Low-Grade Gliomas in Patients with Noonan Syndrome: Case-Based Review of the Literature



 ,
,  , , ,
, , ,  ,
,  , , , , ,
, , , , , 
Abstract
:1. Introduction
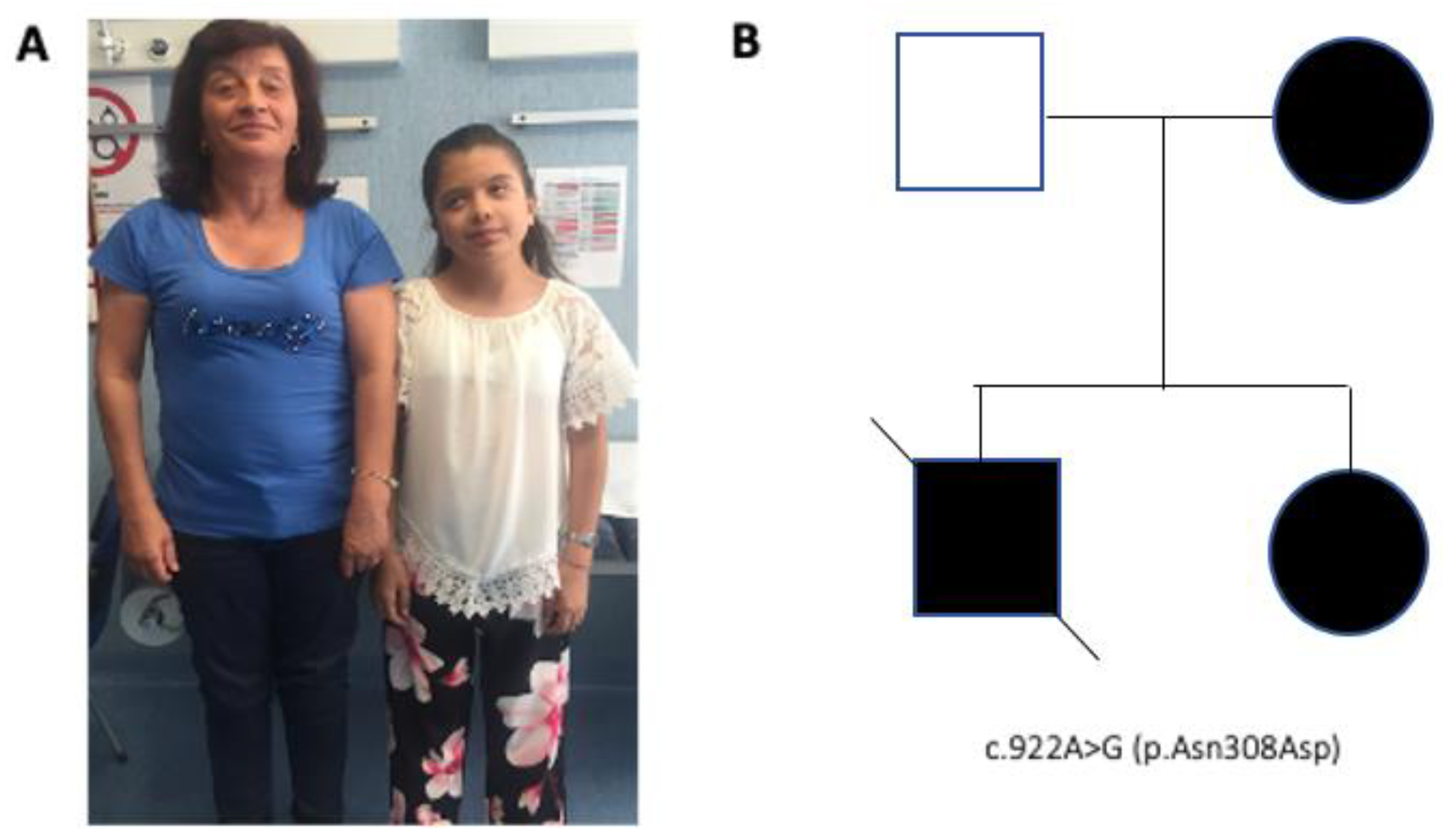
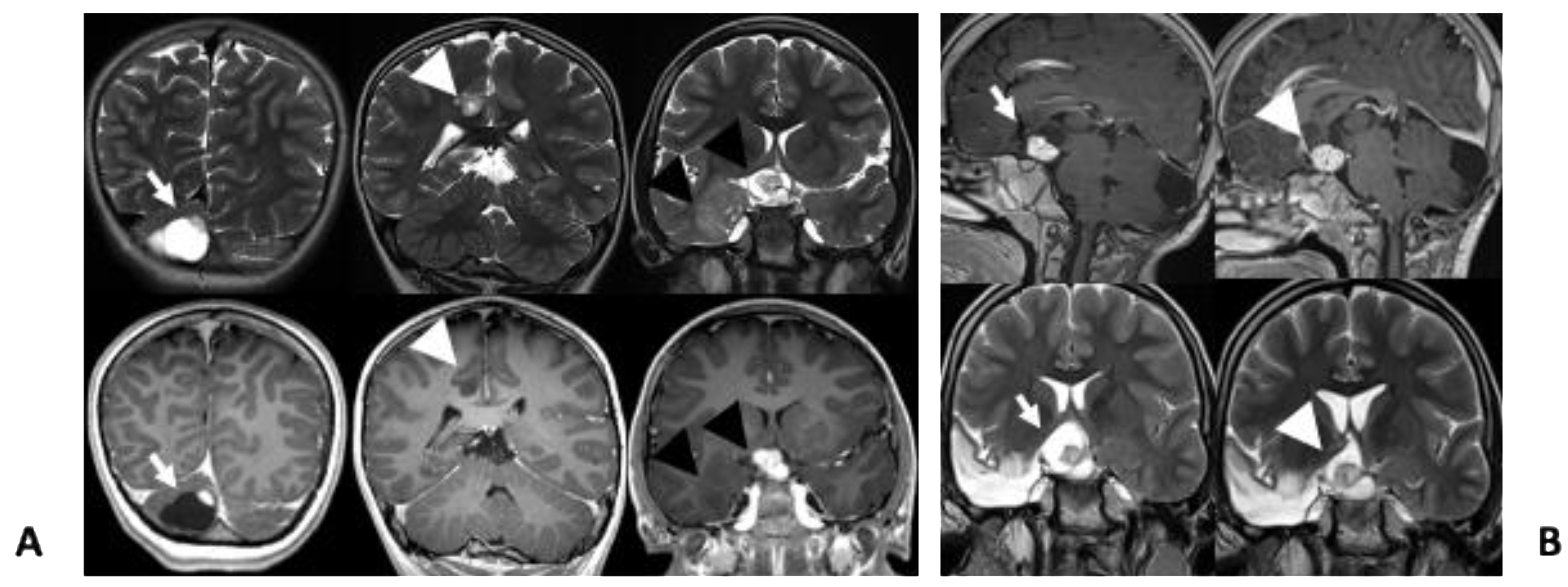
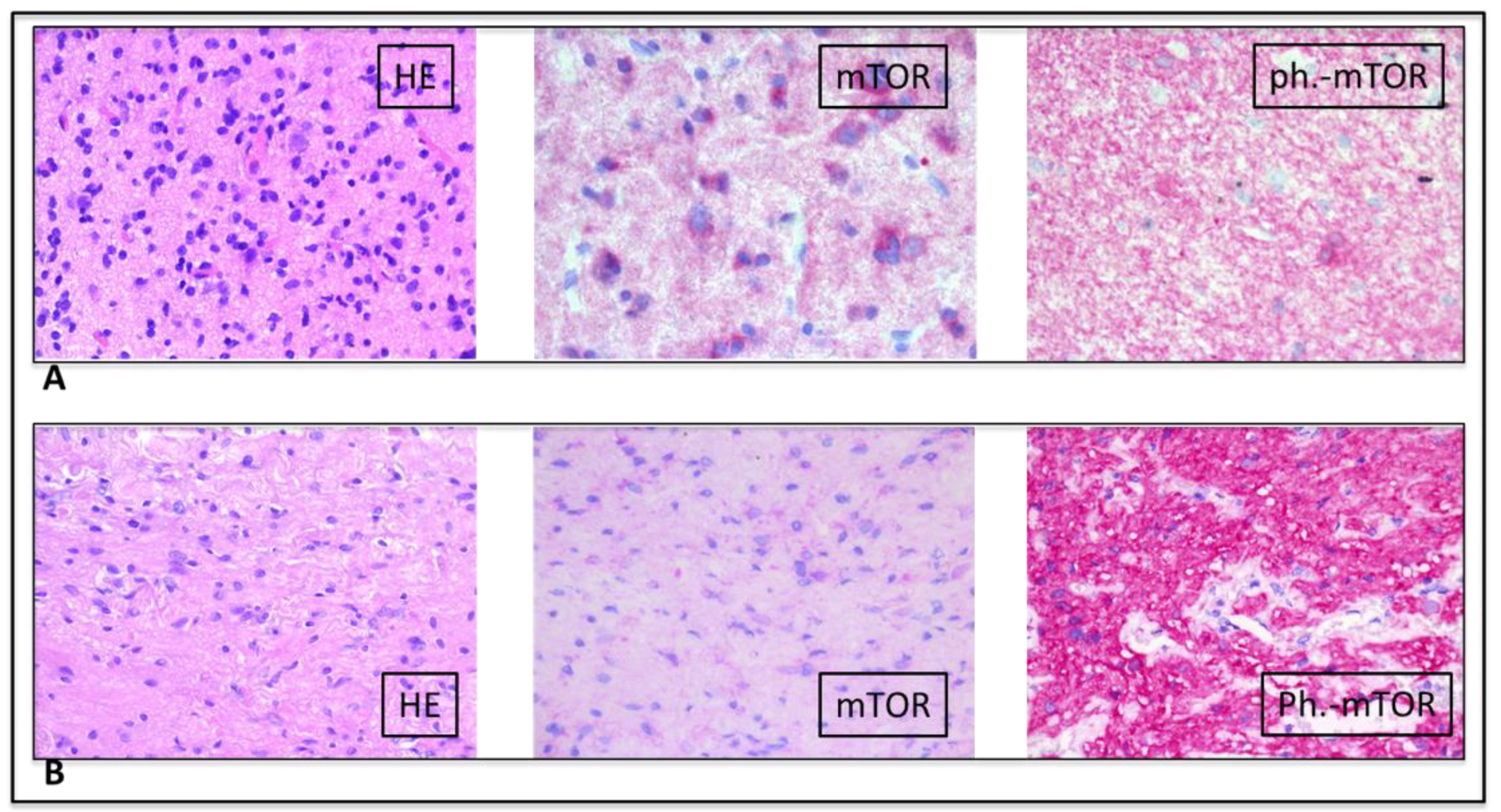
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roberts, A.E.; Allanson, J.E.; Tartaglia, M.; Gelb, B.D. Noonan syndrome. Lancet 2013, 381, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, S.; de Luca, A.; Stellacci, E.; Rossi, C.; Checquolo, S.; Lepri, F.; Caputo, V.; Silvano, M.; Buscherini, F.; Consoli, F.; et al. Heterozygous germline mutations in the CBL tumor-suppressor gene cause a Noonan syndrome-like phenotype. Am. J. Hum. Genet. 2010, 87, 250–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athota, J.P.; Bhat, M.; Nampoothiri, S.; Gowrishankar, K.; Narayanachar, S.G.; Puttamallesh, V.; Farooque, M.O.; Shetty, S. Molecular and clinical studies in 107 Noonan syndrome affected individuals with PTPN11 mutations. BMC Med. Genet. 2020, 21, 50. [Google Scholar] [CrossRef] [PubMed]
- Franz, D.N.; Belousova, E.; Sparagana, S.P.; Bebin, E.M.; Frost, M.; Kuperman, R.; Witt, O.; Kohrman, M.; Flamini, J.R.; Wu, J.Y.; et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): A multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2013, 381, 125–132. [Google Scholar] [CrossRef]
- Smpokou, P.; Zand, D.J.; Rosenbaum, K.N.; Summar, M.L. Malignancy in Noonan syndrome and related disorders. Clin. Genet. 2015, 88, 516–522. [Google Scholar] [CrossRef]
- Jongmans, M.C.J.; van der Burgt, I.; Hoogerbrugge, P.M.; Noordam, K.; Yntema, H.G.; Nillesen, W.M.; Kuiper, R.P.; Ligtenberg, M.J.L.; van Kessel, A.G.; van Krieken, J.H.; et al. Cancer risk in patients with Noonan syndrome carrying a PTPN11 mutation. Eur. J. Hum. Genet. 2011, 19, 870–874. [Google Scholar] [CrossRef] [Green Version]
- Kratz, C.P.; Rapisuwon, S.; Reed, H.; Hasle, H.; Rosenberg, P.S. Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am. J. Med. Genet. C Semin Med. Genet. 2011, 157C, 83–89. [Google Scholar] [CrossRef]
