Exploring the Spectrum of Kidney Ciliopathies


Abstract
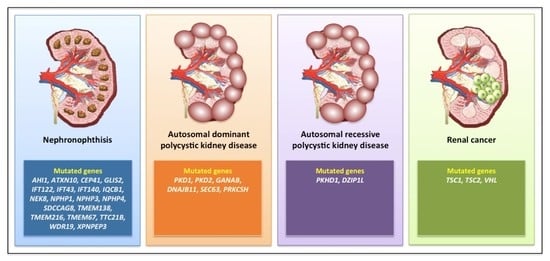
:
{kind=link}
{kind=link}
{kind=link}
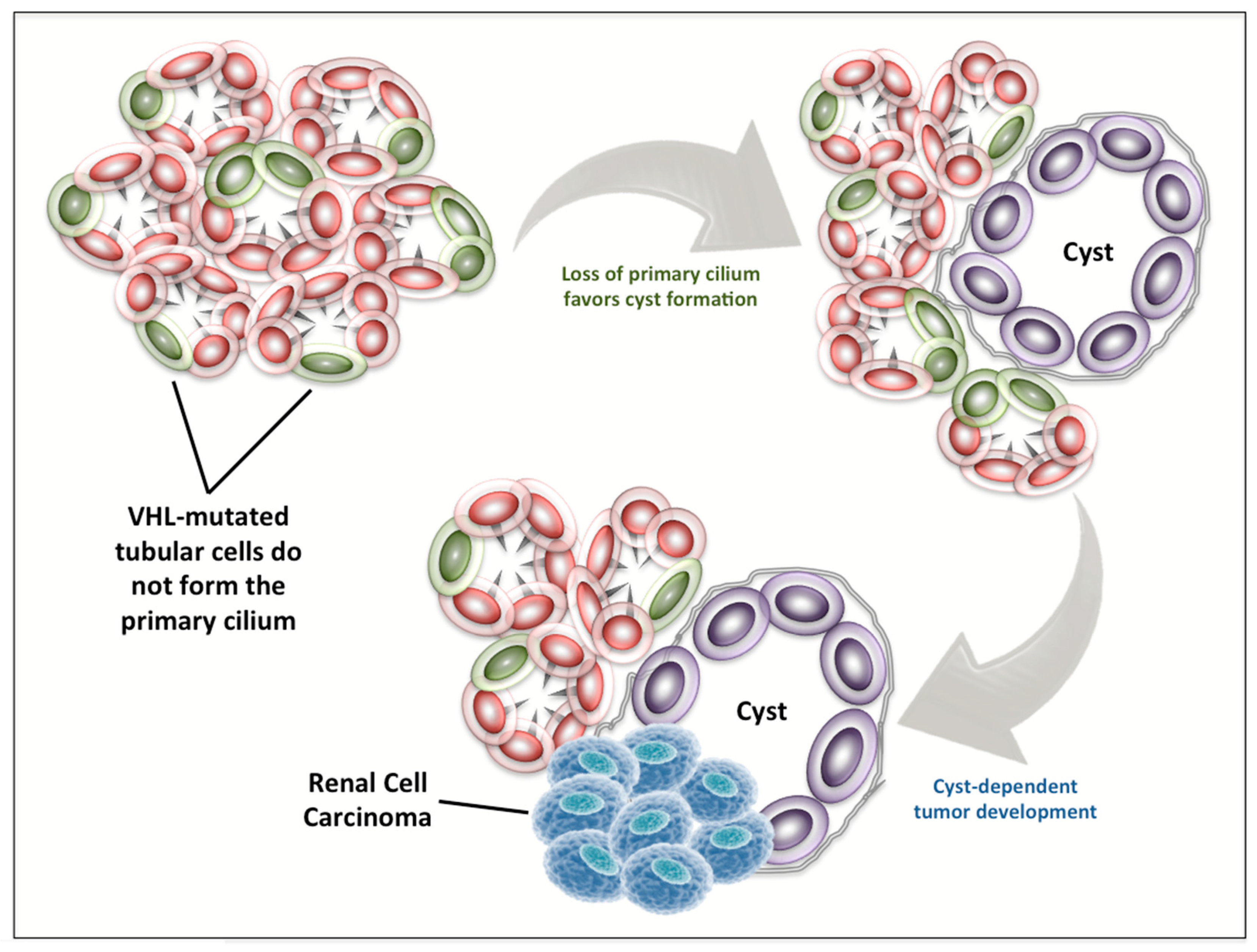{kind=link}
1. Introduction
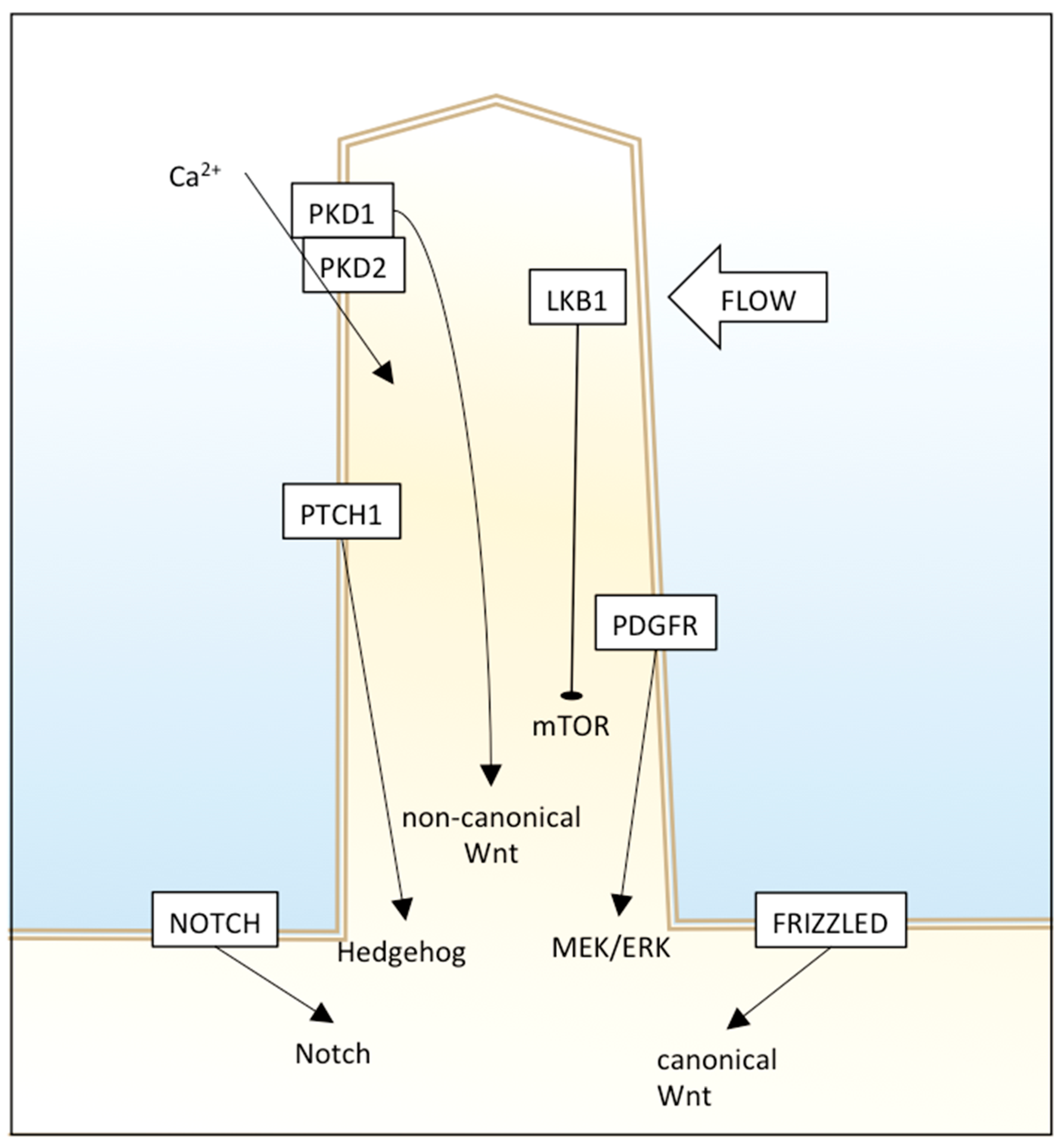
2. Genes and Signaling Pathways in the Primary Cilia
3. Renal Ciliopathies
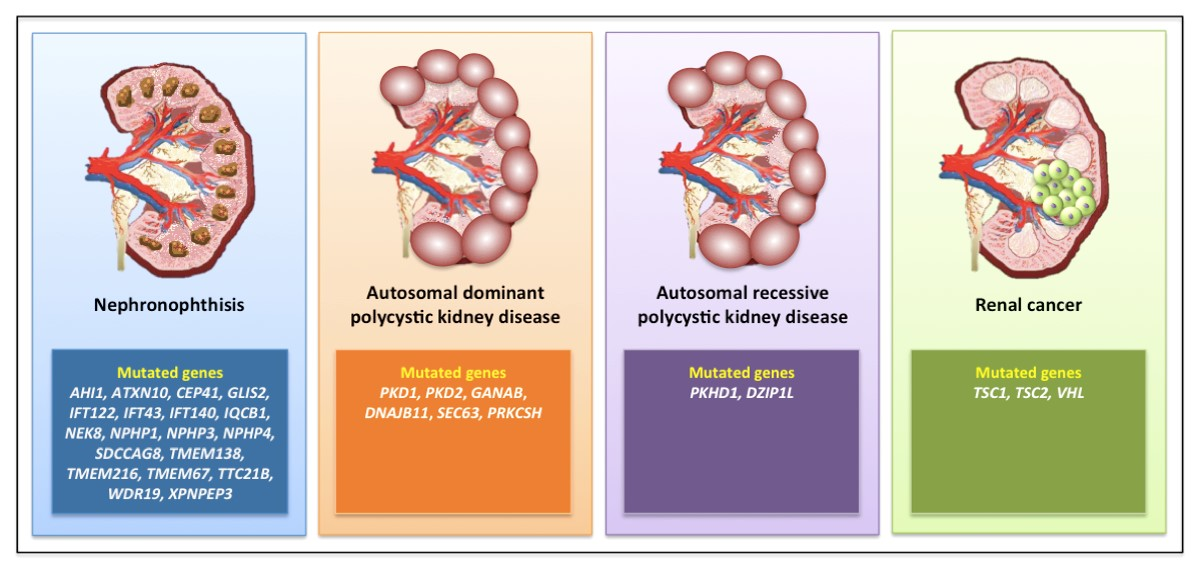
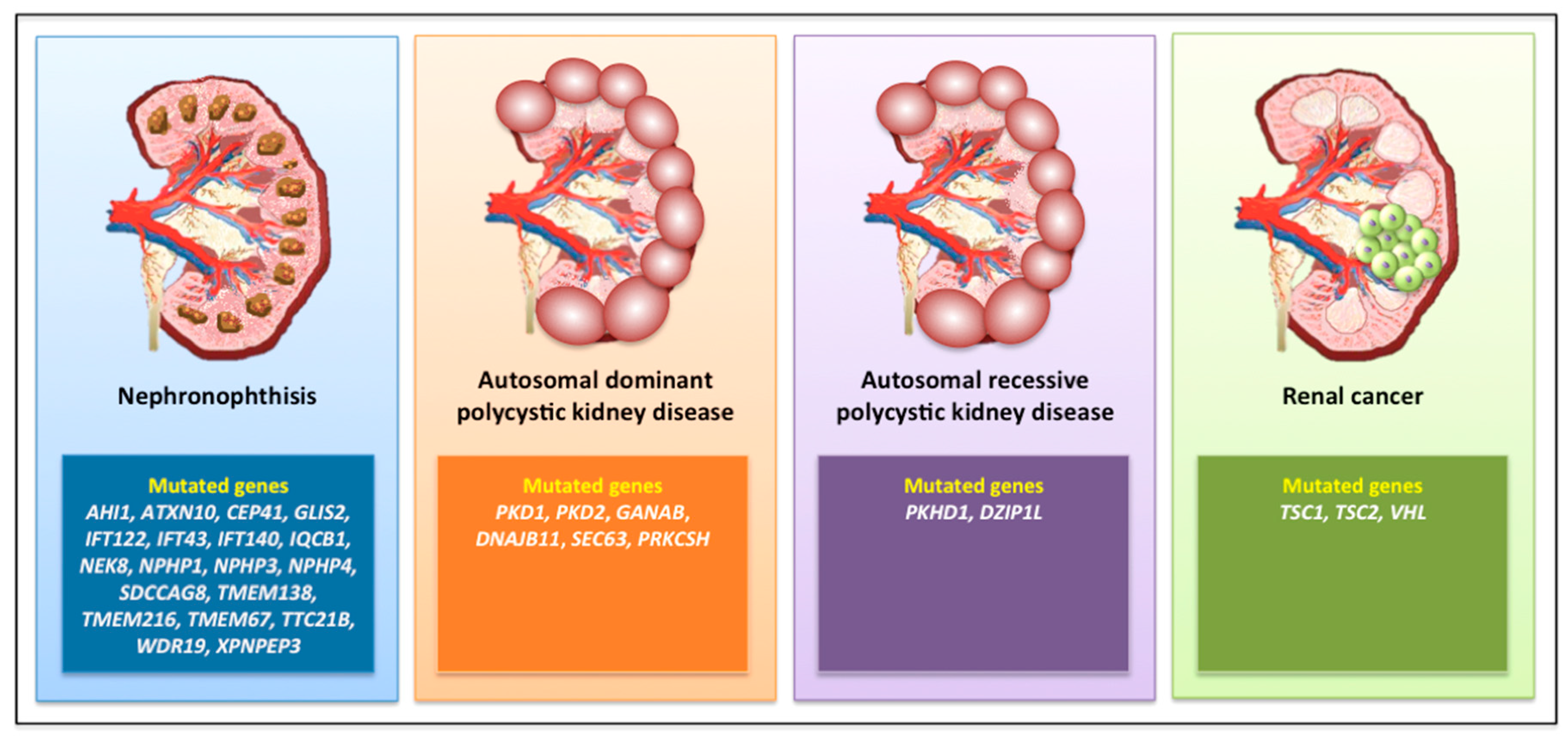
3.1. Nephronophthisis
3.2. Autosomal Dominant Polycystic Kidney Disease
3.3. Autosomal Recessive Polycystic Kidney Disease
3.4. Renal Cystic Dysplasia
3.5. Rare Renal Phenotypes
3.6. Renal Cell Carcinoma
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Adams, M. The Primary Cilium: An Orphan Organelle Finds a Home. Nat. Educ. 2010, 3, 54. [Google Scholar]
- Adams, G.M.W.; Huang, B.; Piperno, G.; Luck, D.J.L. Central-pair microtubular complex of Chlamydomonas flagella: Polypeptide composition as revealed by analysis of mutants. J. Cell Biol. 1981, 91, 69–76. [Google Scholar] [CrossRef]
- Satir, P.; Pedersen, L.B.; Christensen, S.T. The primary cilium at a glance. J. Cell Sci. 2010, 123, 499–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huangfu, D.; Liu, A.; Rakeman, A.S.; Murcia, N.S.; Niswander, L.; Anderson, K.V. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 2003, 426, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.; Clement, C.A.; Teilmann, S.C.; Pazour, G.J.; Hoffmann, E.K.; Satir, P.; Christensen, S.T. PDGFRαα signaling is regulated through the primary cilium in fibroblasts. Curr. Biol. 2005, 15, 1861–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, M.; Gloy, J.; Ganner, A.; Bullerkotte, A.; Bashkurov, M.; Krönig, C.; Schermer, B.; Benzing, T.; Cabello, O.A.; Jenny, A.; et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat. Genet. 2005, 37, 537–543. [Google Scholar] [CrossRef] [Green Version]
- Gherman, A.; Davis, E.E.; Katsanis, N. The ciliary proteome database: An integrated community resource for the genetic and functional dissection of cilia. Nat. Genet. 2006, 38, 961–962. [Google Scholar] [CrossRef]
- Inglis, P.N.; Boroevich, K.A.; Leroux, M.R. Piecing together a ciliome. Trends Genet. 2006, 22, 491–500. [Google Scholar] [CrossRef]
- Cervesato, A.; Raucci, R.; Buononato, D.; Marchese, E.; Capolongo, G.; Perna, A.; Capasso, G.; Zacchia, M. Application of proteomics and metabolomics to study inherited kidney disorders: From big data to precision medicine. Giornale Ital. Nefrol. 2020, 37, 1853–1861. [Google Scholar]
