
Nationwide Newborn Screening Program for Mucopolysaccharidoses in Taiwan and an Update of the “Gold Standard” Criteria Required to Make a Confirmatory Diagnosis

 ,
, 
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. NBS for MPSs and Samples Referred
2.2. Ethical Approval
2.3. Urinary First-Line Biochemistry Examinations
2.3.1. Total GAG Quantification (Dimethylene Blue/Creatinine Ratio; DMB/Cre Ratio)
2.3.2. GAG-Derived Disaccharide Quantification by Tandem Mass Spectrometry Assay
2.4. Leukocyte Enzyme Activity by Fluorometric Assay
2.5. Molecular DNA Analysis
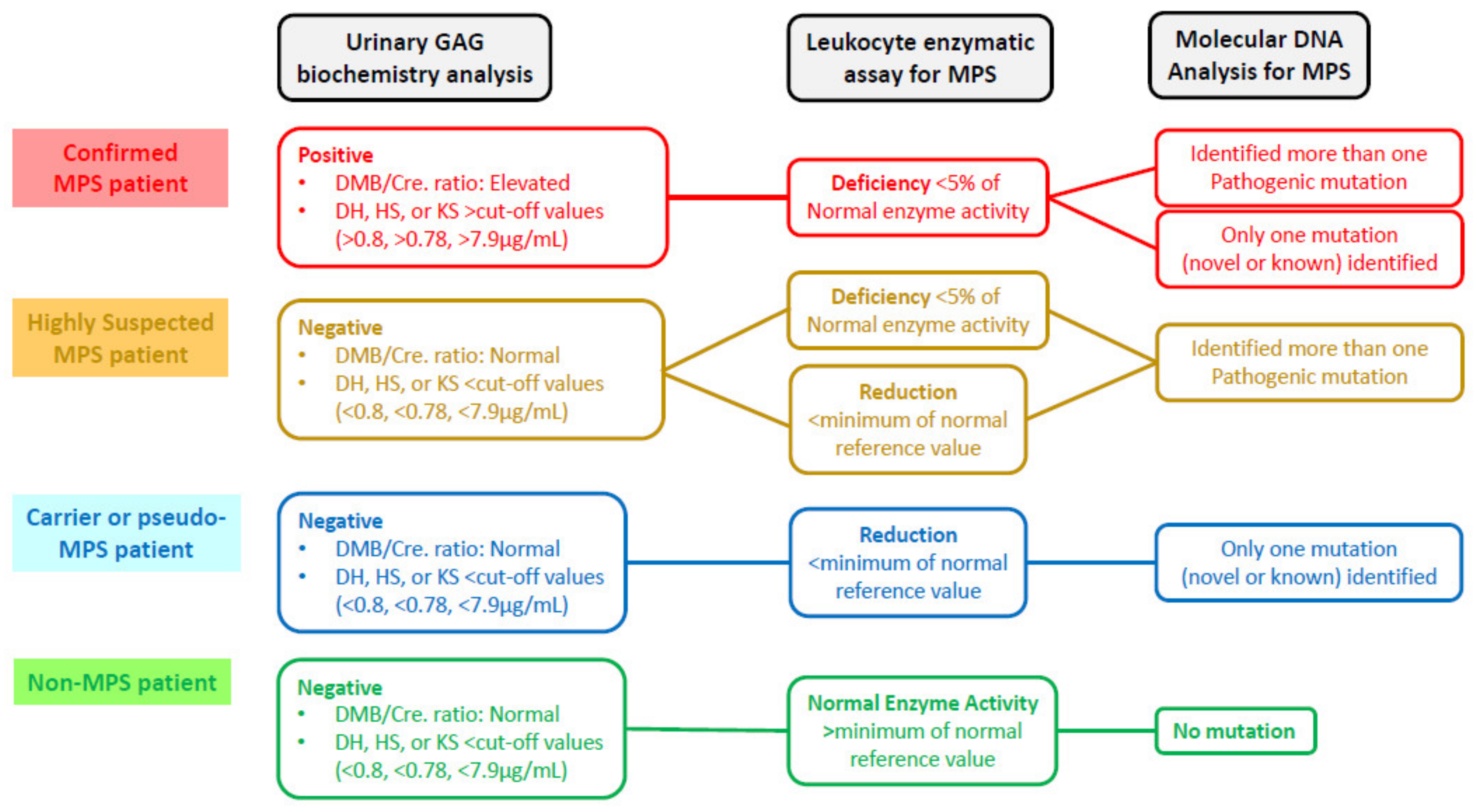
2.6. Verification of a New “Gold Standard” for MPS Confirmation
3. Results
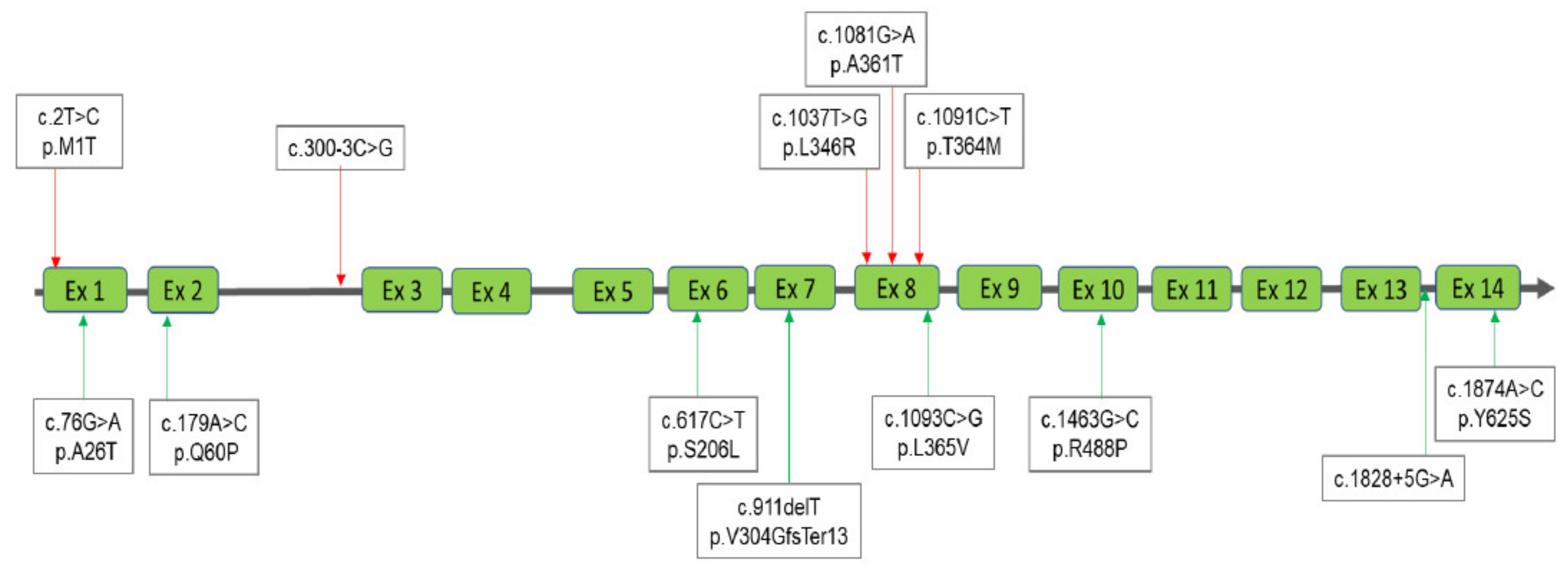
3.1. Interpretation of the Results for MPS I
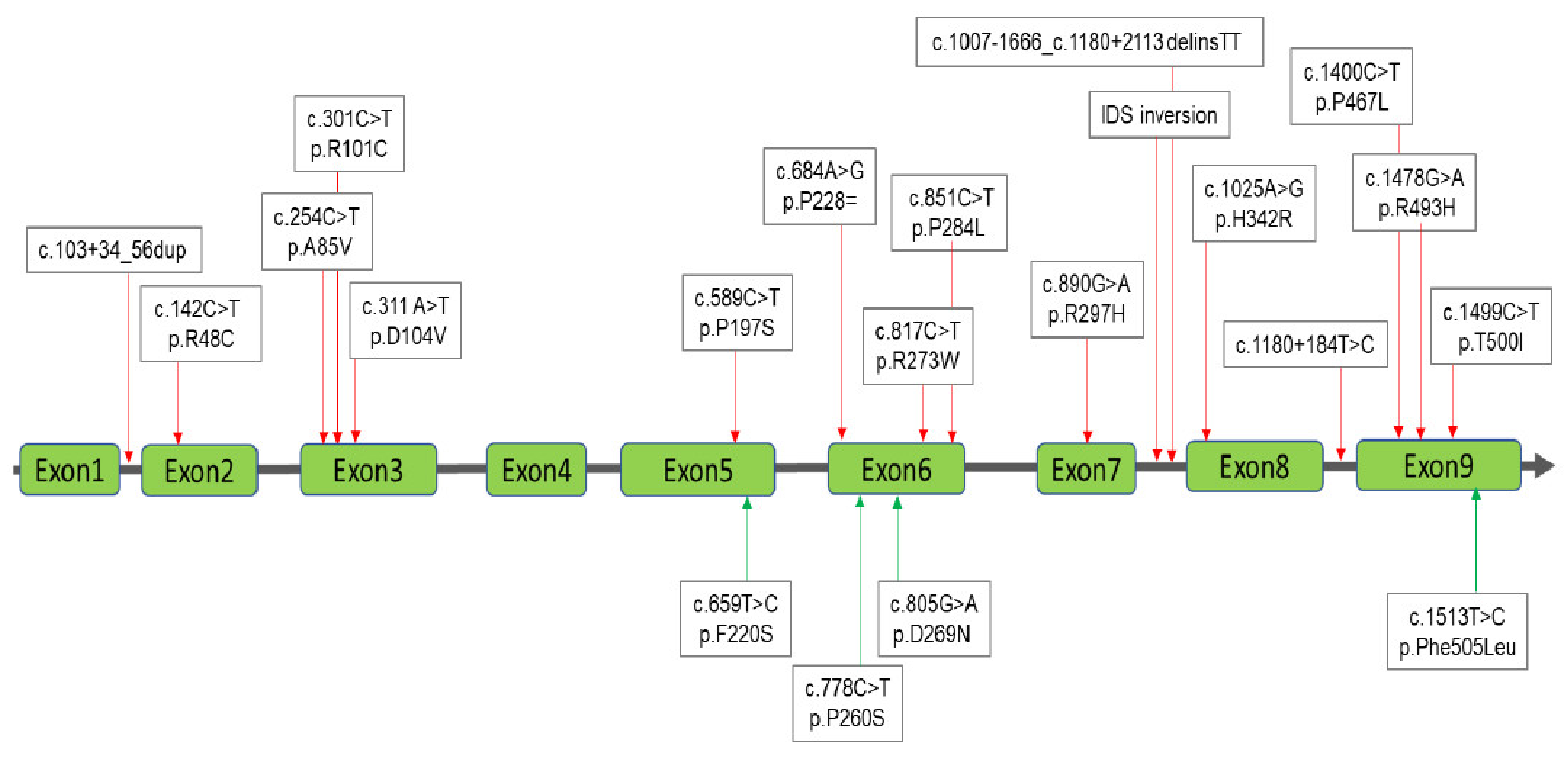
3.2. Interpretation of the Results for MPS II
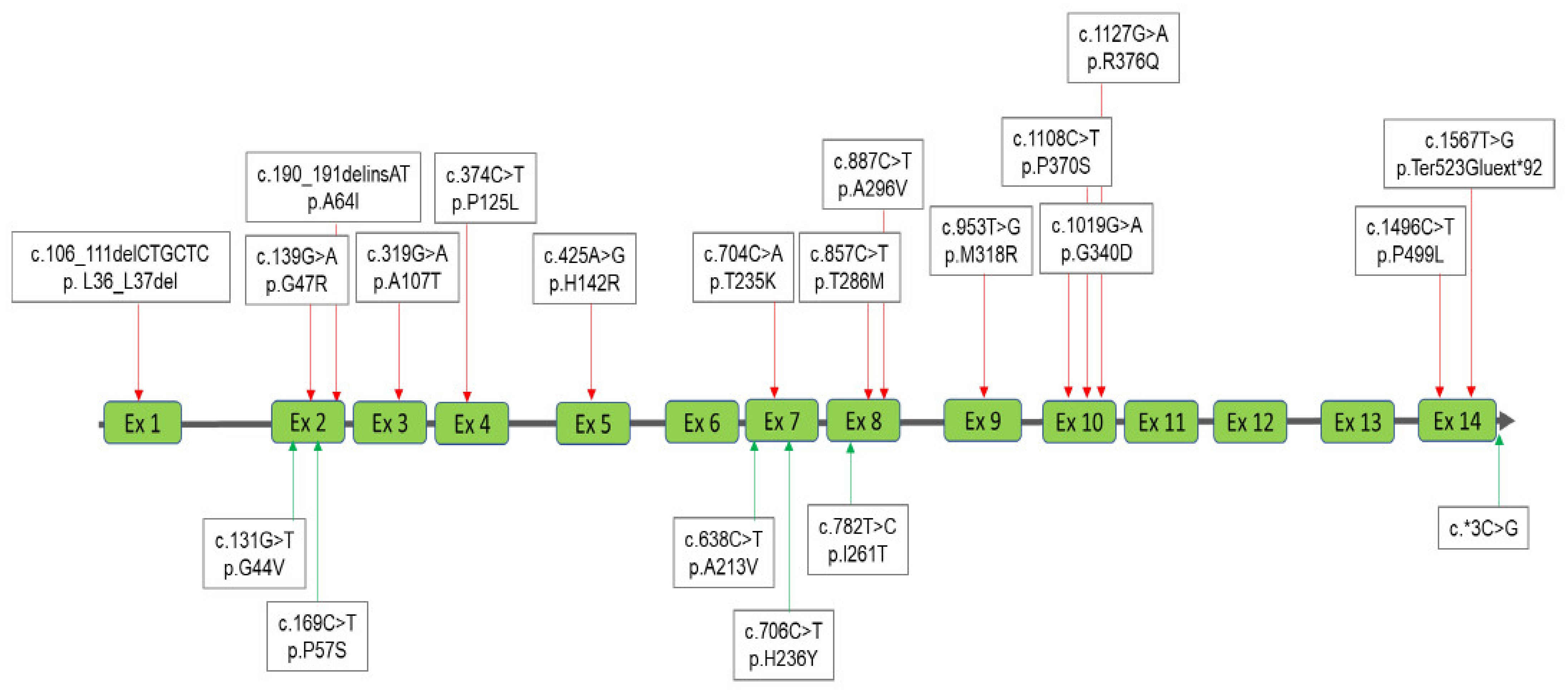
3.3. Interpretation of the Results for MPS IVA
3.4. Interpretation of the Results for MPS VI
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Neufeld, E.F.; Muenzer, J. The Mucopolysaccharidoses. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill, Inc. Health Profession Division: New York, NY, USA, 1995; pp. 2465–2494. [Google Scholar]
- Applegarth, D.A.; Dimmick, J.E.; Hall, J.G. Organelle Diseases: Clinical features, diagnosis, pathogenesis and management. Chapman. Hall Med. 1997, 45–97. [Google Scholar]
- Besley, G.T.N.; Wraith, J.E. Lysosomal Disorders. Curr. Paediatr. 1997, 7, 128–134. [Google Scholar] [CrossRef]
- Wraith, J.E. Mucopolysaccharidoses. Curr. Paediatr. 1996, 6, 74–79. [Google Scholar] [CrossRef]
- Lin, H.Y.; Lee, C.L.; Chang, C.Y.; Chiu, P.C.; Chien, Y.H.; Niu, D.M.; Tsai, F.J.; Hwu, W.L.; Lin, S.J.; Lin, J.-L.; et al. Survival and diagnostic age of 175 Taiwanese patients with mucopolysaccharidoses (1985–2019). Orphanet J. Rare Dis. 2020, 15, 314. [Google Scholar] [CrossRef]
- Chuang, C.K.; Lin, H.Y.; Wang, T.J.; Huang, Y.H.; Chan, M.J.; Liao, H.C.; Lo, Y.T.; Wang, L.Y.; Tu, R.Y.; Fang, Y.Y.; et al. Status of newborn screening and follow up investigations for Mucopolysaccharidoses I and II in Taiwan. Orphanet J. Rare Dis. 2018, 13, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mashima, R.; Sakai, E.; Tanaka, M.; Kosuga, M.; Okuyama, T. The levels of urinary glycosaminoglycans of patients with attenuated and severe type of mucopolysaccharidosis II determined by liquid chromatography-tandem mass spectrometry. Mol. Genet. Metab. Rep. 2016, 7, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Fujii, T.; Fukushi, M.; Oguma, T.; Shimada, T.; Maeda, M.; Kida, K.; Shibata, Y.; Futatsumori, H.; Montaño, A.M.; et al. Newborn screening and diagnosis of mucopolysaccharidoses. Mol. Genet. Metab. 2013, 110, 42–53. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.Y.; Lee, C.L.; Lo, Y.T.; Tu, R.Y.; Chang, Y.H.; Chang, C.Y.; Chiu, P.C.; Chang, T.M.; Tsai, W.H.; Niu, D.M.; et al. An At-Risk Population Screening Program for Mucopolysaccharidoses by Measuring Urinary Glycosaminoglycans in Taiwan. Diagnostics 2019, 9, 140. [Google Scholar] [CrossRef] [Green Version]
- Chuang, C.K.; Lin, S.P.; Chung, S.F. Diagnostic screening for mucopolysaccharidoses by the dimethylmethylene blue method and twodimensional electrophoresis. Chin. Med. J. 2001, 64, 15–22. [Google Scholar]