- Kratz, C.P.; Franke, L.; Peters, H.; Kohlschmidt, N.; Kazmierczak, B.; Finckh, U.; Bier, A.; Eichhorn, B.; Blank, C.; Kraus, C.; et al. Cancer spectrum and frequency among children with Noonan, Costello, and cardio-facio-cutaneous syndromes. Br. J. Cancer 2015, 112, 1392–1397. [Google Scholar] [CrossRef] [Green Version]
- Loh, M.L.; Vattikuti, S.; Schubbert, S.; Reynolds, M.G.; Carlson, E.; Lieuw, K.H.; Cheng, J.W.; Lee, C.M.; Stokoe, D.; Bonifas, J.M.; et al. Mutations in PTPN11 implicate the SHP-2 phosphatase in leukemogenesis. Blood 2004, 103, 2325–2331. [Google Scholar] [CrossRef]
- Tartaglia, M.; Martinelli, S.; Iavarone, I.; Cazzaniga, G.; Spinelli, M.; Giarin, E.; Petrangeli, V.; Carta, C.; Masetti, R.; Arico, M.; et al. Somatic PTPN11 mutations in childhood acute myeloid leukaemia. Br. J. Haematol. 2005, 129, 333–339. [Google Scholar] [CrossRef]
- Miyamoto, D.; Miyamoto, M.; Takahashi, A.; Yomogita, Y.; Higashi, H.; Kondo, S.; Hatakeyama, M. Isolation of a distinct class of gain-of-function SHP-2 mutants with oncogenic RAS-like transforming activity from solid tumors. Oncogene 2008, 27, 3508–3515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentiresalj, M.; Paez, J.G.; David, F.S.; Keilhack, H.; Halmos, B.; Naoki, K.; Maris, J.M.; Richardson, A.L.; Bardelli, A.; Sagarbaker, D.J.; et al. Activating mutations of the noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 2004, 64, 8816–8820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tartaglia, M.; Mehler, E.L.; Goldberg, R.; Zampino, G.; Brunner, H.G.; Kremer, H.; van der Burgt, I.; Crosby, A.H.; Ion, A.; Jeffery, S.; et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 2001, 29, 465–468. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L. Ras oncogenes in human cancer: A review. Cancer Res. 1989, 49, 4682–4689. [Google Scholar] [PubMed]
- Tartaglia, M.; Martinelli, S.; Stella, L.; Bocchinfuso, G.; Flex, E.; Cordeddu, V.; Zampino, G.; van der Burgt, I.; Palleschi, A.; Petrucci, T.C.; et al. Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease. Am. J. Hum. Genet. 2006, 78, 279–290. [Google Scholar] [CrossRef]