- Arnaiz, O.; Malinowska, A.; Klotz, C.; Sperling, L.; Dadlez, M.; Koll, F.; Cohen, J. Cildb: A knowledgebase for centrosomes and cilia. Database 2009, 2009. [Google Scholar] [CrossRef]
- Swoboda, P.; Adler, H.T.; Thomas, J.H. The RFX-type transcription factor DAF-19 regulates sensory neuron cilium formation in C. Elegans. Mol. Cell 2000, 5, 411–421. [Google Scholar] [CrossRef]
- Piasecki, B.P.; Burghoorn, J.; Swoboda, P. Regulatory Factor X (RFX)-mediated transcriptional rewiring of ciliary genes in animals. Proc. Natl. Acad. Sci. USA 2010, 107, 12969–12974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badano, J.L.; Leitch, C.C.; Ansley, S.J.; May-Simera, H.; Lawson, S.; Lewis, R.A.; Beales, P.L.; Dietz, H.C.; Fisher, S.; Katsanis, N. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature 2006, 439, 326–330. [Google Scholar] [CrossRef]
- Chiang, A.P.; Nishimura, D.; Searby, C.; Elbedour, K.; Carmi, R.; Ferguson, A.L.; Secrist, J.; Braun, T.; Casavant, T.; Stone, E.M.; et al. Comparative genomic analysis identifies an ADP-ribosylation factor-like gene as the cause of Bardet-Biedl syndrome (BBS3). Am. J. Hum. Genet. 2004, 75, 475–484. [Google Scholar] [CrossRef] [Green Version]
- Kyttälä, M.; Tallila, J.; Salonen, R.; Kopra, O.; Kohlschmidt, N.; Paavola-Sakki, P.; Peltonen, L.; Kestilä, M. MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat. Genet. 2006, 38, 155–157. [Google Scholar] [CrossRef]
- Li, J.B.; Gerdes, J.M.; Haycraft, C.J.; Fan, Y.; Teslovich, T.M.; May-Simera, H.; Li, H.; Blacque, O.E.; Li, L.; Leitch, C.C.; et al. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell 2004, 117, 541–552. [Google Scholar] [CrossRef] [Green Version]
- Eley, L.; Gabrielides, C.; Adams, M.; Johnson, C.A.; Hildebrandt, F.; Sayer, J.A. Jouberin localizes to collecting ducts and interacts with nephrocystin-1. Kidney Int. 2008, 74, 1139–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorden, N.T.; Arts, H.H.; Parisi, M.A.; Coene, K.L.M.; Letteboer, S.J.F.; van Beersum, S.E.C.; Mans, D.A.; Hikida, A.; Eckert, M.; Knutzen, D.; et al. CC2D2A Is Mutated in Joubert Syndrome and Interacts with the Ciliopathy-Associated Basal Body Protein CEP290. Am. J. Hum. Genet. 2008, 83, 559–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConnachie, D.J.; Stow, J.L.; Mallett, A.J. Ciliopathies and the Kidney: A Review. Am. J. Kidney Dis. 2020. [Google Scholar] [CrossRef]