- Auray-Blais, C.; Bhérer, P.; Gagnon, R.; Young, S.P.; Zhang, H.H.; An, Y.; Clarke, J.T.; Millington, D.S. Efficient analysis of urinary glycosaminoglycans by LC-MS/MS in mucopolysaccharidoses type I, II and VI. Mol. Genet. Metab. 2011, 102, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.K.; Lin, H.Y.; Wang, T.J.; Tsai, C.C.; Liu, H.L.; Lin, S.P. A modified liquid chromatography/tandem mass spectrometry method for predominant disaccharide units of urinary glycosaminoglycans in patients with mucopolysaccharidoses. Orphanet J. Rare Dis. 2014, 9, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubaski, F.; Osago, H.; Mason, R.W.; Yamaguchi, S.; Kobayashi, H.; Tsuchiya, M.; Orii, T.; Tomatsu, S. Glycosaminoglycans detection methods: Applications of mass spectrometry. Mol. Genet. Metab. 2017, 120, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.Y.; Lo, Y.T.; Wang, T.J.; Huang, S.F.; Tu, R.Y.; Chen, T.L.; Lin, S.P.; Chuang, C.K. Normalization of glycosaminoglycan-derived disaccharides detected by tandem mass spectrometry assay for the diagnosis of mucopolysaccharidosis. Sci. Rep. 2019, 9, 10755. [Google Scholar] [CrossRef] [Green Version]
- Hopwood, J.J.; Muller, V.; Smithson, A.; Baggett, N. A fluorometric assay using 4-methylumbelliferyl α-L-iduronide for the estimation of α-L-iduronidase activity and the detection of hurler and Scheie syndromes. Clin. Chim. Acta. 1979, 92, 257–265. [Google Scholar] [CrossRef]
- Voznyi, Y.V.; Keulemans, J.L.; van Diggelen, O.P. A fluorimetric enzyme assay for the diagnosis of MPS II (hunter disease). J. Inherit. Metab. Dis. 2001, 24, 675–680. [Google Scholar] [CrossRef]
- Van Diggelen, O.P.; Zhao, H.; Kleijer, W.J.; Janse, H.C.; Poorthuis, B.J.; van Pelt, J.; Kamerling, J.P.; Galjaard, H. A fluorimetric enzyme assay for the diagnosis of Morquiodisease type A (MPS IV A). Clin. Chim. Acta 1990, 187, 131–139. [Google Scholar] [CrossRef]
- Lin, H.Y.; Tu, R.Y.; Chern, S.R.; Lo, Y.T.; Fran, S.; Wei, F.J.; Huang, S.F.; Tsai, S.Y.; Chang, Y.H.; Lee, C.L.; et al. Identification and Functional Characterization of IDS Gene Mutations Underlying Taiwanese Hunter Syndrome (Mucopolysaccharidosis Type II). Int. J. Mol. Sci. 2019, 21, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhang, W.; Shi, H.; Qiu, Z.; Meng, Y.; Yao, F.; Wei, M. Mucopolysaccharidosis I mutations in Chinese patients: Identification of 27 novel mutations and 6 cases involving prenatal diagnosis. Clin. Genet. 2012, 81, 443–452. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Savost’anov, K.V.; Kuzenkova, L.M.; Gevorkyan, A.K.; Pushkov, A.A.; Nikitin, A.G.; Pakhomov, A.V.; Vashakmadze, N.D.; Zhurkova, N.V.; Podkletnova, T.V.; et al. Molecular characteristics of patients with glycosaminoglycan storage disorders in Russia. Clin. Chim. Acta. 2014, 436, 112–120. [Google Scholar] [CrossRef]
- Teng, Y.N.; Wang, T.R.; Hwu, W.L.; Lin, S.P.; Lee-Chen, G.J. Identification and characterization of -3c-g acceptor splice site mutation in human alpha-L-iduronidase associated with mucopolysaccharidosis type IH/S. Clin. Genet. 2000, 57, 131–136. [Google Scholar] [CrossRef]