- McWilliams, G.; SantaCruz, K.; Hart, B.; Clericuzio, C. Occurrence of DNET and other brain tumors in Noonan syndrome warrants caution with growth hormone therapy. Am. J. Med. Genet. 2015, 170 Pt A, 195–201. [Google Scholar] [CrossRef]
- Siegfried, A.; Cances, C.; Denuelle, M.; Loukh, N.; Tauber, M.; Cave, H.; Delisle, M. Noonan syndrome, PTPN11 mutations, and brain tumors. A clinical report and review of the literature. Am. J. Med. Genet. 2017, 173, 1061–1065. [Google Scholar] [CrossRef]
- Bangalore Krishna, K.; Pagan, P.; Escobar, O.; Popovic, J. Occurrence of Cranial Neoplasms in Pediatric Patients with Noonan Syndrome Receiving Growth Hormone: Is Screening with Brain MRI prior to Initiation of Growth Hormone Indicated? Horm. Res. Paediatr. 2017, 88, 423–426. [Google Scholar] [CrossRef]
- Elayadi, M.; Ansari, M.; Kuhnol, C.D.; Bendel, A.; Sturm, D.; Pietsch, T.; Kramm, C.M.; von Bueren, A.O. Occurrence of high-grade glioma in Noonan syndrome: Report of two cases. Pediatr. Blood Cancer 2019, 66, e27625. [Google Scholar] [CrossRef]
- Rickert, C.H.; Paulus, W. Epidemiology of central nervous system tumors in childhood and adolescence based on the new WHO classification. Childs Nerv. Syst. 2001, 17, 503–511. [Google Scholar] [CrossRef]
- Bessis, D.; Miquel, J.; Bourrat, E.; Chiaverini, C.; Moricepicard, F.; Abadie, C.; Manna, F.; Baumann, C.; Best, M.; Blanchet, P.; et al. Dermatological manifestations in Noonan syndrome: A prospective multicentric study of 129 patients positive for mutation. Br. J. Dermatol. 2019, 180, 1438–1448. [Google Scholar] [CrossRef] [PubMed]
- Digilio, M.C.; Sarkozy, A.; de Zorzi, A.; Pacileo, G.; Limongelli, G.; Mingarelli, R.; Calabro, R.; Marino, B.; Dallapiccola, B. Leopard syndrome: Clinical diagnosis in the first year of life. Am. J. Med. Genet. A 2006, 140, 740–746. [Google Scholar] [CrossRef] [PubMed]