- Pazour, G.J.; Dickert, B.L.; Vucica, Y.; Seeley, E.S.; Rosenbaum, J.L.; Witman, G.B.; Cole, D.G. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene Tg737, are required for assembly of cilia and flagella. J. Cell Biol. 2000, 151, 709–718. [Google Scholar] [CrossRef]
- Dell, K.M. The role of cilia in the pathogenesis of cystic kidney disease. Curr. Opin. Pediatr. 2015, 27, 212–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Husson, H.; Moreno, S.; Smith, L.A.; Smith, M.M.; Russo, R.J.; Pitstick, R.; Sergeev, M.; Ledbetter, S.R.; Bukanov, N.O.; Lane, M.; et al. Reduction of ciliary length through pharmacologic or genetic inhibition of CDK5 attenuates polycystic kidney disease in a model of nephronophthisis. Hum. Mol. Genet. 2016, 25, 2245–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, A.; Margall-Ducos, G.; Guidotti, J.E.; Brégerie, O.; Celati, C.; Bréchot, C.; Desdouets, C. The intraflagellar transport component IFT88/polaris is a centrosomal protein regulating G1-S transition in non-ciliated cells. J. Cell Sci. 2007, 120 Pt 4, 628–637. [Google Scholar] [CrossRef] [Green Version]
- Maskey, D.; Marlin, M.C.; Kim, S.; Kim, S.; Ong, E.; Li, G.; Tsiokas, L. Cell cycle-dependent ubiquitylation and destruction of NDE 1 by CDK 5- FBW 7 regulates ciliary length. EMBO J. 2015, 34, 2424–2440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inaba, H.; Goto, H.; Kasahara, K.; Kumamoto, K.; Yonemura, S.; Inoko, A.; Yamano, S.; Wanibuchi, H.; He, D.; Goshima, N.; et al. Ndel1 suppresses ciliogenesis in proliferating cells by regulating the trichoplein-Aurora A pathway. J. Cell Biol. 2016, 212, 409–423. [Google Scholar] [CrossRef] [Green Version]
- Arjumand, W.; Sultana, S. Role of VHL gene mutation in human renal cell carcinoma. Tumor Biol. 2012, 33, 9–16. [Google Scholar] [CrossRef]
- Yuan, K.; Frolova, N.; Xie, Y.; Wang, D.; Cook, L.; Kwon, Y.J.; Steg, A.D.; Serra, R.; Frost, A.R. Primary cilia are decreased in breast cancer: Analysis of a collection of human breast cancer cell lines and tissues. J. Histochem. Cytochem. 2010, 58, 857–870. [Google Scholar] [CrossRef] [Green Version]
- Malicki, J.J.; Johnson, C.A. The Cilium: Cellular Antenna and Central Processing Unit. Trends Cell Biol. 2017, 27, 126–140. [Google Scholar] [CrossRef] [Green Version]
- Wheway, G.; Nazlamova, L.; Hancock, J.T. Signaling through the primary cilium. Front. Cell Dev. Biol. 2018, 6, 8. [Google Scholar] [CrossRef]