- Lee-Chen, G.J.; Wang, T.R. Mucopolysaccharidosis type I: Identification of novel mutations that cause Hurler/Scheie syndrome in Chinese families. J. Med. Genet. 1997, 34, 939–941. [Google Scholar] [CrossRef] [Green Version]
- Keeratichamroen, S.; Cairns, J.R.; Wattanasirichaigoon, D.; Wasant, P.; Ngiwsara, L.; Suwannarat, P.; Pangkanon, S.; Kuptanon, J.; Tanpaiboon, P.; Rujirawat, T.; et al. Molecular analysis of the iduronate-2-sulfatase gene in Thai patients with Hunter syndrome. J. Inherit. Metab. Dis. 2008, 31 (Suppl. 2), S303–S311. [Google Scholar] [CrossRef] [PubMed]
- Lagerstedt, K.; Karsten, S.L.; Carlberg, B.M.; Kleijer, W.J.; Tönnesen, T.; Pettersson, U.; Bondeson, M.L. Double-strand breaks may initiate the inversion mutation causing the Hunter syndrome. Hum. Mol. Genet. 1997, 6, 627–633. [Google Scholar] [CrossRef] [Green Version]
- Tajima, G.; Sakura, N.; Kosuga, M.; Okuyama, T.; Kobayashi, M. Effects of idursulfase enzyme replacement therapy for Mucopolysaccharidosis type II when started in early infancy: Comparison in two siblings. Mol. Genet. Metab. 2013, 108, 172–177. [Google Scholar] [CrossRef]
- Ogawa, T.; Tomatsu, S.; Fukuda, S.; Yamagishi, A.; Rezvi, G.M.; Sukegawa, K.; Kondo, N.; Suzuki, Y.; Shimozawa, N.; Orü, T. Mucopolysaccharidosis IVA: Screening and identification of mutations of the N-acetylgalactosamine-6-sulfate sulfatase gene. Hum. Mol. Genet. 1995, 4, 341–349. [Google Scholar] [CrossRef]
- Chien, Y.H.; Lee, N.C.; Chen, P.W.; Yeh, H.Y.; Gelb, M.H.; Chiu, P.C.; Chu, S.Y.; Lee, C.H.; Lee, A.R.; Hwu, W.L. Newborn screening for Morquio disease and other lysosomal storage diseases: Results from the 8-plex assay for 70,000 newborns. Orphanet J. Rare Dis. 2020, 15, 38. [Google Scholar] [CrossRef]
- Yamada, N.; Fukuda, S.; Tomatsu, S.; Muller, V.; Hopwood, J.J.; Nelson, J.; Kato, Z.; Yamagishi, A.; Sukegawa, K.; Kondo, N.; et al. Molecular heterogeneity in mucopolysaccharidosis IVA in Australia and Northern Ireland: Nine novel mutations including T312S, a common allele that confers a mild phenotype. Hum. Mutat. 1998, 11, 202–208. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.M.; Nishioka, T.; Gutierrez, M.A.; Peña, O.M.; TrandaFirescu, G.G.; Lopez, P.; Yamaguchi, S.; Noguchi, A.; Orii, T. Mutation and polymorphism spectrum of the GALNS gene in mucopolysaccharidosis IVA (Morquio A). Hum. Mutat. 2005, 26, 500–512. [Google Scholar] [CrossRef]
- Rivera-Colón, Y.; Schutsky, E.K.; Kita, A.Z.; Garman, S.C. The structure of human GALNS reveals the molecular basis for mucopolysaccharidosis IV A. J. Mol. Biol. 2012, 423, 736–751. [Google Scholar] [CrossRef] [Green Version]
- Tomatsu, S.; Filocamo, M.; Orii, K.O.; Sly, W.S.; Gutierrez, M.A.; Nishioka, T.; Serrato, O.P.; Di Natale, P.; Montaño, A.M.; Yamaguchi, S.; et al. Mucopolysaccharidosis IVA (Morquio A): Identification of novel common mutations in the N-acetylgalactosamine-6-sulfate sulfatase (GALNS) gene in Italian patients. Hum. Mutat. 2004, 24, 187–188. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Fukuda, S.; Cooper, A.; Wraith, J.E.; Ferreira, P.; Di Natale, P.; Tortora, P.; Fujimoto, A.; Kato, Z.; Yamada, N.; et al. Fourteen novel mucopolysaccharidosis IVA producing mutations in GALNS gene. Hum. Mutat. 1997, 10, 368–375. [Google Scholar] [CrossRef]