- Digilio, M.C.; Lepri, F.; Baban, A.; Dentici, M.L.; Versacci, P.; Capolino, R.; Ferese, R.; de Luca, A.; Tartaglia, M.; Marino, B.; et al. RASopathies: Clinical Diagnosis in the First Year of Life. Mol. Syndromol. 2011, 1, 282–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villani, A.; Greer, M.C.; Kalish, J.M.; Nakagawara, A.; Nathanson, K.L.; Pajtler, K.W.; Pfister, S.M.; Walsh, M.F.; Wasserman, J.D.; Zelley, K.; et al. Recommendations for Cancer Surveillance in Individuals with RASopathies and Other Rare Genetic Conditions with Increased Cancer Risk. Clin. Cancer Res. 2017, 23, e83–e90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selter, M.; Dresel, R.; Althaus, J.; Bartels, M.B.; Dittrich, S.; Geb, S.; Hoche, F.; Qirshi, M.; Vlaho, S.; Zielen, S.; et al. Dysembryoplastic neuroepithelial tumor (DNET) in a patient with Noonan syndrome. Neuropediatrics 2010, 41, P1356. [Google Scholar] [CrossRef]
- Bendel, A.; Hansen, M.; Dugan, S.; Mendelsohn, N. Dysembyoplastic Neuroepithelial Tumor in Two Relatives with Noonan Syndrome And A PTPN 11 Mutation. Neuro. Oncol. 2012, 14, 156. [Google Scholar] [CrossRef] [Green Version]
- Delisle, M.; Siegfried, A.; Tauber, M.; Cave, H.; Loukh, N.; Boetto, S.; Bertozzi, A.; Urocoste, E.; Cances, C. Dysembryoplastic neuroepithelial tumor (DNET) and Noonan syndrome. A case report. In Proceedings of the Brain Pathology. Abstracts of the XVIII International Congress of Neuropathology, Rio de Janeiro, Brazil, 14–18 September 2014. [Google Scholar]
- Sherman, C.B.; Ali-Nazir, A.; Gonzales-Gomez, I.; Finlay, J.L.; Dhall, G. Primary mixed glioneuronal tumor of the central nervous system in a patient with noonan syndrome: A case report and review of the literature. J. Pediatr. Hematol. Oncol. 2009, 31, 61–64. [Google Scholar] [CrossRef]
- Schuettpelz, L.G.; Mcdonald, S.; Whitesell, K.; Desruisseau, D.M.; Grange, D.K.; Gurnett, C.A.; Wilson, D.B. Pilocytic astrocytoma in a child with Noonan syndrome. Pediatr. Blood Cancer 2009, 53, 1147–1149. [Google Scholar] [CrossRef]
