
Systemic Sclerosis-Associated Pulmonary Hypertension: Spectrum and Impact


Abstract
:1. Introduction
2. Epidemiology
3. Pathogenesis and Clinical Features
3.1. Autoimmune Dysfunction
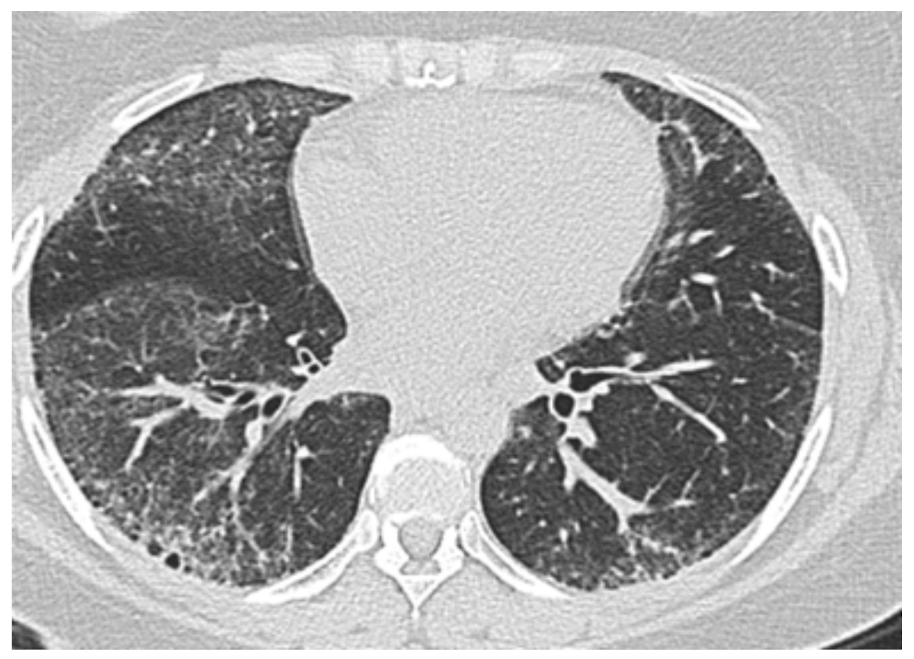
3.2. Interstitial Lung Disease-Associated PH in SSc
3.3. Cardiac Involvement in SSc-PAH
3.4. Clinical Features
4. Evaluation
4.1. Screening for SSc-PAH
4.2. Echocardiography
4.3. Laboratory
4.4. Pulmonary Function Test
4.5. Cardiac MRI
4.6. RHC and Vasodilator Testing
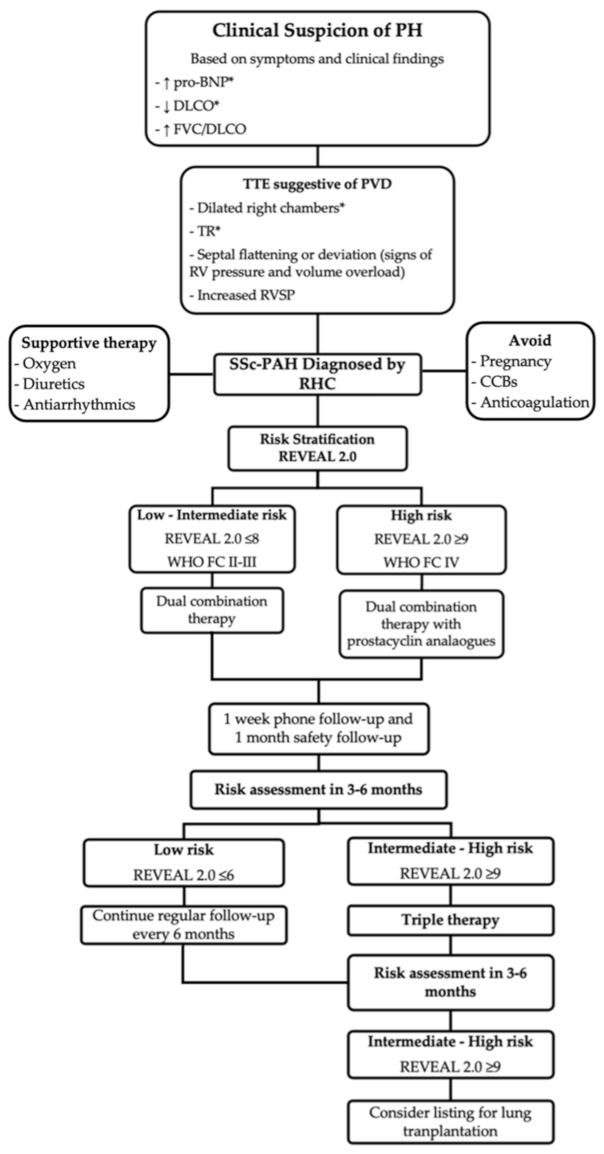
5. Management
5.1. Therapy
5.1.1. Prostacyclin Analogs
5.1.2. Phosphodiesterase Inhibitors
5.1.3. Endothelin Receptor Antagonists
5.1.4. Guanylate Cyclase Stimulator
5.1.5. Combination Therapy
5.2. Adjunct Therapies
5.3. Investigational Therapies
5.4. Lung Transplant
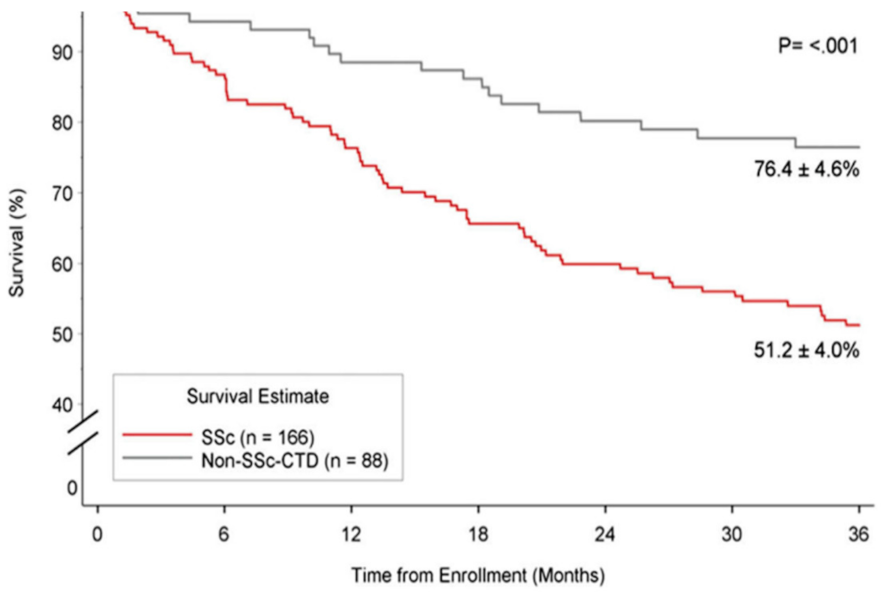
6. Survival and Prognosis
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Denton, C.P.; Khanna, D. Systemic sclerosis. Lancet 2017, 390, 1685–1699. [Google Scholar] [CrossRef]
- Jimenez, S.A.; Derk, C.T. Following the molecular pathways toward an understanding of the pathogenesis of systemic sclerosis. Ann. Intern Med. 2004, 140, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Allanore, Y.; Simms, R.; Distler, O.; Trojanowska, M.; Pope, J.; Denton, C.P.; Varga, J. Systemic sclerosis. Nat. Rev. Dis. Prim. 2015, 1, 15002. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Clements, P.J.; Tan, M.; McLaughlin, V.V.; Oudiz, R.J.; Tapson, V.F.; Channick, R.N.; Rubin, L.J.; Langer, A. Pulmonary Arterial Hyper-tension Quality Enhancement Research Initiative, I. The pulmonary arterial hypertension quality enhancement research ini-tiative: Comparison of patients with idiopathic PAH to patients with systemic sclerosis-associated PAH. Ann. Rheum. Dis. 2012, 71, 249–252. [Google Scholar] [CrossRef]
- Fisher, M.R.; Mathai, S.C.; Champion, H.C.; Girgis, R.E.; Housten-Harris, T.; Hummers, L.; Krishnan, J.A.; Wigley, F.; Hassoun, P.M. Clinical differences between idiopathic and scleroderma-related pulmonary hypertension. Arthritis Rheum. 2006, 54, 3043–3050. [Google Scholar] [CrossRef]
- Avouac, J.; Airo, P.; Meune, C.; Beretta, L.; Dieude, P.; Caramaschi, P.; Tiev, K.; Cappelli, S.; Diot, E.; Vacca, A.; et al. Prevalence of pul-monary hypertension in systemic sclerosis in European Caucasians and metaanalysis of 5 studies. J. Rheumatol. 2010, 37, 2290–2298. [Google Scholar] [CrossRef]
- Humbert, M.; Sitbon, O.; Chaouat, A.; Bertocchi, M.; Habib, G.; Gressin, V.; Yaici., A.; Weitzenblum., E.; Cordier, J.F.; Chabot, F.; et al. Pulmonary arterial hypertension in France: Results from a national registry. Am. J. Respir. Crit. Care Med. 2006, 173, 1023–1030. [Google Scholar] [CrossRef] [Green Version]
- Mukerjee, D.; George, D.S.; Coleiro, B.; Knight, C.; Denton, C.P.; Davar, J.; Black, C.M.; Coghlan, J.G. Prevalence and outcome in systemic sclerosis associated pulmonary arterial hypertension: Application of a registry approach. Ann. Rheum. Dis. 2003, 62, 1088–1093. [Google Scholar] [CrossRef] [Green Version]
- Hsu, V.M.; Chung, L.; Hummers, L.K.; Wigley, F.; Simms, R.; Bolster, M.; Silver, R.; Fischer, A.; Hinchcliff, M.E.; Varga, J.; et al. Development of pulmonary hypertension in a high-risk population with systemic sclerosis in the Pulmonary Hypertension Assessment and Recognition of Outcomes in Scleroderma (PHAROS) cohort study. Semin. Arthritis Rheum. 2014, 44, 55–62. [Google Scholar] [CrossRef]
- Badesch, D.B.; Raskob, G.E.; Elliott, C.G.; Krichman, A.M.; Farber, H.W.; Frost, A.E.; Barst, R.J.; Benza, R.L.; Liou, T.G.; Turner, M.; et al. Pul-monary arterial hypertension: Baseline characteristics from the REVEAL Registry. Chest 2010, 137, 376–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, D.; Gladue, H.; Channick, R.; Chung, L.; Distler, O.; Furst, D.E.; Hachulla, E.; Humbert, M.; Langleben, D.; Mathai, S.C.; et al. Recommendations for Screening and Detection of Connective Tissue Disease–Associated Pulmonary Arterial Hypertension. Arthritis Rheum. 2013, 65, 3194–3201. [Google Scholar] [CrossRef] [Green Version]
- Pasarikovski, C.R.; Granton, J.T.; Roos, A.M.; Sadeghi, S.; Kron, A.T.; Thenganatt, J.; Moric, J.; Chau, C.; Johnson, S.R. Sex disparities in systemic sclerosis-associated pulmonary arterial hypertension: A cohort study. Arthritis Res. 2016, 18, 30. [Google Scholar] [CrossRef] [Green Version]