- Wong, S.Y.; Seol, A.D.; So, P.L.; Ermilov, A.N.; Bichakjian, C.K.; Epstein, E.H.; Dlugosz, A.A.; Reiter, J.F. Primary cilia can both mediate and suppress Hedgehog pathway-dependent tumorigenesis. Nat. Med. 2009, 15, 1055–1061. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, M.A.; Louie, C.M.; Silhavy, J.L.; Sintasath, L.; Decambre, M.; Nigam, S.K.; Willert, K.; Gleeson, J.G. Impaired Wnt-Β-catenin signaling disrupts adult renal homeostasis and leads to cystic kidney ciliopathy. Nat. Med. 2009, 15, 1046–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferland, R.J.; Walsh, C.A. Joubert Syndrome. In Encyclopedia of Neuroscience; Elsevier: Amsterdam, The Netherlands, 2009; pp. 249–256. ISBN 9780080450469. [Google Scholar]
- Kim, S.; Nie, H.; Nesin, V.; Tran, U.; Outeda, P.; Bai, C.X.; Keeling, J.; Maskey, D.; Watnick, T.; Wessely, O.; et al. The polycystin complex mediates Wnt/Ca2+ signalling. Nat. Cell Biol. 2016, 18, 752–764. [Google Scholar] [CrossRef] [PubMed]
- Radford, R.; Slattery, C.; Jennings, P.; Blaque, O.; Pfaller, W.; Gmuender, H.; van Delft, J.; Ryan, M.P.; McMorrow, T. Carcinogens induce loss of the primary cilium in human renal proximal tubular epithelial cells independently of effects on the cell cycle. Am. J. Physiol. Ren. Physiol. 2012, 302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pala, R.; Alomari, N.; Nauli, S.M. Primary cilium-dependent signaling mechanisms. Int. J. Mol. Sci. 2017, 18, 2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgins, M.; Obaidi, I.; McMorrow, T. Primary cilia and their role in cancer (Review). Oncol. Lett. 2019, 17, 3041–3047. [Google Scholar] [CrossRef]
- Arts, H.H.; Knoers, N.V.A.M. Current insights into renal ciliopathies: What can genetics teach us? Pediatr. Nephrol. 2013, 28, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.; Tao, Y.H. Nephronophthisis: A review of genotype–phenotype correlation. Nephrology 2018, 23, 904–911. [Google Scholar] [CrossRef] [Green Version]
- Simms, R.J.; Eley, L.; Sayer, J.A. Nephronophthisis. Eur. J. Hum. Genet. 2009, 17, 406–416. [Google Scholar] [CrossRef] [Green Version]
- Novarino, G.; Akizu, N.; Gleeson, J.G. Modeling human disease in humans: The ciliopathies. Cell 2011, 147, 70–79. [Google Scholar] [CrossRef] [Green Version]
- Hildebrandt, F.; Otto, E.; Rensing, C.; Nothwang, H.G.; Vollmer, M.; Adolphs, J.; Hanusch, H.; Brandis, M. A novel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Nat. Genet. 1997, 17, 149–153. [Google Scholar] [CrossRef]