- Fryssira, H.; Leventopoulos, G.; Psoni, S.; Kitsioutzeli, S.; Stavrianeas, N.G.; Kanavakis, E. Tumor development in three patients with Noonan syndrome. Eur. J. Pediatr. 2008, 167, 1025–1031. [Google Scholar] [CrossRef]
- de Jong, M.; Schieving, J.; Goraj, B. Remarkable intra-cerebral lesions on MRI in a patient with Noonan syndrome. Eur. J. Radiol. Extra. 2011, 78, e17–e19. [Google Scholar] [CrossRef]
- Karafin, M.; Jallo, G.I.; Ayars, M.; Eberhart, C.G.; Rodriguez, F.J. Rosette forming glioneuronal tumor in association with Noonan syndrome: Pathobiological implications. Clin. Neuropathol. 2011, 30, 297–300. [Google Scholar] [PubMed]
- Takagi, M.; Miyashita, Y.; Koga, M.; Ebara, S.; Arita, N.; Kasayama, S. Estrogen deficiency is a potential cause for osteopenia in adult male patients with Noonan’s syndrome. Calcif. Tissue Int. 2000, 66, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Sanford, R.A.; Bowman, R.; Tomita, T.; De Leon, G.; Palka, P. A 16-year-old male with Noonan’s syndrome develops progressive scoliosis and deteriorating gait. Pediatr. Neurosurg. 1999, 30, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Fort, J.A.; Yachnis, A.T.; Williams, C.A. Optic nerve pilomyxoid astrocytoma in a patient with Noonan syndrome. Pediatr. Blood Cancer 2015, 62, 1084–1086. [Google Scholar] [CrossRef]
- Martinelli, S.; Carta, C.; Flex, E.; Binni, F.; Cordisco, E.L.; Moretti, S.; Puxeddu, E.; Tonacchera, M.; Pinchera, A.; Mcdowell, H.P.; et al. Activating PTPN11 mutations play a minor role in pediatric and adult solid tumors. Cancer Genet. Cytogenet. 2006, 166, 124–129. [Google Scholar] [CrossRef]
- Gnekow, A.; Kandels, D.; van Tilburg, C.M.; Azizi, A.; Opocher, E.; Stokland, T.; Driever, P.H.; Meeteren, A.Y.N.S.; Thomale, U.W.; Schuhmann, M.U.; et al. SIOP-E-BTG and GPOH Guidelines for Diagnosis and Treatment of Children and Adolescents with Low Grade Glioma. Klin. Padiatr. 2019, 231, 107–135. [Google Scholar]