- Morrone, A.; Tylee, K.L.; Al-Sayed, M.; Brusius-Facchin, A.C.; Caciotti, A.; Church, H.J.; Coll, M.J.; Davidson, K.; Fietz, M.J.; Gort, L.; et al. Molecular testing of 163 patients with Morquio A (Mucopolysaccharidosis IVA) identifies 39 novel GALNS mutations. Mol. Genet. Metab. 2014, 112, 160–170. [Google Scholar] [CrossRef]
- Cozma, C.; Eichler, S.; Wittmann, G.; Flores Bonet, A.; Kramp, G.J.; Giese, A.K.; Rolfs, A. Diagnosis of Morquio Syndrome in Dried Blood Spots Based on a New MRM-MS Assay. PLoS ONE 2015, 10, e0131228. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhang, W.; Wang, Y.; Meng, Y.; Su, L.; Shi, H.; Huang, S. Mucopolysaccharidosis IVA mutations in Chinese patients: 16 novel mutations. J. Hum. Genet. 2010, 55, 534–540. [Google Scholar] [CrossRef] [Green Version]
- Tomatsu, S.; Nishioka, T.; Montaño, A.M.; Gutierrez, M.A.; Pena, O.S.; Orii, K.O.; Sly, W.S.; Yamaguchi, S.; Orii, T.; Paschke, E.; et al. Mucopolysaccharidosis IVA: Identification of mutations and methylation study in GALNS gene. J. Med. Genet. 2004, 41, e98. [Google Scholar] [CrossRef]
- Bunge, S.; Kleijer, W.J.; Tylki-Szymanska, A.; Steglich, C.; Beck, M.; Tomatsu, S.; Fukuda, S.; Poorthuis, B.J.; Czartoryska, B.; Orii, T.; et al. Identification of 31 novel mutations in the N-acetylgalactosamine-6-sulfatase gene reveals excessive allelic heterogeneity among patients with Morquio A syndrome. Hum. Mutat. 1997, 10, 223–232. [Google Scholar] [CrossRef]
- Yang, C.F.; Tsai, F.J.; Lin, S.P.; Lee, C.C.; Wu, J.Y. A novel in-frame deletion mutation (c106-111del) identified in a Taiwan Chinese patient with type IVA mucopolysaccharidosis. Hum. Mutat. 2001, 18, 254. [Google Scholar] [CrossRef]
- Lin, W.D.; Lin, S.P.; Wang, C.H.; Hwu, W.L.; Chuang, C.K.; Lin, S.J.; Tsai, Y.; Chen, C.P.; Tsai, F.J. Genetic analysis of mucopolysaccharidosis type VI in Taiwanese patients. Clin. Chim. Acta 2008, 394, 89–93. [Google Scholar] [CrossRef]
- Garrido, E.; Cormand, B.; Hopwood, J.J.; Chabás, A.; Grinberg, D.; Vilageliu, L. Maroteaux-Lamy syndrome: Functional characterization of pathogenic mutations and polymorphisms in the arylsulfatase B gene. Mol. Genet. Metab. 2008, 94, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Voskoboeva, E.; Isbrandt, D.; von Figura, K.; Krasnopolskaya, X.; Peters, C. Four novel mutant alleles of the arylsulfatase B gene in two patients with intermediate form of mucopolysaccharidosis VI (Maroteaux-Lamy syndrome). Hum. Genet. 1994, 93, 259–264. [Google Scholar] [CrossRef]
- Kantaputra, P.N.; Kayserili, H.; Guven, Y.; Kantaputra, W.; Balci, M.C.; Tanpaiboon, P.; Tananuvat, N.; Uttarilli, A.; Dalal, A. Clinical manifestations of 17 patients affected with mucopolysaccharidosis type VI and eight novel ARSB mutations. Am. J. Med. Genet. A 2014, 164A, 1443–1453. [Google Scholar] [CrossRef]
- Yang, C.F.; Wu, J.Y.; Lin, S.P.; Tsai, F.J. Mucopolysaccharidosis type VI: Report of two Taiwanese patients and identification of one novel mutation. J. Formos. Med. Assoc. 2001, 100, 820–823. [Google Scholar]