- Caridi, G.; Dagnino, M.; Rossi, A.; Valente, E.M.; Bertini, E.; Fazzi, E.; Emma, F.; Murer, L.; Verrina, E.; Ghiggeri, G.M. Nephronophthisis type 1 deletion syndrome with neurological symptoms: Prevalence and significance of the association. Kidney Int. 2006, 70, 1342–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delvallée, C.; Nicaise, S.; Antin, M.; Leuvrey, A.S.; Nourisson, E.; Leitch, C.C.; Kellaris, G.; Stoetzel, C.; Geoffroy, V.; Scheidecker, S.; et al. A BBS1 SVA F retrotransposon insertion is a frequent cause of Bardet-Biedl syndrome. Clin. Genet. 2020. [Google Scholar] [CrossRef]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef] [Green Version]
- Walczak-Sztulpa, J.; Eggenschwiler, J.; Osborn, D.; Brown, D.A.; Emma, F.; Klingenberg, C.; Hennekam, R.C.; Torre, G.; Garshasbi, M.; Tzschach, A.; et al. Cranioectodermal Dysplasia, Sensenbrenner Syndrome, Is a Ciliopathy Caused by Mutations in the IFT122 Gene. Am. J. Hum. Genet. 2010, 86, 949–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartill, V.; Szymanska, K.; Sharif, S.M.; Wheway, G.; Johnson, C.A. Meckel-Gruber syndrome: An update on diagnosis, clinical management, and research advances. Front. Pediatr. 2017, 5, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, V.E.; Harris, P.C.; Pirson, Y. Autosomal dominant polycystic kidney disease. Lancet 2007, 369, 1287–1301. [Google Scholar] [CrossRef]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Prim. 2018, 4, 1–24. [Google Scholar] [CrossRef]
- Nauli, S.M.; Alenghat, F.J.; Luo, Y.; Williams, E.; Vassilev, P.; Li, X.; Elia, A.E.H.; Lu, W.; Brown, E.M.; Quinn, S.J.; et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 2003, 33, 129–137. [Google Scholar] [CrossRef]
- Yoder, B.K.; Hou, X.; Guay-Woodford, L.M. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J. Am. Soc. Nephrol. 2002, 13, 2508–2516. [Google Scholar] [CrossRef] [Green Version]
- Heyer, C.M.; Sundsbak, J.L.; Abebe, K.Z.; Chapman, A.B.; Torres, V.E.; Grantham, J.J.; Bae, K.T.; Schrier, R.W.; Perrone, R.D.; Braun, W.E.; et al. Predicted mutation strength of nontruncating PKD1 mutations AIDS genotype-phenotype correlations in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2016, 27, 2872–2884. [Google Scholar] [CrossRef] [Green Version]