- Goodden, J.; Pizer, B.; Pettorini, B.; Williams, D.; Blair, J.; Didi, M.; Thorp, N.; Mallucci, C. The role of surgery in optic pathway/hypothalamic gliomas in children. J. Neurosurg. Pediatr. 2014, 13, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Yu, W.; Zhang, J.; Chan, R.J.; Loh, M.L.; Zhang, Z.; Bunting, K.D.; Qu, C. Inhibition of the Gab2/PI3K/mTOR signaling ameliorates myeloid malignancy caused by Ptpn11 (Shp2) gain-of-function mutations. Leukemia 2017, 31, 1415–1422. [Google Scholar] [CrossRef] [Green Version]
- Catanzaro, G.; Besharat, Z.M.; Miele, E.; Chiacchiarini, M.; Po, A.; Carai, A.; Marras, C.E.; Antonelli, M.; Badiali, M.; Raso, A.; et al. The miR-139-5p regulates proliferation of supratentorial paediatric low-grade gliomas by targeting the PI3K/AKT/mTORC1 signalling. Neuropath. Appl. Neuro. 2018, 44, 687–706. [Google Scholar] [CrossRef] [Green Version]
- Krueger, D.A.; Care, M.M.; Holland, K.D.; Agricola, K.; Tudor, C.; Mangeshkar, P.; Wilson, K.; Byars, A.W.; Sahmoud, T.; Franz, D.N. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N. Engl. J. Med. 2010, 363, 1801–1811. [Google Scholar] [CrossRef]
- Tartaglia, M.; Gelb, B.D.; Zenker, M. Noonan syndrome and clinically related disorders. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 161–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| No. | Reference | Age | Gender | Noonan Syndrome Diagnosis | Brain Tumor Diagnosis | Location |
|---|---|---|---|---|---|---|
| 1 | McWilliams et al. [16] | 8 | M | PTPN11 p.Glu139Asp | DNET | Temporal lobe and cerebellum |
| 2 | Jongmans et al. [6] | 10 | Ukn | PTPN11 c.179G > C; p.Gly60Ala | DNET | Temporal lobe |
| 3 | Selter et al. [25] Pellegrin et al. [17] | 13 | M | PTPN11 exon 3 ** | DNET | Left parietal lobe |
| 4 | Bendel et al. [26] | 17 | M | PTPN11 c.174C > G; p.Asn58Lys | DNET | Occipital cortex |
| 5 | Bendel et al. [26] | 37 | M | Maternal uncle of Case 4 | DNET | Unknown |
| 6 | Delisle et al. [27] | 12 | M | PTPN11 mutation ** | DNET | Temporal lobe and thalamus |
| 7 | Krishna et al. [18] | 9 | M | PTPN11 p.Asp61Gly | DNET | Temporal lobe and cerebellum |
| 8 | Pellegrin et al. [17] | 13 | M | N/A | DNET | Right parieto-occipital cortex |