- Zanetti, A.; D’Avanzo, F.; Rigon, L.; Rampazzo, A.; Concolino, D.; Barone, R.; Volpi, N.; Santoro, L.; Lualdi, S.; Bertola, F.; et al. Molecular diagnosis of patients affected by mucopolysaccharidosis: A multicenter study. Eur. J. Pediatr. 2019, 178, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Jurecka, A.; Zakharova, E.; Cimbalistiene, L.; Gusina, N.; Malinova, V.; Różdżyńska-Świątkowska, A.; Golda, A.; Kulpanovich, A.; KaldenovnaAbdilova, G.; Voskoboeva, E.; et al. Mucopolysaccharidosis type VI in Russia, Kazakhstan, and Central and Eastern Europe. Pediatr. Int. 2014, 56, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.J.; Liao, H.C.; Gelb, M.H.; Chuang, C.K.; Liu, M.Y.; Chen, H.J.; Kao, S.M.; Lin, H.Y.; Huang, Y.H.; Kumar, A.B.; et al. Taiwan NationalNewborn Screening Program by Tandem Mass Spectrometry for MucopolysaccharidosesTypes I, II, and VI. J. Pediatr. 2019, 205, 176–182. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.M.; Oguma, T.; Dung, V.C.; Oikawa, H.; de Carvalho, T.G.; Gutiérrez, M.L.; Yamaguchi, S.; Suzuki, Y.; Fukushi, M.; et al. Dermatan sulfate and heparan sulfate as a biomarker for mucopolysaccharidosis I. J. Inherit. Metab. Dis. 2010, 33, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Menkovic, I.; Lavoie, P.; Boutin, M.; Auray-Blais, C. Distribution of heparan sulfate and dermatan sulfate in mucopolysaccharidosis type II mouse tissues pre- and post-enzyme-replacement therapy determined by UPLC-MS/MS. Bioanalysis 2019, 11, 727–740. [Google Scholar] [CrossRef]
- Stapleton, M.; Kubaski, F.; Mason, R.W.; Shintaku, H.; Kobayashi, H.; Yamaguchi, S.; Taketani, T.; Suzuki, Y.; Orii, K.; Orii, T.; et al. Newborn screening for mucopolysaccharidoses: Measurement of glycosaminoglycans by LC-MS/MS. Mol. Genet. Metab. Rep. 2020, 22, 100563. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Infant No. | Sex | DMB/Cre. Ratio | DS | HS | KS | Enzyme Activity | Nucleotide Variations | Protein Variations | ACMG Classification |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Female | 37.86 | 0.27 | 0.10 | 5.69 | 1.20 | [c.343G/A]+ [c.2T/C] [19] | [p.D115N]+[p.M1T] | Uncertain SignificanceLP(VUS with minor Pathogenic evidence)+ Pathogenic |
| 2 | Male | 34.02 | 0.11 | 0.17 | 0.53 | 1.50 | [WT/c.1395delC]+ [WT/c.1081G>A] [20]+ [WT/c.1816G>T] | [p.G466AfsTer59]+ [p.A361T]+[p.V606L] | Pathogenic + Benign + Uncertain SignificanceLP(VUS with minor Pathogenic evidence) |
| 3 | Male | 24.99 | 0.11 | 0.17 | 0.83 | 2.10 | [WT/c.355G>T]+ [WT/c.617C>T] | [p.D119Y]+[p.S206L] | Uncertain SignificanceLP(VUS with minor Pathogenic evidence) + Uncertain SignificanceLP(VUS with minor Pathogenic evidence) |
| 4 | Male | 24.89 | 0.05 | 0.29 | 0.16 | 3.88 | [WT/c.179A>C] | [p.Q60P] | Uncertain SignificanceLP(VUS with minor Pathogenic evidence) |
| 5 | Female | 36.34 | 0.01 | 0.04 | 2.57 | 3.82 | [−] | [−] | - |
| 6 | Male | 30.99 | 0.04 | 0.06 | 0.10 | 9.62 | [−] | [−] | - |
| 7 | Female | 26.82 | 0.20 | 0.01 | 6.49 | 21.60 | [c.76G>A]+ [c.911delT] | [p.A26T ]+ [p.V304Gfs*13] | Uncertain SignificanceLP(VUS with minor Pathogenic evidence) + Pathogenic |
| 8 | Male | 17.97 | 21.18 | 11.34 | 2.0 | 1.60 | [c.300-3C>G] [21]+ [c.1874A>C] | [−]+[p.Y625S] | Uncertain Significance + Likely Pathogenic |
| 9 | Female | 33.61 | 5.91 | 9.38 | 0.28 | 0.46 | [c.300-3C>G] [21]+ [c.1874A>C] | [−]+[p.Y625S] | Uncertain Significance + Likely Pathogenic |
| 10 | Female | 204.70 | 176.96 | 28.51 | 1.78 | 0.70 | [c.1037T>G] [21]+ [c.1091C>T] [22] | [p.L346R]+[p.364M] | Pathogenic + Likely Pathogenic |