- Girgis, R.E.; Mathai, S.C.; Krishnan, J.A.; Wigley, F.M.; Hassoun, P.M. Long-term outcome of bosentan treatment in idiopathic pul-monary arterial hypertension and pulmonary arterial hypertension associated with the scleroderma spectrum of diseases. J. Heart Lung Transplant. 2005, 24, 1626–1631. [Google Scholar] [CrossRef]
- Badesch, D.B.; Champion, H.C.; Sanchez, M.A.G.; Hoeper, M.M.; Loyd, J.E.; Manes, A.; McGoon, M.; Naeije, R.; Olschewski, H.; Oudiz, R.J.; et al. Diagnosis and Assessment of Pulmonary Arterial Hypertension. J. Am. Coll. Cardiol. 2009, 54, S55–S66. [Google Scholar] [CrossRef] [PubMed]
- Chaisson, N.F.; Hassoun, P.M. Systemic Sclerosis-Associated Pulmonary Arterial Hypertension. Chest 2013, 144, 1346–1356. [Google Scholar] [CrossRef] [Green Version]
- Farber, H.W.; Loscalzo, J. Pulmonary arterial hypertension. N. Engl. J. Med. 2004, 351, 1655–1665. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.R.; Granton, J.T. Pulmonary hypertension in systemic sclerosis and systemic lupus erythematosus. Eur. Respir. Rev. 2011, 20, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Kherbeck, N.; Tamby, M.C.; Bussone, G.; Dib, H.; Perros, F.; Humbert, M.; Mouthon, L. The Role of Inflammation and Autoimmunity in the Pathophysiology of Pulmonary Arterial Hypertension. Clin. Rev. Allergy Immunol. 2013, 44, 31–38. [Google Scholar] [CrossRef]
- Bourji, K.I.; Hassoun, P.M. Right ventricle dysfunction in pulmonary hypertension: Mechanisms and modes of detection. Curr. Opin. Pulm. Med. 2015, 21, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Campo, A.; Mathai, S.C.; Le Pavec, J.; Zaiman, A.L.; Hummers, L.K.; Boyce, D.; Housten, T.; Champion, H.C.; Lechtzin, N.; Wigley, F.M.; et al. Hemodynamic Predictors of Survival in Scleroderma-related Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2010, 182, 252–260. [Google Scholar] [CrossRef]
- El Hajj, M.C.; Viray, M.C.; Tedford, R.J. Right Heart Failure: A Hemodynamic Review. Cardiol. Clin. 2020, 38, 161–173. [Google Scholar] [CrossRef]
- Hassoun, P.M. The right ventricle in scleroderma (2013 Grover Conference Series). Pulm. Circ. 2015, 5, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Hsu, S.; Houston, B.A.; Tampakakis, E.; Bacher, A.C.; Rhodes, P.S.; Mathai, S.C.; Damico, R.L.; Kolb, T.M.; Hummers, L.K.; Shah, A.A.; et al. Right Ventricular Functional Reserve in Pulmonary Arterial Hypertension. Circulation 2016, 133, 2413–2422. [Google Scholar] [CrossRef] [Green Version]
- Morrisroe, K.; The Australian Scleroderma Interest Group (ASIG); Huq, M.; Stevens, W.; Rabusa, C.; Proudman, S.M.; Nikpour, M. Risk factors for development of pulmonary arterial hypertension in Australian systemic sclerosis patients: Results from a large multicenter cohort study. Bmc Pulm. Med. 2016, 16, 134. [Google Scholar] [CrossRef] [Green Version]
- Steen, V.; Medsger, T.A., Jr. Predictors of isolated pulmonary hypertension in patients with systemic sclerosis and limited cu-taneous involvement. Arthritis Rheum. 2003, 48, 516–522. [Google Scholar] [CrossRef]
- Steen, V.D. Autoantibodies in Systemic Sclerosis. Semin. Arthritis Rheum. 2005, 35, 35–42. [Google Scholar] [CrossRef]
- Riemekasten, G.; Philippe, A.; Näther, M.; Slowinski, T.; Müller, D.N.; Heidecke, H.; Matucci-Cerinic, M.; Czirják, L.; Lukitsch, I.; Becker, M.; et al. Involvement of functional autoantibodies against vascular receptors in systemic sclerosis. Ann. Rheum. Dis. 2010, 70, 530–536. [Google Scholar] [CrossRef]
- Mouthon, L.; Tamby, M.C.; Guillevin, L.; Servettaz, A.; Tamas, N.; Caux, F.; Meyer, O.; Allanore, Y.; Kahan, A. IgG from patients with systemic sclerosis bind to DNA antitopoisomerase 1 in normal human fibroblasts extracts. Biol. Targets Ther. 2008, 2, 583–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinchcliff, M.; Fischer, A.; Schiopu, E.; Steen, V.D.; PHAROS Investigators; Alkassab, F.; Bolster, M.B.; Chung, L.; Csuka, M.E.; Derk, C.T.; et al. Pulmonary Hypertension Assessment and Recognition of Outcomes in Scleroderma (PHAROS): Baseline Characteristics and Description of Study Population. J. Rheumatol. 2011, 38, 2172–2179. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.H.; Krowka, M.J.; Pellikka, P.A.; Swanson, K.L.; McGoon, M.D. Pulmonary hypertension in patients with interstitial lung diseases. Mayo Clin. Proc. 2007, 82, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Hurdman, J.; Condliffe, R.; Elliot, C.; Davies, C.; Hill, C.; Wild, J.; Capener, D.; Sephton, P.; Hamilton, N.; Armstrong, I.; et al. ASPIRE registry: Assessing the Spectrum of Pulmonary hypertension Identified at a REferral centre. Eur. Respir. J. 2011, 39, 945–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacGregor, A.J.; Canavan, R.; Knight, C.; Denton, C.P.; Davar, J.; Coghlan, J.; Black, C.M. Pulmonary hypertension in systemic sclerosis: Risk factors for progression and consequences for survival. Rheumatology 2001, 40, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Le Pavec, J.; Girgis, R.E.; Lechtzin, N.; Mathai, S.C.; Launay, D.; Hummers, L.K.; Zaiman, A.; Sitbon, O.; Simonneau, G.; Humbert, M.; et al. Systemic sclerosis-related pulmonary hypertension associated with interstitial lung disease: Impact of pulmonary arterial hypertension therapies. Arthritis Rheum. 2011, 63, 2456–2464. [Google Scholar] [CrossRef]
- Mathai, S.C.; Hummers, L.K.; Champion, H.C.; Wigley, F.M.; Zaiman, A.; Hassoun, P.M.; Girgis, R.E. Survival in pulmonary hypertension associated with the scleroderma spectrum of diseases: Impact of interstitial lung disease. Arthritis Rheum. 2009, 60, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Waxman, A.; Restrepo-Jaramillo, R.; Thenappan, T.; Ravichandran, A.; Engel, P.; Bajwa, A.; Allen, R.; Feldman, J.; Argula, R.; Smith, P.; et al. Inhaled Treprostinil in Pulmonary Hypertension Due to Interstitial Lung Disease. N. Engl. J. Med. 2021, 384, 325–334. [Google Scholar] [CrossRef]
- Boueiz, A.; Mathai, S.C.; Hummers, L.K.; Hassoun, P.M. Cardiac complications of systemic sclerosis: Recent progress in diagnosis. Curr. Opin. Rheumatol. 2010, 22, 696–703. [Google Scholar] [CrossRef]
- Fernandez-Codina, A.; Simeon-Aznar, C.P.; Pinal-Fernandez, I.; Rodriguez-Palomares, J.; Pizzi, M.N.; Hidalgo, C.E.; Guillen-Del Castillo, A.; Prado-Galbarro, F.J.; Sarria-Santamera, A.; Fonollosa-Pla, V.; et al. Cardiac involvement in systemic sclerosis: Dif-ferences between clinical subsets and influence on survival. Rheumatol. Int. 2017, 37, 75–84. [Google Scholar] [CrossRef]
- Lambova, S. Cardiac manifestations in systemic sclerosis. World J. Cardiol. 2014, 6, 993–1005. [Google Scholar] [CrossRef] [PubMed]
- de Groote, P.; Gressin, V.; Hachulla, E.; Carpentier, P.; Guillevin, L.; Kahan, A.; Cabane, J.; Frances, C.; Lamblin, N.; Diot, E.; et al. Evaluation of cardiac abnormalities by Doppler echocardiography in a large nationwide multicentric cohort of patients with systemic sclerosis. Ann. Rheum. Dis. 2008, 67, 31–36. [Google Scholar] [CrossRef]
- Sutendra, G.; Dromparis, P.; Paulin, R.; Zervopoulos, S.; Haromy, A.; Nagendran, J.; Michelakis, E.D. A metabolic remodeling in right ventricular hypertrophy is associated with decreased angiogenesis and a transition from a compensated to a decompensated state in pulmonary hypertension. J. Mol. Med. 2013, 91, 1315–1327. [Google Scholar] [CrossRef]
- Natarajan, R.; Drake, J.I.; Bogaard, H.J.; Fawcett, P.M.; Clifton, B.; Gao, Y.; Voelkel, N.F. The Molecular Signature Of The Right Heart Failure Program In Chronic Severe Pulmonary Hypertension. In Proceedings of the American Thoracic Society 2010 International Conference, New Orleans, LA, USA, 14–19 May 2010; Volume 45, pp. 1239–1247. [Google Scholar] [CrossRef]
- Vogel-Claussen, J.; Skrok, J.; Shehata, M.L.; Singh, S.; Sibley, C.T.; Boyce, D.M.; Lechtzin, N.; Girgis, R.E.; Mathai, S.C.; Goldstein, T.A.; et al. Right and left ventricular myocardial perfusion reserves correlate with right ventricular function and pulmonary hemody-namics in patients with pulmonary arterial hypertension. Radiology 2011, 258, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Gashouta, M.A.; Humbert, M.; Hassoun, P.M. Update in systemic sclerosis-associated pulmonary arterial hypertension. La Presse Médicale 2014, 43, e293–e304. [Google Scholar] [CrossRef] [PubMed]
- Mathai, S.C.; Bueso, M.; Hummers, L.K.; Boyce, D.; Lechtzin, N.; Le Pavec, J.; Campo, A.; Champion, H.C.; Housten, T.; Forfia, P.R.; et al. Disproportionate elevation of N-terminal pro-brain natriuretic peptide in scleroderma-related pulmonary hypertension. Eur. Respir. J. 2009, 35, 95–104. [Google Scholar] [CrossRef] [Green Version]