- Besse, W.; Dong, K.; Choi, J.; Punia, S.; Fedeles, S.V.; Choi, M.; Gallagher, A.R.; Huang, E.B.; Gulati, A.; Knight, J.; et al. Isolated polycystic liver disease genes define effectors of polycystin-1 function. J. Clin. Investig. 2017, 127, 1772–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornec-Le Gall, E.; Olson, R.J.; Besse, W.; Heyer, C.M.; Gainullin, V.G.; Smith, J.M.; Audrézet, M.P.; Hopp, K.; Porath, B.; Shi, B.; et al. Monoallelic Mutations to DNAJB11 Cause Atypical Autosomal-Dominant Polycystic Kidney Disease. Am. J. Hum. Genet. 2018, 102, 832–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drenth, J.P.H.; TeMorsche, R.H.M.; Smink, R.; Bonifacino, J.S.; Jansen, J.B.M.J. Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat. Genet. 2003, 33, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Beales, P.L. Ciliopathies: An expanding disease spectrum. Pediatr. Nephrol. 2011, 26, 1039–1056. [Google Scholar] [CrossRef] [Green Version]
- Chebib, F.T.; Torres, V.E. Autosomal Dominant Polycystic Kidney Disease: Core Curriculum 2016. Am. J. Kidney Dis. 2016, 67, 792–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, C.J.; Yuan, D.; Masyuk, T.V.; Wang, X.; Punyashthiti, R.; Whelan, S.; Bacallao, R.; Torra, R.; LaRusso, N.F.; Torres, V.E.; et al. Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum. Mol. Genet. 2003, 12, 2703–2710. [Google Scholar] [CrossRef]
- Zhang, M.Z.; Mai, W.; Li, C.; Cho, S.Y.; Hao, C.; Moeckel, G.; Zhao, R.; Kim, I.; Wang, J.; Xiong, H.; et al. PKHD1 protein encoded by the gene for autosomal recessive polycystic kidney disease associates with basal bodies and primary cilia in renal epithelial cells. Proc. Natl. Acad. Sci. USA 2004, 101, 2311–2316. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, W.E.; Avner, E.D. Molecular and cellular pathophysiology of autosomal recessive polycystic kidney disease (ARPKD). Cell Tissue Res. 2006, 326, 671–685. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Galeano, M.C.R.; Ott, E.; Kaeslin, G.; Kausalya, P.J.; Kramer, C.; Ortiz-Brüchle, N.; Hilger, N.; Metzis, V.; Hiersche, M.; et al. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat. Genet. 2017, 49, 1025–1034. [Google Scholar] [CrossRef]
- Bergmann, C. Educational paper; ciliopathies. Eur. J. Pediatr. 2012, 171, 1285–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiel, C.; Kessler, K.; Giessl, A.; Dimmler, A.; Shalev, S.A.; Von Der Haar, S.; Zenker, M.; Zahnleiter, D.; Stöss, H.; Beinder, E.; et al. NEK1 mutations cause short-rib polydactyly syndrome type majewski. Am. J. Hum. Genet. 