| 11 | Female | 239.25 | 296.10 | 33.55 | 1.37 | 0.80 | [c.1037T>G] [21]+ [c.1091C>T] [22] | [p.L346R]+[p.364M] | Pathogenic + Likely Pathogenic |
| 12 | Male | 18.53 | 0.09 | 0.17 | 0.78 | 0.95 | [WT/c.1093C>G]+ [WT/c.1463G>C]+ [WT/c.1828+5G>A] | [p.L365V]+[p.R488P]+ [−] | Uncertain Significance + Uncertain Significance + Uncertain Significance |
| Infant No. | Sex | DMB/Cre. Ratio | DS | HS | KS | Enzyme Activity | Nucleotide Variations | Protein Variations | ACMG Classification |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Male | 78.58 | 11.59 | 12.36 | 0.25 | 0.83 | [c.254C>T] [18] | [p.A85V] | Likely Pathogenic |
| 2-23 | Male | 30.3 ± 10.8 | 0.09 ± 0.06 | 0.1 ± 0.09 | 0.36 ± 0.26 | 25 ± 10.8 | [c.1499C>T] [18] | [p.T500I] | Benign |
| 24–31 | Male | 29 ± 11.1 | 0.16 ± 0.1 | 0.06 ± 0.04 | 0.79 ± 0.6 | 23.9 ± 11.1 | [c.1478G>A] [18] | [p.R493H] | |
| 32 | Male | 27.69 | 0.18 | 0.2 | 0.08 | 5.93 | [c.1513T>C] | [p.F505L] | Likely Pathogenic |
| 33 | Male | 46.09 | 0.17 | 0.53 | 1.32 | 17.68 | [c.805G>A] | [p.D269N] | |
| 34 | Male | 73.20 | 0.21 | 3.69 | 4.03 | 10.36 | [c.659T>C] | [p.F220S] | |
| 35–40 | Male | 18.1 ± 6.4 | 0.06 ± 0.04 | 0.14 ± 0.04 | 4.92 ± 1.9 | 26.1 ± 9.9 | [c.301C>T] [23] | [p.R101C] | Benign |
| 41 | Male | 69.67 | 0.08 | 0.04 | 3.47 | 9.20 | [c.890G>A] [18] | [p.R297H] | Likely Pathogenic |
| 42–43 | Male | 77.3/16.73 | 7.39/5.48 | 1.83/24.9 | 6.13/0.58 | 0.20/0.41 | [c.817C>T] [18] | [p.R273W] | Likely Pathogenic |
| 44 | Male | 63.82 | 0.38 | 1.46 | 6.35 | 7.80 | [c.589C>T] [18] | [p.P197S] | Likely Pathogenic |
| 45–46 | Male | 59.46/70.9 | 1.18/21.21 | 8.22/12.06 | 1.49/6.47 | 1.26/0.40 | [c.1025A>G] [18] | [p.H342R] | Likely Pathogenic |
| 47 | Male | 113.95 | 15.62 | 103.44 | 1.42 | 0.32 | [c.311A>T] [18] | [p.D104V] | Likely Pathogenic |
| 48–51 | Male | 27.9 ± 5.5 | 0.03 | 0.12 ± 0.05 | 0.22 ± 0.02 | 8.7 ± 8 | [c.851C>T] [18] | [p.P284L] | Uncertain Significance |
| 52 | Male | 153.16 | 21.4 | 30.01 | 0.11 | 0.27 | [c.1400C>T] [18] | [p.P467L] | Likely Pathogenic |
| 53 | Male | 55.87 | 0.14 | 0.38 | 0.32 | 2.93 | [c.851C>T] [18]+ [c.1180+184T>C] [18] | [p.P284L]+[−] | Uncertain Significance + [−] |
| 54–55 | Male | 38.73/26.2 | 0.01/0.08 | 0.75/0.38 | 0.03/0.59 | 16.27/23.78 | [c.142C>T] [18] | [p.R48C] | Likely Pathogenic |
| 56 | Male | 177.96 | 30.77 | 203.35 | 0.31 | 0.99 | [c.1007-1666_c.1180+2113 delinsTT] [18] | Pathogenic | |
| 57 | Male | 104.84 | 11.93 | 175.36 | 0.15 | 0.13 | IDS inversion [24,25] | ||
| 58–186 | Male | 29.7 ± 12.5 | 0.16 ± 0.15 | 0.15 ± 0.13 | 0.43 ± 0.29 | 5.5 ± 4.8 | [c.103+34_56dup] [18]+ [c.684A>G] [18]+ [c.851C>T] [18]+ [c.1180+184T>C] [18] | [−]+[p.Pro228=]+ [p.P284L] +[−] | Uncertain Significance + Benign + Uncertain Significance + [−] |
| Infant No. | Sex | DMB/Cre. Ratio | DS | HS | KS | Enzyme Activity | Nucleotide Variations | Protein Variations | ACMG Classification |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Male | 86.81 | 0.12 | 0.01 | 10.64 | 0.00 | [WT/c.953T>G] [26] | [p.M318R] | Likely Pathogenic |
| 2–5 | Male | 33.6 ± 7.8 | 0.17 ± 0.1 | 0.09 ± 0.07 | 2.01 ± 1.5 | 1.63 ± 1.4 | [WT/c.953T>G] [26] | [p.M318R] | Likely Pathogenic |