- Forfia, P.R.; Mathai, S.C.; Fisher, M.R.; Housten-Harris, T.; Hemnes, A.R.; Champion, H.C.; Girgis, R.E.; Hassoun, P.M. Hyponatremia Predicts Right Heart Failure and Poor Survival in Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2008, 177, 1364–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, M.; Coghlan, J.G.; Khanna, D. Early detection and management of pulmonary arterial hypertension. Eur. Respir. Rev. 2012, 21, 306–312. [Google Scholar] [CrossRef]
- Morrisroe, K.; The Australian Scleroderma Interest Group (ASIG); Stevens, W.; Huq, M.; Prior, D.; Sahhar, J.; Ngian, G.-S.; Celermajer, D.; Zochling, J.; Proudman, S.; et al. Survival and quality of life in incident systemic sclerosis-related pulmonary arterial hypertension. Arthritis Res. 2017, 19, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Almaaitah, S.; Highland, K.B.; Tonelli, A.R. Management of Pulmonary Arterial Hypertension in Patients with Systemic Sclerosis. Integr. Blood Press. Control 2020, 13, 15–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coghlan, J.G.; Denton, C.P.; Grünig, E.; Bonderman, D.; Distler, O.; Khanna, D.; Müller-Ladner, U.; Pope, J.E.; Vonk, M.C.; Doelberg, M.; et al. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: The DETECT study. Ann. Rheum. Dis. 2013, 73, 1340–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respir-atory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [PubMed]
- Kowal-Bielecka, O.; Fransen, J.; Avouac, J.; Becker, M.; Kulak, A.; Allanore, Y.; Distler, O.; Clements, P.J.; Cutolo, M.; Czirjak, L.; et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann. Rheum. Dis. 2017, 76, 1327–1339. [Google Scholar] [CrossRef] [Green Version]
- Hachulla, E.; Carpentier, P.; Gressin, V.; Diot, E.; Allanore, Y.; Sibilia, J.; Launay, D.; Mouthon, L.; Jego, P.; Cabane, J.; et al. Risk factors for death and the 3-year survival of patients with systemic sclerosis: The French ItinerAIR-Sclerodermie study. Rheumatology 2009, 48, 304–308. [Google Scholar] [CrossRef] [Green Version]
- Hachulla, E.; De Groote, P.; Gressin, V.; Sibilia, J.; Diot, E.; Carpentier, P.; Mouthon, L.; Hatron, P.-Y.; Jego, P.; Allanore, Y.; et al. The three-year incidence of pulmonary arterial hypertension associated with systemic sclerosis in a multicenter nationwide longitudinal study in France. Arthritis Rheum. 2009, 60, 1831–1839. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.F.; Zhou, L.; Ma, G.F.; Shao, F.C.; Wu, X.H.; Ying, K.J. Diagnostic value of transthoracic Doppler echocardiography in pul-monary hypertension: A meta-analysis. Am. J. Hypertens. 2010, 23, 1261–1264. [Google Scholar] [CrossRef] [Green Version]
- Janda, S.; Shahidi, N.; Gin, K.; Swiston, J. Diagnostic accuracy of echocardiography for pulmonary hypertension: A systematic review and meta-analysis. Heart 2011, 97, 612–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, J.R.; Yan, P.J.; Liu, S.D.; Hu, Y.; Yang, K.H.; Song, B.; Lei, J.Q. Diagnostic accuracy of transthoracic echocardiography for pulmonary hypertension: A systematic review and meta-analysis. BMJ Open 2019, 9, e033084. [Google Scholar] [CrossRef] [Green Version]
- Forfia, P.R.; Fisher, M.R.; Mathai, S.C.; Housten-Harris, T.; Hemnes, A.R.; Borlaug, B.A.; Chamera, E.; Corretti, M.C.; Champion, H.C.; Abraham, T.P.; et al. Tricuspid Annular Displacement Predicts Survival in Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2006, 174, 1034–1041. [Google Scholar] [CrossRef]
- Mathai, S.C.; Sibley, C.T.; Forfia, P.R.; Mudd, J.O.; Fisher, M.R.; Tedford, R.J.; Lechtzin, N.; Boyce, D.; Hummers, L.K.; Housten, T.; et al. Tri-cuspid annular plane systolic excursion is a robust outcome measure in systemic sclerosis-associated pulmonary arterial hypertension. J. Rheumatol. 2011, 38, 2410–2418. [Google Scholar] [CrossRef]
- Cavagna, L.; Caporali, R.; Klersy, C.; Ghio, S.; Albertini, R.; Scelsi, L.; Moratti, R.; Bonino, C.; Montecucco, C. Comparison of brain natriuretic peptide (BNP) and NT-proBNP in screening for pulmonary arterial hypertension in patients with systemic scle-rosis. J. Rheumatol. 2010, 37, 2064–2070. [Google Scholar] [CrossRef]