| 9 | Pellegrin et al. [17] | 13 | M | PTPN11 exon 3 mutation ** | DNET (MRI) | Left parietal lobe |
| 10 | Kratz et al. [8] | 6 | F | PTPN11 p.Asn308Asp | DNET | Unknown |
| 11 | Siegfried et al. [17] | 16 | M | PTPN11 c.922A > G; p.Asn308Asp | DNET | Left temporal and frontal lobe, right thalamus |
| 12 | Rankin et al. [26] | 10 | M | PTPN11 c.1403C > T; p.Thr468Met | Medulloblastoma | Cerebellum |
| 13 | Jongmans el al [6] | 18 | F | PTPN11 c.417G > C; p.Glu139Asp | Oligodendroglioma | Hypothalamus |
| 14 | Sherman et al. [28] | 6 | M | PTPN11 c.172A > G; p.Asn58Asp | Low-grade mixed glioneuronal tumor | Suprasellar cisterns, sella turcica and hypothalamus and diffuse leptomeningeal disease |
| 15 | Schuettpelz et al. [29] | 8 | M | PTPN11 c.1471C > T and c.1472C > T; p.Pro491Phe | Pilocytic astrocytoma | Sellar/suprasellar with extension to prepontine region to the level of the pontomedullary junction |
| 16 | Fryssira et al. [30] | 11 | F | Clinical diagnosis | Pilocytic astrocytoma | Sellar/suprasellar |
| 17 | De Jongo et al. [31] | 21 | M | PTPN11 mutation ** | Multiple indeterminate lesions on MRI | Multiple: supratentorial, infratentorial, cortical and subortical, thalamus |
| 18 | Karafin et al. [32] | 18 | M | Clinical diagnosis | Rosette forming glioneuronal tumor | Fourth ventricle |
| 19, 20, 21 | Rush et al. * [17] | Ukn | M | PTPN11 mutation ** | Low grade astrocytoma | Suprasellar and thalamic region |
| 22 | Kratz et al. [19] | 7 | M | PTPN11 p.Gly60Ala | Pilocytic astrocytoma | Right optic nerve |
| 23 | Takagi at al. [33] | 20 | M | Clinical diagnosis | Glioma | Unknown |
| 24 | Standford et al. [34] | 16 | M | Clinical diagnosis | Pilocytic astrocytoma | Intramedullary spinal cord lesion involving the cervical medullary junction and descending to the C2-C3 disc space level |
| 25 | Nair et al. [35] | 14 | M | PTPN11 c.417G > C in exon 4 | Pilomyxoid astrocytoma | Right optic nerve |
| 26 | Bendel and Pond. [19] | 14 | F | PTPN11 c.922A > G; p.Asn308Asp | High grade glioma | Left brainstem/cerebellar peduncle |