2011, 88, 106–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slavotinek, A.M.; Stone, E.M.; Mykytyn, K.; Heckenlively, J.R.; Green, J.S.; Heon, E.; Musarella, M.A.; Parfrey, P.S.; Sheffield, V.C.; Biesecker, L.G. Mutations in MKKS cause Bardet-Biedl syndrome. Nat. Genet. 2000, 26, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Beales, P.L.; Warner, A.M.; Hitman, G.A.; Thakker, R.; Flinter, F.A. Bardet-Biedl syndrome: A molecular and phenotypic study of 18 families. J. Med. Genet. 1997, 34, 92–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Esmail, M.A.; Ansley, S.J.; Blacque, O.E.; Boroevich, K.; Ross, A.J.; Moore, S.J.; Badano, J.L.; May-Simera, H.; Compton, D.S.; et al. Mutations in a member of the Ras superfamily of small GTP-binding proteins causes Bardet-Biedl syndrome. Nat. Genet. 2004, 36, 989–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piva, F.; Santoni, M.; Matrana, M.R.; Satti, S.; Giulietti, M.; Occhipinti, G.; Massari, F.; Cheng, L.; Lopez-Beltran, A.; Scarpelli, M.; et al. BAP1, PBRM1 and SETD2 in clear-cell renal cell carcinoma: Molecular diagnostics and possible targets for personalized therapies. Expert Rev. Mol. Diagn. 2015, 15, 1201–1210. [Google Scholar] [CrossRef]
- Montironi, R.; Santoni, M.; Scarpelli, M.; Piva, F.; Lopez-Beltran, A.; Cheng, L.; Briganti, A.; Montorsi, F. Re: Epithelial-to-mesenchymal transition in renal neoplasms. Eur. Urol. 2015, 68, 736–737. [Google Scholar] [CrossRef]
- Massari, F.; Ciccarese, C.; Santoni, M.; Brunelli, M.; Piva, F.; Modena, A.; Bimbatti, D.; Fantinel, E.; Santini, D.; Cheng, L.; et al. Metabolic alterations in renal cell carcinoma. Cancer Treat. Rev. 2015, 41, 767–776. [Google Scholar] [CrossRef]
- Kim, W.Y.; Kaelin, W.G. Role of VHL gene mutation in human cancer. J. Clin. Oncol. 2004, 22, 4991–5004. [Google Scholar] [CrossRef]
- Peña-Llopis, S.; Vega-Rubín-De-Celis, S.; Liao, A.; Leng, N.; Pavía-Jiménez, A.; Wang, S.; Yamasaki, T.; Zhrebker, L.; Sivanand, S.; Spence, P.; et al. BAP1 loss defines a new class of renal cell carcinoma. Nat. Genet. 2012, 44, 751–759. [Google Scholar] [CrossRef]
- Piva, F.; Giulietti, M.; Occhipinti, G.; Santoni, M.; Massari, F.; Sotte, V.; Iacovelli, R.; Burattini, L.; Santini, D.; Montironi, R.; et al. Computational analysis of the mutations in BAP1, PBRM1 and SETD2 genes reveals the impaired molecular processes in Renal Cell Carcinoma. Oncotarget 2015, 6, 32161–32168. [Google Scholar] [CrossRef]
- Sabanovic, B.; Giulietti, M.; Piva, F. Role of primary cilium in pancreatic ductal adenocarcinoma (Review). Int. J. Oncol. 2020, 57, 1095–1102. [Google Scholar] [CrossRef]