- Allanore, Y.; Borderie, D.; Avouac, J.; Zerkak, D.; Meune, C.; Hachulla, E.; Mouthon, L.; Guillevin, L.; Meyer, O.; Ekindjian, O.G.; et al. High N-terminal pro–brain natriuretic peptide levels and low diffusing capacity for carbon monoxide as independent predictors of the occurrence of precapillary pulmonary arterial hypertension in patients with systemic sclerosis. Arthritis Rheum. 2007, 58, 284–291. [Google Scholar] [CrossRef]
- Mukerjee, D.; George, D.S.; Knight, C.; Davar, J.; Wells, A.U.; Du Bois, R.M.; Black, C.M.; Coghlan, J.G. Echocardiography and pulmonary function as screening tests for pulmonary arterial hypertension in systemic sclerosis. Rheumatology 2004, 43, 461–466. [Google Scholar] [CrossRef] [Green Version]
- York, M.; Farber, H.W. Pulmonary hypertension: Screening and evaluation in scleroderma. Curr. Opin. Rheumatol. 2011, 23, 536–544. [Google Scholar] [CrossRef]
- Bezante, G.P.; Rollando, D.; Sessarego, M.; Panico, N.; Setti, M.; Filaci, G.; Molinari, G.; Balbi, M.; Cutolo, M.; Barsotti, A.; et al. Cardiac magnetic resonance imaging detects subclinical right ventricular impairment in systemic sclerosis. J. Rheumatol. 2007, 34, 2431–2437. [Google Scholar]
- Hachulla, A.-L.; Launay, D.; Gaxotte, V.; De Groote, P.; Lamblin, N.; Devos, P.; Hatron, P.-Y.; Beregi, J.-P.; Hachulla, E. Cardiac magnetic resonance imaging in systemic sclerosis: A cross-sectional observational study of 52 patients. Ann. Rheum. Dis. 2008, 68, 1878–1884. [Google Scholar] [CrossRef]
- Hagger, D.; Condliffe, R.; Woodhouse, N.; Elliot, C.A.; Armstrong, I.J.; Davies, C.; Hill, C.; Akil, M.; Wild, J.M.; Kiely, D.G. Ventricular mass index correlates with pulmonary artery pressure and predicts survival in suspected systemic sclerosis-associated pulmonary arterial hypertension. Rheumatology 2009, 48, 1137–1142. [Google Scholar] [CrossRef] [Green Version]
- Sundaram, S.M.; Chung, L. An Update on Systemic Sclerosis-Associated Pulmonary Arterial Hypertension: A Review of the Current Literature. Curr. Rheumatol. Rep. 2018, 20, 10. [Google Scholar] [CrossRef]
- Hassoun, P.M.; Zamanian, R.T.; Damico, R.; Lechtzin, N.; Khair, R.; Kolb, T.M.; Tedford, R.J.; Hulme, O.L.; Housten, T.; Pisanello, C.; et al. Ambrisentan and Tadalafil Up-front Combination Therapy in Scleroderma-associated Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 1102–1110. [Google Scholar] [CrossRef] [Green Version]
- Weatherald, J.; Boucly, A.; Launay, D.; Cottin, V.; Prevot, G.; Bourlier, D.; Dauphin, C.; Chaouat, A.; Savale, L.; Jais, X.; et al. Haemody-namics and serial risk assessment in systemic sclerosis associated pulmonary arterial hypertension. Eur. Respir. J 2018, 52, 1340–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitbon, O.; Humbert, M.; Jaìs, X.; Ioos, V.; Hamid, A.M.; Provencher, S.; Garcia, G.; Parent, F.; Hervé, P.; Simonneau, G. Long-Term Response to Calcium Channel Blockers in Idiopathic Pulmonary Arterial Hypertension. Circulation 2005, 111, 3105–3111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, S. Pulmonary Hypertension in Systemic Sclerosis. Semin. Arthritis Rheum. 2011, 41, 19–37. [Google Scholar] [CrossRef] [PubMed]
- Montani, D.; Savale, L.; Natali, D.; Jais, X.; Herve, P.; Garcia, G.; Humbert, M.; Simonneau, G.; Sitbon, O. Long-term response to cal-cium-channel blockers in non-idiopathic pulmonary arterial hypertension. Eur. Heart J. 2010, 31, 1898–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandecasteele, E.; Drieghe, B.; Melsens, K.; Thevissen, K.; De Pauw, M.; Deschepper, E.; Decuman, S.; Bonroy, C.; Piette, Y.; De Keyser, F.; et al. Screening for pulmonary arterial hypertension in an unselected prospective systemic sclerosis cohort. Eur. Respir. J. 2017, 49, 1602275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galie, N.; McLaughlin, V.V.; Rubin, L.J.; Simonneau, G. An overview of the 6th World Symposium on Pulmonary Hypertension. Eur. Respir. J. 2019, 53, 1802148. [Google Scholar] [CrossRef] [Green Version]
- Galie, N.; Channick, R.N.; Frantz, R.P.; Grunig, E.; Jing, Z.C.; Moiseeva, O.; Preston, I.R.; Pulido, T.; Safdar, Z.; Tamura, Y.; et al. Risk strat-ification and medical therapy of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801889. [Google Scholar] [CrossRef] [PubMed]