| 27 | Martinelli et al. [36] | 24 | Ukn | PTPN11 c.64A > G; pThr22Ala | Oligodendroglioma grade II | Unknown |
| 28 | El Ayadi et al. [19] | 14 | F | PTPN11 c.922A > G; p.Asn308Asp | Anaplastic astocytoma | Left brainstem/cerebellar peduncle |
| 29 | El Ayadi et al. [19] | 9 | M | PTPN11 c.5C > T; p.Thr2lle | Anaplastic astocytoma | Third ventricle, cerebellum and fornix |
| 30 | Our case | 9 | F | PTPN11 c.922A > G; p.Asn308Asp | Pilocytic astrocitoma and glioneuronal tumor | Cerebellum Right temporal lobe |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lodi, M.; Boccuto, L.; Carai, A.; Cacchione, A.; Miele, E.; Colafati, G.S.; Diomedi Camassei, F.; De Palma, L.; De Benedictis, A.; Ferretti, E.; et al. Low-Grade Gliomas in Patients with Noonan Syndrome: Case-Based Review of the Literature. Diagnostics 2020, 10, 582. https://doi.org/10.3390/diagnostics10080582
Lodi M, Boccuto L, Carai A, Cacchione A, Miele E, Colafati GS, Diomedi Camassei F, De Palma L, De Benedictis A, Ferretti E, et al. Low-Grade Gliomas in Patients with Noonan Syndrome: Case-Based Review of the Literature. Diagnostics. 2020; 10(8):582. https://doi.org/10.3390/diagnostics10080582
Chicago/Turabian StyleLodi, Mariachiara, Luigi Boccuto, Andrea Carai, Antonella Cacchione, Evelina Miele, Giovanna Stefania Colafati, Francesca Diomedi Camassei, Luca De Palma, Alessandro De Benedictis, Elisabetta Ferretti, and et al. 2020. "Low-Grade Gliomas in Patients with Noonan Syndrome: Case-Based Review of the Literature" Diagnostics 10, no. 8: 582. https://doi.org/10.3390/diagnostics10080582
APA StyleLodi, M., Boccuto, L., Carai, A., Cacchione, A., Miele, E., Colafati, G. S., Diomedi Camassei, F., De Palma, L., De Benedictis, A., Ferretti, E., Catanzaro, G., Pò, A., De Luca, A., Rinelli, M., Lepri, F. R., Agolini, E., Tartaglia, M., Locatelli, F., & Mastronuzzi, A. (2020). Low-Grade Gliomas in Patients with Noonan Syndrome: Case-Based Review of the Literature. Diagnostics, 10(8), 582. https://doi.org/10.3390/diagnostics10080582