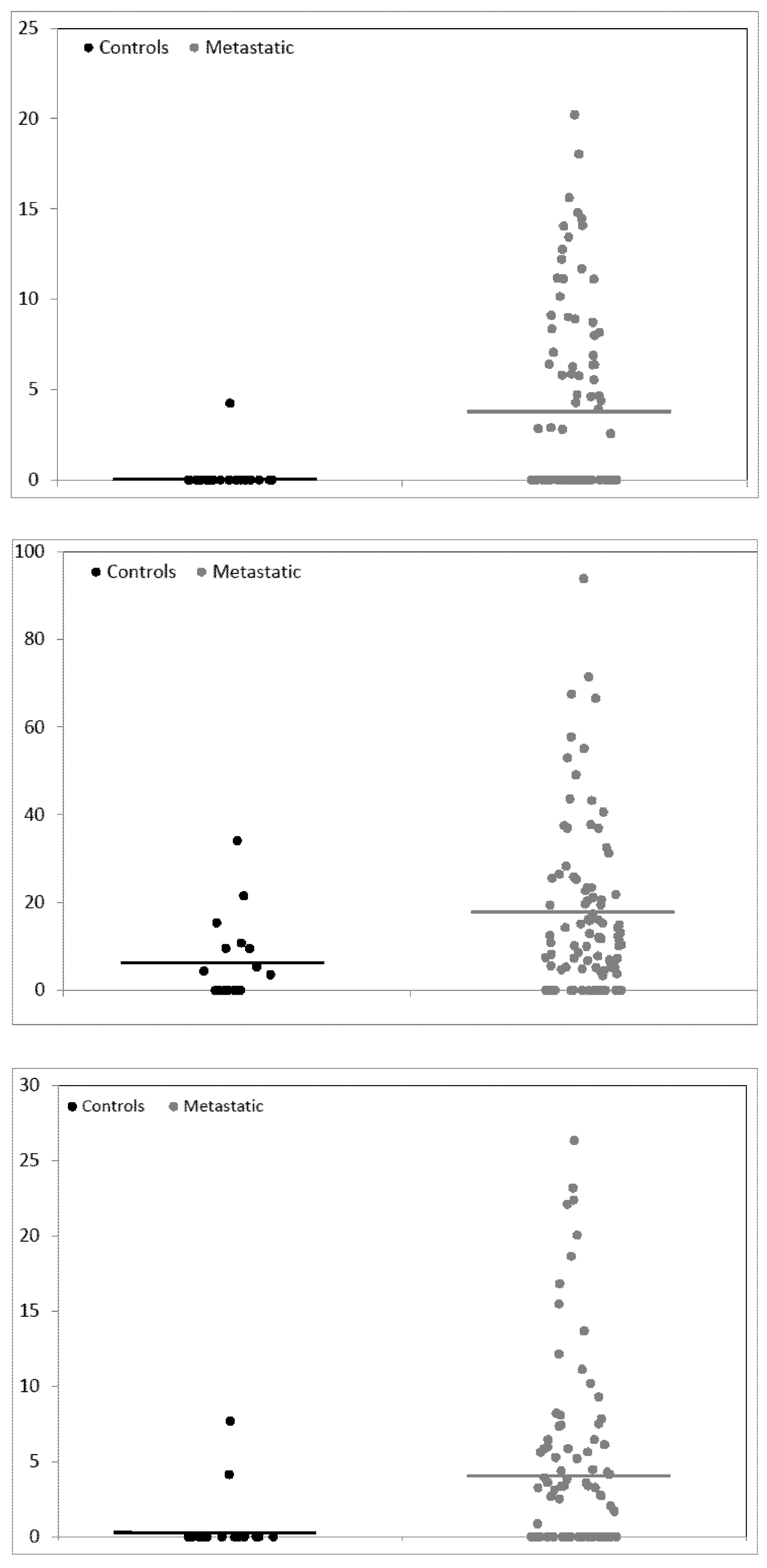
| 1561.720 | 0.22 (0.22) | 3.67 (0.61) | 2.371 | 16.408 | *0.04490 |
| 2021.084 | 6.01 (2.12) | 15.80 (2.54) | 1.772 | 2.628 | 0.10264 |
| 2399.165 | 0.62 (0.45) | 4.17 (0.79) | 1.938 | 6.705 | 0.10264 |
| Controls vs. Metastatic | |||||
| Signal | Controls (n = 19) Mean (SE) | Cases (n = 92) Mean (SE) | Score | Fold Change | q-Value |
| 1561.720 | 0.22 (0.22) | 3.97 (0.55) | 2.634 | 17.786 | * 0.03754 |
| 2021.084 | 6.01 (2.12) | 17.52 (1.95) | 2.489 | 2.915 | * 0.03754 |
| 2399.165 | 0.62 (0.45) | 4.28 (0.64) | 2.246 | 6.877 | * 0.03754 |
| Controls vs. Non Metastatic | Controls vs. Metastatic | ||||
|---|---|---|---|---|---|
| Signal | q-Value | Bootstrap | Signal | q-Value | Bootstrap |
| 1561.720 | * 0.04490 | 70/100 | 1561.720 | * 0.03754 | 95/100 |
| 2021.084 | 0.10264 | 35/100 | 2021.084 | * 0.03754 | 95/100 |
| 2399.165 | 0.10264 | 46/100 | 2399.165 | * 0.03754 | 85/100 |
| Adjusted by Age | ||||||
| Non Metastatic vs. Controls | Metastatic vs. Controls | |||||
| Signal | Mean Difference | 95% Confidence Interval | p-Value | Mean Difference | 95% Confidence Interval | p-Value |
| 1561.720 | 2.58 | 0.13–5.02 | 0.039 | 4.85 | 2.68–7.03 | <0.001 |
| 2021.084 | Untested because not significant by SAM | 9.51 | 1.12–17.91 | 0.026 | ||
| 2399.165 | Untested because not significant by SAM | 3.54 | 0.65–6.43 | 0.016 | ||
| Adjusted by Gender | ||||||
| Non Metastatic vs. Controls | Metastatic vs. Controls | |||||
| Signal | Mean Difference | 95% Confidence Interval | p-Value | Mean Difference | 95% Confidence Interval | p-Value |
| 1561.720 | 3.40 | 2.04–4.77 | <0.001 | 3.75 | 2.55–4.95 | <0.001 |