- Boucly, A.; Weatherald, J.; Savale, L.; Jaïs, X.; Cottin, V.; Prevot, G.; Picard, F.; De Groote, P.; Jevnikar, M.; Bergot, E.; et al. Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. Eur. Respir. J. 2017, 50, 1700889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benza, R.L.; Gomberg-Maitland, M.; Elliott, C.G.; Farber, H.W.; Foreman, A.J.; Frost, A.E.; McGoon, M.D.; Pasta, D.J.; Selej, M.; Burger, C.D.; et al. Predicting Survival in Patients With Pulmonary Arterial Hypertension: The REVEAL Risk Score Calculator 2.0 and Comparison With ESC/ERS-Based Risk Assessment Strategies. Chest 2019, 156, 323–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badesch, D.B.; Tapson, V.F.; McGoon, M.D.; Brundage, B.H.; Rubin, L.J.; Wigley, F.M.; Rich, S.; Barst, R.J.; Barrett, P.S.; Kral, K.M.; et al. Con-tinuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann. Intern. Med. 2000, 132, 425–434. [Google Scholar] [CrossRef]
- Oudiz, R.J.; Schilz, R.J.; Barst, R.J.; Galie, N.; Rich, S.; Rubin, L.J.; Simonneau, G.; Treprostinil Study Group. Treprostinil, a prostacyclin ana-logue, in pulmonary arterial hypertension associated with connective tissue disease. Chest 2004, 126, 420–427. [Google Scholar] [CrossRef] [Green Version]
- Simonneau, G.; Barst, R.J.; Galie, N.; Naeije, R.; Rich, S.; Bourge, R.C.; Keogh, A.; Oudiz, R.; Frost, A.; Blackburn, S.D.; et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: A dou-ble-blind, randomized, placebo-controlled trial. Am. J. Respir Crit Care Med. 2002, 165, 800–804. [Google Scholar] [CrossRef]
- Caravita, S.; Wu, S.C.; Secchi, M.B.; Dadone, V.; Bencini, C.; Pierini, S. Long-term effects of intermittent Iloprost infusion on pulmonary arterial pressure in connective tissue disease. Eur. J. Intern. Med. 2011, 22, 518–521. [Google Scholar] [CrossRef]
- Gaine, S.; Chin, K.; Coghlan, G.; Channick, R.; Di Scala, L.; Galiè, N.; Ghofrani, H.-A.; Lang, I.M.; McLaughlin, V.; Preiss, R.; et al. Selexipag for the treatment of connective tissue disease-associated pulmonary arterial hypertension. Eur. Respir. J. 2017, 50, 1602493. [Google Scholar] [CrossRef]
- Sitbon, O.; Channick, R.; Chin, K.M.; Frey, A.; Gaine, S.; Galiè, N.; Ghofrani, H.-A.; Hoeper, M.M.; Lang, I.M.; Preiss, R.; et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 2522–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badesch, D.B.; Hill, N.S.; Burgess, G.; Rubin, L.J.; Barst, R.J.; Galie, N.; Simonneau, G.; SUPER Study Group. Sildenafil for pulmonary arterial hy-pertension associated with connective tissue disease. J. Rheumatol. 2007, 34, 2417–2422. [Google Scholar]
- Galiè, N.; Ghofrani, H.A.; Torbicki, A.; Barst, R.J.; Rubin, L.J.; Badesch, D.; Fleming, T.; Parpia, T.; Burgess, G.; Branzi, A.; et al. Sildenafil Citrate Therapy for Pulmonary Arterial Hypertension. N. Engl. J. Med. 2005, 353, 2148–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galiè, N.; Brundage, B.H.; Ghofrani, H.A.; Oudiz, R.J.; Simonneau, G.; Safdar, Z.; Shapiro, S.; White, R.J.; Chan, M.; Beardsworth, A.; et al. Tadalafil Therapy for Pulmonary Arterial Hypertension. Circulation 2009, 119, 2894–2903. [Google Scholar] [CrossRef] [Green Version]
- Galiè, N.; Denton, C.P.; Dardi, F.; Manes, A.; Mazzanti, G.; Li, B.; Varanese, L.; Esler, A.; Harmon, C.; Palazzini, M. Tadalafil in idiopathic or heritable pulmonary arterial hypertension (PAH) compared to PAH associated with connective tissue disease. Int. J. Cardiol. 2017, 235, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.J.; Badesch, D.B.; Barst, R.J.; Galiè, N.; Black, C.M.; Keogh, A.; Pulido, T.; Frost, A.; Roux, S.; Leconte, I.; et al. Bosentan Therapy for Pulmonary Arterial Hypertension. N. Engl. J. Med. 2002, 346, 896–903. [Google Scholar] [CrossRef]
- Denton, C.P.; Pope, J.E.; Peter, H.-H.; Gabrielli, A.; Boonstra, A.; Hoogen, F.H.J.V.D.; Riemekasten, G.; De Vita, S.; Morganti, A.; Dolberg, M.; et al. Long-term effects of bosentan on quality of life, survival, safety and tolerability in pulmonary arterial hypertension related to connective tissue diseases. Ann. Rheum. Dis. 2007, 67, 1222–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, A.; Denton, C.P.; Matucci-Cerinic, M.; Gillies, H.; Blair, C.; Tislow, J.; Nathan, S.D. Ambrisentan response in connective tissue disease-associated pulmonary arterial hypertension (CTD-PAH)—A subgroup analysis of the ARIES-E clinical trial. Respir. Med. 2016, 117, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Galie, N.; Olschewski, H.; Oudiz, R.J.; Torres, F.; Frost, A.; Ghofrani, H.A.; Badesch, D.B.; McGoon, M.D.; McLaughlin, V.V.; Roecker, E.B.; et al. Ambrisentan for the treatment of pulmonary arterial hypertension: Results of the ambrisentan in pulmonary arterial hy-pertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008, 117, 3010–3019. [Google Scholar] [CrossRef]