| 6–7 | Male, Female | 33.37 ± 11.7 | 0.06 ± 0.01 | 0.41 ± 0.5 | 0.81 ± 0.2 | 1.75 ± 0.07 | [WT/c.887C>T] [27]+ [WT/c.953T>G] [26] | [p.A296V]+ [p.M318R] | Uncertain Significance + Likely Pathogenic |
| 8–9 | Male | 39.61 ± 0.2 | 0.12 ± 0.05 | 0.03 ± 0.01 | 0.6 ± 0.4 | 3.0 ± 0.1 | [WT/c.782T>C] | [p.I261T] | Likely Pathogenic |
| 10–11 | Female | 33.67 ± 3.9 | 0.19 ± 0.1 | 0.02 ± 0.01 | 0.56 ± 0.06 | 0.5 ± 0.3 | [WT/c.857C>T] [27]+ [WT/c.953T>G] [26] | [p.T286M]+ [p.M318R] | Likely Pathogenic + Likely Pathogenic |
| 12–18 | 3 Male, 4 Female | 29.9 ± 5.9 | 0.27 ± 0.08 | 0.06 ± 0.05 | 0.97 ± 0.7 | 2.0 ± 0.4 | [WT/c.857C>T] [27] | [p.T286M] | Likely Pathogenic |
| 19 | Male | 48.17 | 0.02 | 0.05 | 3.54 | 1.40 | [WT/c.857C>T] [27]+ [WT/c.1127G>A] [28,29,30] | [p.T286M]+ [p.R376Q] | Likely Pathogenic + Likely Pathogenic |
| 20 | Female | 32.67 | 0.64 | 0.04 | 0.71 | 0.40 | [WT/c.638C>T]+ [WT/c.953T>G] [26] | [p.A213V]+ [p.M318R] | Likely Pathogenic + Likely Pathogenic |
| 21 | Male | 38.14 | 0.17 | 0.02 | 0.06 | 1.50 | [c.857C>T] [27] | [p.T286M] | Likely Pathogenic |
| 22 | Female | 34.21 | 0.13 | 0.36 | 8.18 | 1.70 | [WT/c.319G>A] [31] | [p.A107T] | Likely Pathogenic |
| 23 | Male | 15.32 | 0.04 | 0.46 | 0.87 | 2.40 | [WT/c.857C>T] [27]+ [c.*3C>G] | [p.T286M]+[−] | Likely Pathogenic + Uncertain Significance |
| 24–25 | Female | 17.43 ± 0.1 | 0.24 ± 0.1 | 0.21 ± 0.1 | 0.78 ± 0.03 | 1.55 ± 1.0 | [WT/c.374C>T] [32] | [p.P125L] | Likely Pathogenic |
| 26 | Male | 19.02 | 0.03 | 0.16 | 0.90 | 0.80 | [WT/c.131G>T]+ [WT/c.985C>A] | [p.G44V]+ [p.H329N] | Likely Pathogenic + Likely Pathogenic |
| 27–31 | Male | 24.8 ± 7.3 | 0.32 ± 0.2 | 0.07 ± 0.03 | 0.41 ± 0.3 | 3.15 ± 1.0 | [−] | [−] | - |
| 32 | Male | 41.54 | 0.07 | 0.28 | 0.67 | 3.50 | [WT/c.704C>A] [33]+ [WT/c.887C>T] [27] | [p.T235K]+ [p.A296V] | Likely Pathogenic + Uncertain Significance |
| 33 | Female | 21.12 | 0.08 | 0.17 | 0.44 | 4.10 | [WT/c.1496C>T] [34] | [p.P499L] | Likely Pathogenic |
| 34 | Female | 56.83 | 0.11 | 0.05 | 0.23 | 3.80 | [WT/c.425A>G] [35] | [p.H142R] | Likely Pathogenic |
| 35 | Female | 33.87 | 0.14 | 0.16 | 0.43 | 1.40 | [WT/c.706C>T] | [p.H236Y] | Likely Pathogenic |
| 36 | Male | 49.59 | 0.04 | 0.01 | 16.82 | 1.50 | [WT/c.139G>A] [36,37] | [p.G47R] | Pathogenic |
| 37 | Male | 18.45 | 0.04 | 0.03 | 5.15 | 3.60 | [WT/c.887C>T] [27] | [p.A296V] | Uncertain Significance |
| 38 | Female | 33.45 | 0.04 | 0.07 | 9.74 | 0.60 | [WT/c.190_191delinsAT] [27]+[WT/c.1108C>T] [27] | [p.A64I]+ [p.P370S] | Likely Pathogenic + Uncertain Significance |
| 39 | Male | 50.38 | 0.05 | 0.09 | 0.59 | 2.10 | [WT/c.106_111delCTGCTC] [38] | [p.L36_L37del] | Likely Pathogenic |
| 40 | Male | 7.80 | 0.02 | 0.03 | 4.80 | 2.00 | [WT/c.190_191delinsAT] [27] | [p.A64I] | Uncertain Significance |
| 41 | Male | 32.38 | 0.03 | 0.08 | 1.10 | 2.10 | [WT/c.1108C>T] [27] | [p.P370S] | Uncertain Significance |
| 42 | Female | 12.76 | 0.02 | 0.23 | 5.08 | 4.40 | [WT/c.1019G>A] [35,36] | [p.G340D] | Pathogenic |
| Infant No. | Sex | DMB/Cre. Ratio | DS | HS | KS | Enzyme Activity | Nucleotide Variations | Protein Variations | ACMG Classification |
|---|---|---|---|---|---|---|---|---|---|
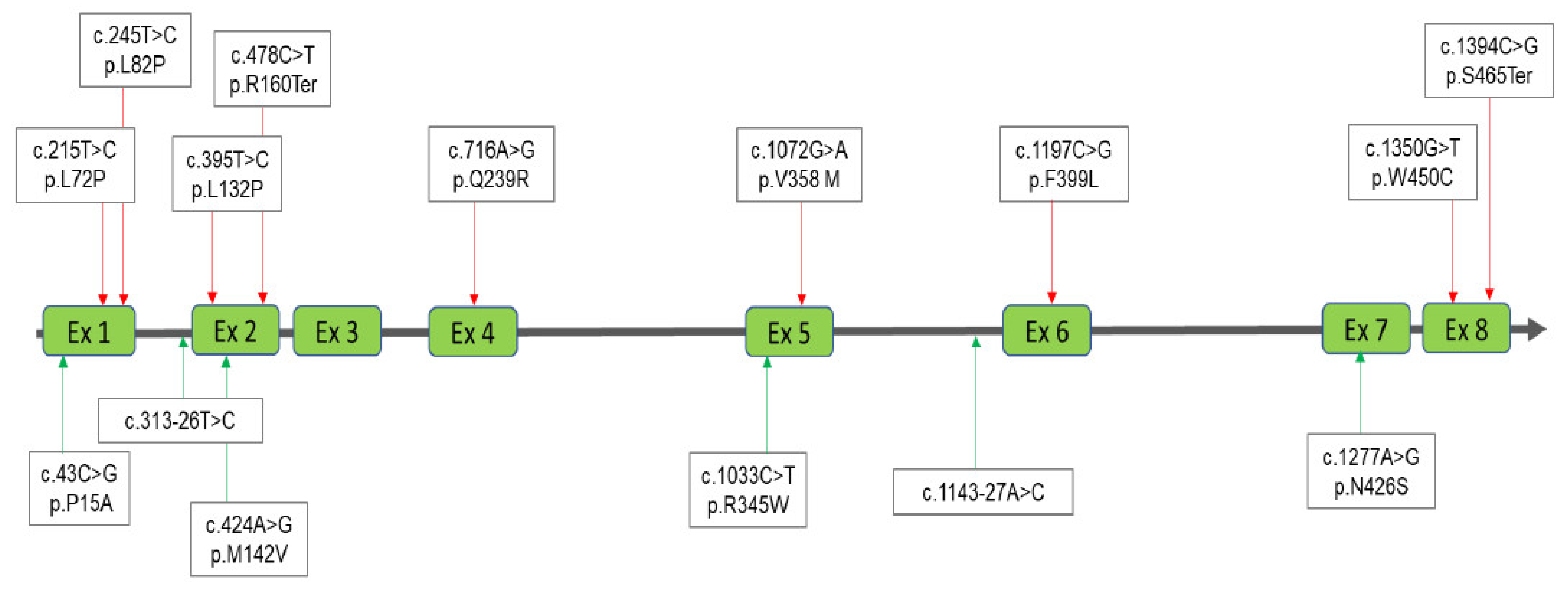
| 1 | Male | 44.41 | 0.49 | 0.10 | 0.68 | 16.30 | [WT/c.1394C>G] [39] | [p.S465Ter] | Pathogenic |