| 2021.084 | Untested because not significant by SAM | 10.84 | 5.57–16.11 | <0.001 | ||
| 2399.165 | Untested because not significant by SAM | 3.33 | 1.86–4.81 | <0.001 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beitia, M.; Romano, P.; Larrinaga, G.; Solano-Iturri, J.D.; Salis, A.; Damonte, G.; Bruzzone, M.; Ceppi, M.; Profumo, A. The Activation of Prothrombin Seems to Play an Earlier Role than the Complement System in the Progression of Colorectal Cancer: A Mass Spectrometry Evaluation. Diagnostics 2020, 10, 1077. https://doi.org/10.3390/diagnostics10121077
Beitia M, Romano P, Larrinaga G, Solano-Iturri JD, Salis A, Damonte G, Bruzzone M, Ceppi M, Profumo A. The Activation of Prothrombin Seems to Play an Earlier Role than the Complement System in the Progression of Colorectal Cancer: A Mass Spectrometry Evaluation. Diagnostics. 2020; 10(12):1077. https://doi.org/10.3390/diagnostics10121077
Chicago/Turabian StyleBeitia, Maider, Paolo Romano, Gorka Larrinaga, Jon Danel Solano-Iturri, Annalisa Salis, Gianluca Damonte, Marco Bruzzone, Marcello Ceppi, and Aldo Profumo. 2020. "The Activation of Prothrombin Seems to Play an Earlier Role than the Complement System in the Progression of Colorectal Cancer: A Mass Spectrometry Evaluation" Diagnostics 10, no. 12: 1077. https://doi.org/10.3390/diagnostics10121077
APA StyleBeitia, M., Romano, P., Larrinaga, G., Solano-Iturri, J. D., Salis, A., Damonte, G., Bruzzone, M., Ceppi, M., & Profumo, A. (2020). The Activation of Prothrombin Seems to Play an Earlier Role than the Complement System in the Progression of Colorectal Cancer: A Mass Spectrometry Evaluation. Diagnostics, 10(12), 1077. https://doi.org/10.3390/diagnostics10121077