- Pulido, T.; Adzerikho, I.; Channick, R.N.; Delcroix, M.; Galiè, N.; Ghofrani, H.-A.; Jansa, P.; Jing, Z.-C.; Le Brun, F.-O.; Mehta, S.; et al. Macitentan and Morbidity and Mortality in Pulmonary Arterial Hypertension. N. Engl. J. Med. 2013, 369, 809–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, R.L.; Gabler, N.B.; Sangani, S.; Praestgaard, A.; Merkel, P.A.; Kawut, S.M. Comparison of Treatment Response in Idiopathic and Connective Tissue Disease–associated Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2015, 192, 1111–1117. [Google Scholar] [CrossRef] [Green Version]
- Ghofrani, H.-A.; Galiè, N.; Grimminger, F.; Grünig, E.; Humbert, M.; Jing, Z.-C.; Keogh, A.M.; Langleben, D.; Kilama, M.O.; Fritsch, A.; et al. Riociguat for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2013, 369, 330–340. [Google Scholar] [CrossRef] [Green Version]
- Humbert, M.; Coghlan, J.G.; Ghofrani, H.-A.; Grimminger, F.; He, J.-G.; Riemekasten, G.; Vizza, C.D.; Boeckenhoff, A.; Meier, C.; Pena, J.D.O.; et al. Riociguat for the treatment of pulmonary arterial hypertension associated with connective tissue disease: Results from PATENT-1 and PATENT-2. Ann. Rheum. Dis. 2016, 76, 422–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marra, A.M.; Halank, M.; Benjamin, N.; Bossone, E.; Cittadini, A.; Eichstaedt, C.A.; Egenlauf, B.; Harutyunova, S.; Fischer, C.; Gall, H.; et al. Right ventricular size and function under riociguat in pulmonary arterial hypertension and chronic thromboembolic pul-monary hypertension (the RIVER study). Respir. Res. 2018, 19, 258. [Google Scholar] [CrossRef]
- Galie, N.; Barbera, J.A.; Frost, A.E.; Ghofrani, H.A.; Hoeper, M.M.; McLaughlin, V.V.; Peacock, A.J.; Simonneau, G.; Vachiery, J.L.; Grunig, E.; et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Ambale-Venkatesh, B.; Lima, J.A.; Zimmerman, S.L.; Tedford, R.J.; Fujii, T.; Hulme, O.L.; Pullins, E.H.; Corona-Villalobos, C.P.; Zamanian, R.T.; et al. The impact of ambrisentan and tadalafil upfront combination therapy on cardiac function in scleroderma associated pulmonary arterial hypertension patients: Cardiac magnetic resonance feature tracking study. Pulm. Circ. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Coghlan, J.G.; Channick, R.; Chin, K.; Di Scala, L.; Galiè, N.; Ghofrani, H.-A.; Hoeper, M.M.; Lang, I.M.; McLaughlin, V.; Preiss, R.; et al. Targeting the Prostacyclin Pathway with Selexipag in Patients with Pulmonary Arterial Hypertension Receiving Double Combination Therapy: Insights from the Randomized Controlled GRIPHON Study. Am. J. Cardiovasc. Drugs 2018, 18, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Jansa, P.; Pulido, T. Macitentan in Pulmonary Arterial Hypertension: A Focus on Combination Therapy in the SERAPHIN Trial. Am. J. Cardiovasc. Drugs 2018, 18, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henkens, I.R.; Hazenoot, T.; Boonstra, A.; Huisman, M.V.; Vonk-Noordegraaf, A. Major bleeding with vitamin K antagonist anti-coagulants in pulmonary hypertension. Eur. Respir. J. 2013, 41, 872–878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, K.M.; Delcroix, M.; Ghofrani, H.A.; Tiede, H.; Huscher, D.; Speich, R.; Grünig, E.; Staehler, G.; Rosenkranz, S.; Halank, M.; et al. Response to Letters Regarding Article, “Anticoagulation and Survival in Pulmonary Arterial Hypertension: Results From the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA)”. Circulation 2014, 130, e110–e112. [Google Scholar] [CrossRef] [Green Version]
- Preston, I.R.; Roberts, K.E.; Miller, D.P.; Sen, G.P.; Selej, M.; Benton, W.W.; Hill, N.S.; Farber, H.W. Effect of Warfarin Treatment on Survival of Patients With Pulmonary Arterial Hypertension (PAH) in the Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL). Circulation 2015, 132, 2403–2411. [Google Scholar] [CrossRef]