| 2-3 | Female | 22.3 ± 19 | 0.06 ± 0.04 | 0.06 ± 0.05 | 2.5 ± 1.4 | 13.7 ± 8.5 | [WT/c.716A>G] [39] | [p.Q239R] | Likely Pathogenic |
| 4 | Female | 45.17 | 0.03 | 0.01 | 0.08 | 5.00 | [WT/c.313-26T>C]+ [WT/c.1143-27A>C]+ [WT/c.1072G>A] [40] | [−]+[−]+[p.V358 M] | Benign + Benign + Benign |
| 5–6 | Male | 38.2 ± 4 | 0.33 ± 0.4 | 0.03 ± 0.01 | 0.29 ± 0.07 | 18.5 ± 11.3 | [−] | [−] | - |
| 7 | Male | 21.48 | 0.07 | 0.11 | 0.17 | 13.60 | [WT/c.424A>G]+ [c.1072G>A] [40] | [p.M142V]+ [p.V358M] | Likely Pathogenic + Benign |
| 8 | Male | 37.54 | 0.26 | 0.21 | 1.65 | 1.10 | [WT/c.478C>T] [41] | [p.R160Ter] | Pathogenic |
| 9 | Male | 14.30 | 0.10 | 0.15 | 0.16 | 23.00 | [WT/c.1350G>T] [42] | [p.W450C] | Pathogenic |
| 10 | Female | 26.97 | 0.15 | 0.09 | 1.22 | 20.80 | [WT/c.395T>C] [39] | [p.L132P] | Likely Pathogenic |
| 11 | Male | 5.50 | 0.58 | 0.04 | 0.87 | 11.00 | [WT/c.1277A>G] | [p.N426S] | Likely Pathogenic |
| 12 | Male | 8.49 | 0.19 | 0.11 | 0.37 | 23.00 | [WT/c.1197C>G] [43] | [ p.F399L ] | Pathogenic |
| 13 | Female | 28.02 | 0.36 | 0.11 | 0.85 | 7.50 | [WT/c.1033C>T] | [p.R345W] | Likely Pathogenic |
| 14 | Female | 29.59 | 0.34 | 0.31 | 0.75 | 17.50 | [WT/c.43C>G]+ [WT/c.245T>C] [44] | [p.P15A]+[p.L82P] | Uncertain Significance LP(VUS with minor Pathogenic evidence) + Likely Pathogenic |
| 15 | Male | 26.23 | 0.03 | 0.36 | 0.98 | 5.00 | [WT/c.215T>C] [45] | [p.L72P] | Likely Pathogenic |
| 1 | Male | 44.41 | 0.49 | 0.10 | 0.68 | 16.30 | [WT/c.1394C>G] [39] | [p.S465Ter] | Pathogenic |
| 2–3 | Female | 22.3 ± 19 | 0.06 ± 0.04 | 0.06 ± 0.05 | 2.5 ± 1.4 | 13.7 ± 8.5 | [WT/c.716A>G] [39] | [p.Q239R] | Likely Pathogenic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chuang, C.-K.; Lee, C.-L.; Tu, R.-Y.; Lo, Y.-T.; Sisca, F.; Chang, Y.-H.; Liu, M.-Y.; Liu, H.-Y.; Chen, H.-J.; Kao, S.-M.; et al. Nationwide Newborn Screening Program for Mucopolysaccharidoses in Taiwan and an Update of the “Gold Standard” Criteria Required to Make a Confirmatory Diagnosis. Diagnostics 2021, 11, 1583. https://doi.org/10.3390/diagnostics11091583
Chuang C-K, Lee C-L, Tu R-Y, Lo Y-T, Sisca F, Chang Y-H, Liu M-Y, Liu H-Y, Chen H-J, Kao S-M, et al. Nationwide Newborn Screening Program for Mucopolysaccharidoses in Taiwan and an Update of the “Gold Standard” Criteria Required to Make a Confirmatory Diagnosis. Diagnostics. 2021; 11(9):1583. https://doi.org/10.3390/diagnostics11091583
Chicago/Turabian StyleChuang, Chih-Kuang, Chung-Lin Lee, Ru-Yi Tu, Yun-Ting Lo, Fran Sisca, Ya-Hui Chang, Mei-Ying Liu, Hsin-Yun Liu, Hsiao-Jan Chen, Shu-Min Kao, and et al. 2021. "Nationwide Newborn Screening Program for Mucopolysaccharidoses in Taiwan and an Update of the “Gold Standard” Criteria Required to Make a Confirmatory Diagnosis" Diagnostics 11, no. 9: 1583. https://doi.org/10.3390/diagnostics11091583
APA StyleChuang, C.-K., Lee, C.-L., Tu, R.-Y., Lo, Y.-T., Sisca, F., Chang, Y.-H., Liu, M.-Y., Liu, H.-Y., Chen, H.-J., Kao, S.-M., Wang, L.-Y., Ho, H.-J., Lin, H.-Y., & Lin, S.-P. (2021). Nationwide Newborn Screening Program for Mucopolysaccharidoses in Taiwan and an Update of the “Gold Standard” Criteria Required to Make a Confirmatory Diagnosis. Diagnostics, 11(9), 1583. https://doi.org/10.3390/diagnostics11091583