- Yoder, B.K. Role of primary cilia in the pathogenesis of polycystic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 1381–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lolkema, M.P.; Mehra, N.; Jorna, A.S.; Van Beest, M.; Giles, R.H.; Voest, E.E. The von Hippel-Lindau tumor suppressor protein influences microtubule dynamics at the cell periphery. Exp. Cell Res. 2004, 301, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Schermer, B.; Ghenoiu, C.; Bartram, M.; Müller, R.U.; Kotsis, F.; Höhne, M.; Kühn, W.; Rapka, M.; Nitschke, R.; Zentgraf, H.; et al. The von Hippel-Lindau tumor suppressor protein controls ciliogenesis by orienting microtubule growth. J. Cell Biol. 2006, 175, 547–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteban, M.A.; Harten, S.K.; Tran, M.G.; Maxwell, P.H. Formation of primary cilia in the renal epithelium is regulated by the von Hippel-Lindau tumor suppressor protein. J. Am. Soc. Nephrol. 2006, 17, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Lutz, M.S.; Burk, R.D. Primary cilium formation requires von Hippel-Lindau gene function in renal-derived cells. Cancer Res. 2006, 66, 6903–6907. [Google Scholar] [CrossRef] [Green Version]
- Lolkema, M.P.; Mans, D.A.; Ulfman, L.H.; Volpi, S.; Voest, E.E.; Giles, R.H. Allele-specific regulation of primary cilia function by the von Hippel-Lindau tumor suppressor. Eur. J. Hum. Genet. 2008, 16, 73–78. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santoni, M.; Piva, F.; Cimadamore, A.; Giulietti, M.; Battelli, N.; Montironi, R.; Cosmai, L.; Porta, C. Exploring the Spectrum of Kidney Ciliopathies. Diagnostics 2020, 10, 1099. https://doi.org/10.3390/diagnostics10121099
Santoni M, Piva F, Cimadamore A, Giulietti M, Battelli N, Montironi R, Cosmai L, Porta C. Exploring the Spectrum of Kidney Ciliopathies. Diagnostics. 2020; 10(12):1099. https://doi.org/10.3390/diagnostics10121099
Chicago/Turabian StyleSantoni, Matteo, Francesco Piva, Alessia Cimadamore, Matteo Giulietti, Nicola Battelli, Rodolfo Montironi, Laura Cosmai, and Camillo Porta. 2020. "Exploring the Spectrum of Kidney Ciliopathies" Diagnostics 10, no. 12: 1099. https://doi.org/10.3390/diagnostics10121099
APA StyleSantoni, M., Piva, F., Cimadamore, A., Giulietti, M., Battelli, N., Montironi, R., Cosmai, L., & Porta, C. (2020). Exploring the Spectrum of Kidney Ciliopathies. Diagnostics, 10(12), 1099. https://doi.org/10.3390/diagnostics10121099