- Calderone, A.; Stevens, W.; Prior, D.; Nandurkar, H.; Gabbay, E.; Proudman, S.M.; Williams, T.; Celermajer, D.; Sahhar, J.; Wong, P.K.; et al. Multicentre randomised placebo-controlled trial of oral anticoagulation with apixaban in systemic sclerosis-related pul-monary arterial hypertension: The SPHInX study protocol. BMJ Open 2016, 6, e011028. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, O.; Sitbon, O.; Jaïs, X.; Simonneau, G.; Humbert, M. Immunosuppressive Therapy in Connective Tissue Diseases-Associated Pulmonary Arterial Hypertension. Chest 2006, 130, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, R.T.; Badesch, D.; Chung, L.; Domsic, R.T.; Medsger, T.; Pinckney, A.; Keyes-Elstein, L.; D’Aveta, C.; Spychala, M.; White, R.J.; et al. Safety and Efficacy of B-Cell Depletion with Rituximab for the Treatment of Systemic Sclerosis Associated Pulmonary Arterial Hypertension: A Multi-center, Double-blind, Randomized, Placebo-controlled Trial. Am. J. Respir. Crit. Care Med. 2021. [Google Scholar] [CrossRef]
- Shah, R.J.; Boin, F. Lung Transplantation in Patients With Systemic Sclerosis. Curr. Rheumatol. Rep. 2017, 19, 723. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, E.J.; Peterson, E.R.; Sell, J.L.; D’Ovidio, F.; Arcasoy, S.M.; Bathon, J.M.; Lederer, D.J. Survival of Adults With Systemic Sclerosis Following Lung Transplantation: A Nationwide Cohort Study. Arthritis Rheumatol. 2015, 67, 1314–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schachna, L.; Medsger, T.A., Jr.; Dauber, J.H.; Wigley, F.M.; Braunstein, N.A.; White, B.; Steen, V.D.; Conte, J.V.; Yang, S.C.; McCurry, K.R.; et al. Lung transplantation in scleroderma compared with idiopathic pulmonary fibrosis and idiopathic pulmonary arterial hy-pertension. Arthritis Rheum. 2006, 54, 3954–3961. [Google Scholar] [CrossRef]
- Chung, L.; Farber, H.W.; Benza, R.; Miller, D.P.; Parsons, L.; Hassoun, P.M.; McGoon, M.; Nicolls, M.R.; Zamanian, R.T. Unique Predictors of Mortality in Patients With Pulmonary Arterial Hypertension Associated With Systemic Sclerosis in the REVEAL Registry. Chest 2014, 146, 1494–1504. [Google Scholar] [CrossRef] [Green Version]
- Lefèvre, G.; Dauchet, L.; Hachulla, E.; Montani, D.; Sobanski, V.; Lambert, M.; Hatron, P.-Y.; Humbert, M.; Launay, D. Survival and Prognostic Factors in Systemic Sclerosis-Associated Pulmonary Hypertension: A Systematic Review and Meta-Analysis. Arthritis Rheum. 2013, 65, 2412–2423. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| REVEAL 2.0 | FPHN | |
|---|---|---|
| Indicators | PAH etiology (CTD-PAH, PoPH, Heritable) | WHO FC (I/II) |
| Demographics (Male > 60 years) | 6MWD (>440 m) | |
| Comorbidities (eGFR < 60 mL/min/1.73 m2) | RAP (<8 mmHg) | |
| NYHA/WHO FC (III, IV) | CI (≥2.5 L/min/m2) | |
| Vital signs (SBP < 100 mmHg, HR > 96 BMP) | ||
| All-cause hospitalizations ≤ 6 months | ||
| 6MWD (<165 m) | ||
| Pro-BNP (>1100 pg/mL) | ||
| Echocardiogram (Pericardial effusion) | ||
| PFT (DLCO < 40% predicted) | ||
| RHC (mPAP > 20 mmHg within 1 year) | ||
| Risk score | Low: ≤6 | Low: 3–4 |
| Intermediate: 7–8 | Intermediate: 2 | |
| High: ≥9 | High: 0–1 |
| Prognostic Factors |
|---|
| Male gender |
| Age > 60 years |
| WHO functional class IV |
| 6MWD < 165 m |
| Anti-U1 ribonucleoprotein (RNP) negative status |
| DLCO < 50% predicted |
| Pericardial effusion |
| RA pressure > 20 mmHg |
| PVR > 32 Wood units |
| SBP <110 mmHg |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naranjo, M.; Hassoun, P.M. Systemic Sclerosis-Associated Pulmonary Hypertension: Spectrum and Impact. Diagnostics 2021, 11, 911. https://doi.org/10.3390/diagnostics11050911
Naranjo M, Hassoun PM. Systemic Sclerosis-Associated Pulmonary Hypertension: Spectrum and Impact. Diagnostics. 2021; 11(5):911. https://doi.org/10.3390/diagnostics11050911
Chicago/Turabian StyleNaranjo, Mario, and Paul M. Hassoun. 2021. "Systemic Sclerosis-Associated Pulmonary Hypertension: Spectrum and Impact" Diagnostics 11, no. 5: 911. https://doi.org/10.3390/diagnostics11050911
APA StyleNaranjo, M., & Hassoun, P. M. (2021). Systemic Sclerosis-Associated Pulmonary Hypertension: Spectrum and Impact. Diagnostics, 11(5), 911. https://doi.org/10.3390/diagnostics11050911